

Supplemental Digital Content is available in the text
Keywords: autoimmune pancreatitis, glucocorticoids, IgG4, IgG4-related disease, rituximab
Abstract
To review the reported evidence on the therapeutic management of IgG4-related disease (IgG4-RD) in clinical practice.
A systematic search of the literature was conducted. The primary outcome measured was the rate of efficacy of first-line therapeutic approaches. Secondary outcomes measured included the rate of disease relapse, the outcome of untreated patients, the rate of patients without drug therapy at the end of follow-up, the rate of side effects, and mortality. The MOOSE, AHRQ, STROBE, and GRACE recommendations/statements were followed.
The results of the systematic search strategy yielded 62 studies that included a total of 3034 patients. Complete information about first-line therapeutic regimens was detailed in 1952 patients, including glucocorticoid-based regimens in 1437 (74%), drug-free regimens in 213 (11%), and other therapies in 38 (2%). No therapy (wait and see management) was reported in 264 (13%) patients. The efficacy of monotherapy with glucocorticoids was specified in 1220 patients, of whom 97% had a therapeutic response. Relapses, however, were reported in 464/1395 (33%) patients despite typically short follow-up periods. Therapeutic efficacy was reported in 219/231 (95%) of relapses treated with glucocorticoids, 56/69 (81%) of those treated with azathioprine, 16/22 (72%) of those treated with other immunosuppressive agents, and in the 9 cases treated with rituximab (100%). In 14 studies, the authors detailed the outcome of 159/246 patients with wait-and-see management; spontaneous improvement or resolution was reported in 68 (43%) cases. Wide heterogeneity was observed with respect to the first-line therapeutic approaches used for the different organ-specific disease subsets, including significant differences in the mean dose of glucocorticoids used.
Nearly 70% of reported IgG4-RD patients are treated with oral glucocorticoids in monotherapy. However, the therapeutic management is heavily influenced by geographical, epidemiological, and clinical factors, especially with respect to the predominant organ affected. The frequency of glucocorticoid failure to induce sustained remissions both during and after treatment and the assessment of glucocorticoid toxicity in IgG4-RD require further study.
1. Introduction
IgG4-related disease (IgG4-RD) is an immune-mediated systemic disease first described in Japan at the beginning of this century.[1] The most-common clinical presentation is the development of a mass lesion or unexplained enlargement of one or more organs. Although IgG4-RD usually presents with a subacute onset, the disease leads to progressive organ failure and even death in some patients. Multiorgan disease is easier to identify at diagnosis but may evolve metachronously, with 1 organ at a time being added over months to years.[1] The organs most frequently involved are the pancreas and biliary tract, the salivary and lachrymal glands, the kidneys, the thyroid gland, the lungs, and the aorta.[2] Diagnosis relies on the coexistence of various clinical, laboratory, radiological, and histopathological findings, although none is pathognomonic in itself.[3]
The optimum therapeutic management of IgG4-RD has not yet been established.[4] In spite of an explosion in observational studies in the last decade, no randomized controlled studies on the treatment of IgG4-RD are yet available. Therefore, in the absence of high-quality scientific evidence, a systematic approach using meta-analyses of observational studies is one of the few ways to assess the degree of efficacy of the therapeutic options currently used in clinical practice. This information may be useful for the design of future prospective/controlled therapeutic trials in IgG4-RD patients.
The objective of this review was to summarize reported evidence on the therapeutic management of IgG4-RD in clinical practice with the aim of providing physicians with the best available therapeutic evidence, tailored when possible to the possible clinical scenarios with which IgG4-RD present.
2. Methods
2.1. Data sources and searches
A systematic search of the literature was conducted on MEDLINE and EMBASE and using the OVID interface to search for evidence-based medicine reviews in the Cochrane Databases of systematic reviews and controlled trials. We also searched for unpublished trials using ClinicalTrials.gov (last day of access to the databases, October 31, 2014). Due to the lack of specific Medical Subject Headings for IgG4-RD, a text-word search was conducted using the free text “IgG4-related disease” as the currently most-accepted term.[1] No restrictions were placed on language or type of publication. We manually searched reference lists of selected articles for relevant citations that our searches missed. We followed the AMSTAR recommendations on data sources and searches.[5]
2.2. Study selection
Study selection was made by independent review. Two independent reviewers (MRC and XB) examined abstracts retrieved by the literature search for potentially eligible articles. Studies marked for possible inclusion by either reviewer underwent dual, independent full-text review. Differences between reviewers were resolved by consensus. Criteria for inclusion were the following: inclusion of at least 5 patients, enrollment of adults (aged ≥18 years), diagnosis of IgG4-RD according to current classification criteria sets,[6] and availability of data on at least one of the following outcomes: therapeutic efficacy; disease outcomes (relapses, maintenance drug therapy, and/or death); and/or adverse drug effects.
2.3. Data extraction and quality assessment
A data extraction form was developed by PBZ prior to manuscript review to gather relevant data from each article. All data extractions were reviewed for completeness and accuracy by PBZ, XB, and MRC. Study design was classified according to the STROBE recommendations for observational studies (case–control, cross-sectional, and cohort studies).[7] Variables collected for each study selected included the mean age of the cohort, gender frequencies, country, inclusion criteria, first-line therapeutic approaches, drug therapies (types, dose, length, and adverse effects), therapeutic efficacy, relapses and therapeutic management, side effects, mean time of follow-up, therapies at the last visit, and death. The primary outcome measured was the rate of efficacy of first-line therapeutic approaches. Secondary outcomes measured included the rate of disease relapse, the outcome of untreated patients, the rate of patients without drug therapy at the end of follow-up, the rate of side effects, and mortality. Relapses were defined as a disease exacerbation following a period of improvement – whether or not the treatment response was complete.
For quality assessment, a prespecified study protocol was developed by BK and XB prior to the literature review, according to the MOOSE,[8] AHRQ,[9] STROBE,[7] and GRACE[10] recommendations/statements. Supplementary Table 1 summarizes the quality domains evaluated for each study. Possible overlapping data were managed by contrasting the following variables between studies: name of authors, participating centers, number of patients, epidemiological features, type of organ involvement, period of patient recruitment, and name of the database/multicenter group. When there was more than 1 report from the same group, we included only the publication having the most detailed therapeutic information for the entire cohort.
The ethical approval was not necessary because the study uses existing data (literature review).
2.4. Statistical analysis
Descriptive data were presented as means and standard deviation (SD) for continuous variables and numbers and percentages for categorical variables. Subset statistical analyses searching for potential sources of heterogeneity in the therapeutic approaches suggested by a previous study[2] were carried out according to the geographical origin and the organ predominantly involved. The Chi-square test for contingency tables was used to compare gender, geographical origin, first-line regimens, therapeutic efficacy, relapses, side effects, mortality, and therapeutic management. Continuous outcomes such as mean age of the cohort, mean time of follow-up, and mean starting doses were compared using the nonparametric Kruskal–Wallis test. Forest plots with the odds ratios (ORs) and their 95% confidence intervals (CIs) were constructed to represent the association between study characteristics and outcomes. All significance tests were 2-tailed, and values of P < 0.05 were considered significant. All analyses were conducted by BK and NAD using the R V.3.2.3 for Windows statistical software package.
3. Results
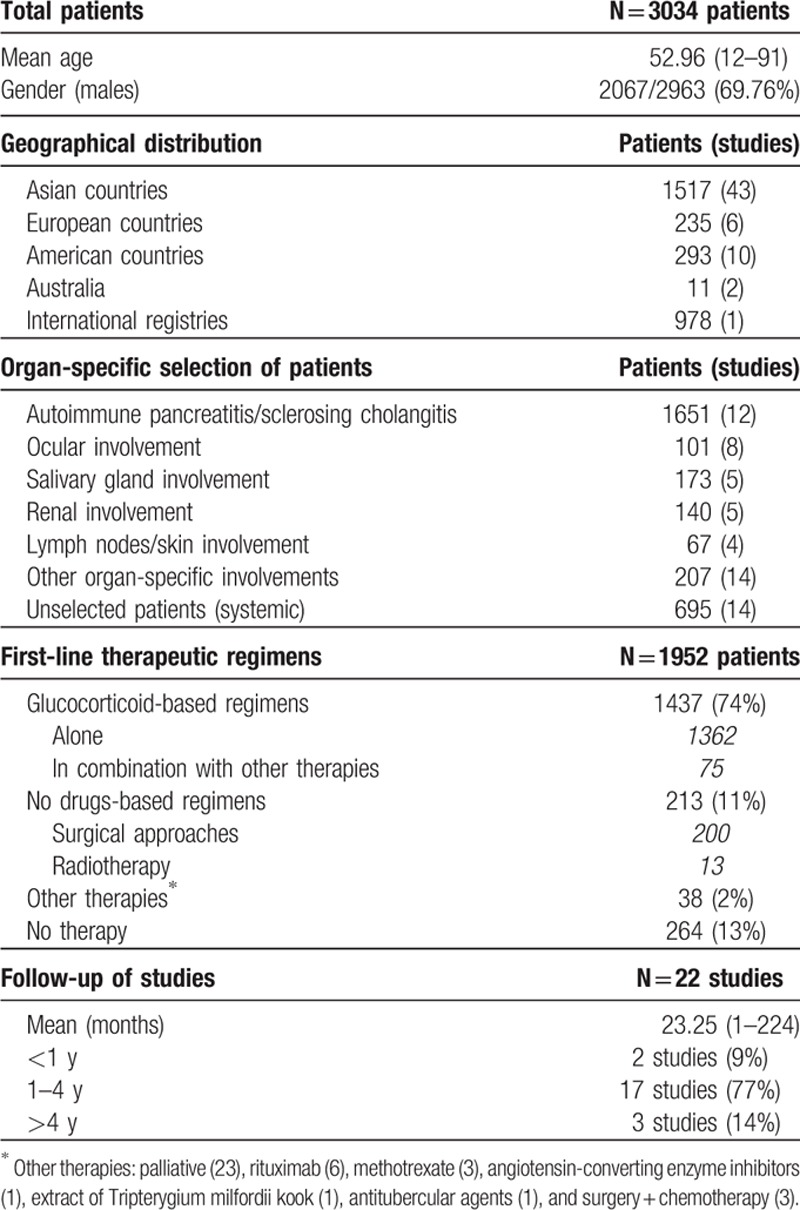
The results of the systematic search strategy are summarized in Fig. 1: 62 studies[11–72] including 3034 patients were analyzed (Supplementary Table 2). Table 1 summarizes the main patient characteristics.
Figure 1.
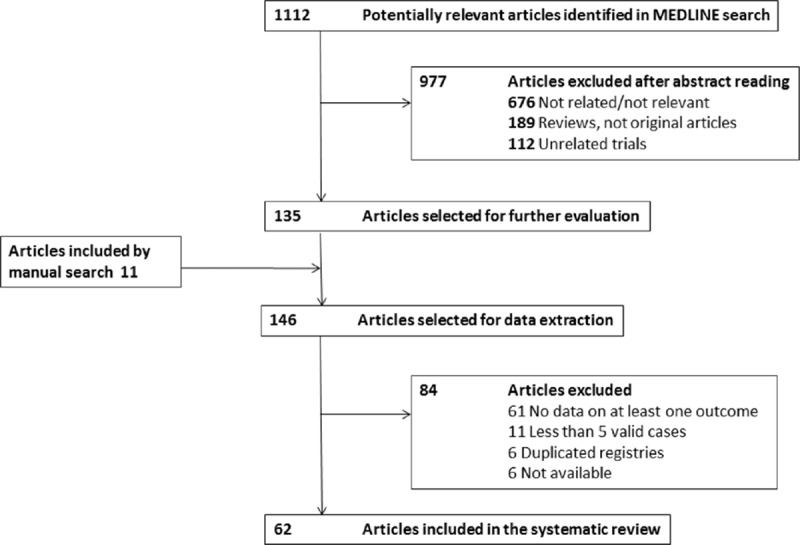
Results of the systematic search strategy.
Table 1.
Results of the systematic search strategy: 62 studies including 3034 patients.
3.1. Outcomes
3.1.1. Efficacy of first-line therapeutic approaches
Complete information about first-line therapeutic regimens was detailed in 1952 patients included in 48 studies (Table 1). First-line therapies included glucocorticoid-based regimens in 1437 (74%) patients, drug-free regimens (surgery or radiotherapy) in 213 (11%) patients, and other therapies in 38 (2%). No therapy, that is, wait-and-see management, was reported in 264 (13%) patients. Glucocorticoids were administered orally in all patients except for 10, who were treated with topical glucocorticoid preparations.
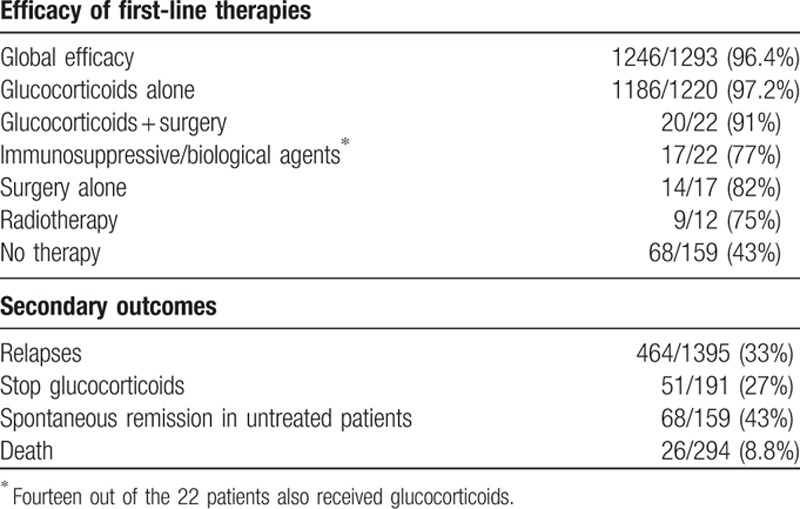
The mean starting dose was ≤0.6 mg/kg/d (equivalent to 40 mg/d ) in 24 (73%) of the 33 studies in which this information was provided. The efficacy of first-line therapies was detailed in 1293 patients, of whom 1246 (96%) were reported as having a therapeutic response. The efficacy of monotherapy with glucocorticoids was specified in 1220 patients, of whom 1186 (97%) were reported as having a therapeutic response. However, the glucocorticoids response was classified as complete in only 84/130 (65%) of the patients, partial/incomplete in 31/130 (24%), and absent (no response) in 15/130 (11%). The efficacy of other therapies was 14/17 (82%) for surgery, 20/22 (91%) for combined glucocorticoids/surgery, 17/22 (77%) for immunosuppressive/biological agents, and 9/12 (75%) for radiotherapy (Table 2).
Table 2.
Efficacy of first-line therapeutic regimens and secondary outcomes (variables were not detailed in all studies, and the prevalence of a specific feature has been stated as number of cases with that feature/number of cases in which the feature was detailed).
Therapeutic failure with first-line therapies was reported in 47 patients (4%). The response to rescue therapies was detailed in only 18 cases and included glucocorticoids in 2 (response in both), surgery in 1 (no response), rituximab in 6 (response in 4), and immunosuppressants in 9 (response in 6) (Supplementary Table 3).
3.1.2. Relapses
Relapses were reported in 464/1395 (33%) patients. The ongoing use of glucocorticoids at relapse was specified in 381 patients. Among these 381, the disease relapse occurred in 245 (64%) after the cessation of glucocorticoids. In more than one third of the patients, however (136; 36%), disease relapses occurred while patients were still receiving glucocorticoids.
Information on the therapeutic management of relapses was detailed in 378 cases and included mainly glucocorticoid use (either as a new course or as an increase/slow tapering of the ongoing dose) in 250 (66%) cases. The use of immunosuppressive agents was reported in 149 (39%) cases, principally in association with glucocorticoids (azathioprine was used in 126/149 cases). Rituximab use was reported in 9 (2%) cases. Therapeutic efficacy was reported in 219/231 (95%) of relapses treated with glucocorticoids, 56/69 (81%) of those treated with azathioprine, 16/22 (72%) of those treated with other immunosuppressive agents, and in all 9 cases treated with rituximab (100%).
3.1.3. Side effects
Only 7 studies detailed the prevalence and/or types of side effects associated with therapies.[12,18,24,49,50,66,70] Ebbo et al[18] reported side effects related to glucocorticoids in 14/21 (67%) patients. Diabetes mellitus (DM) was the most frequently reported side effect in all studies. Other reported side effects included infections (n = 3), osteonecrosis (n = 2), psychosis (n = 1), vertebral fracture (n = 1), weight gain (n = 1), and hypertension (n = 1). Side effects related to azathioprine, reported in 17/49 (35%) patients, pertained primarily to gastrointestinal intolerance.[70] There was also 1 case of azathioprine-induced pancreatitis.[70]
3.1.4. Glucocorticoid cessation
Eleven studies detailed information on how many patients treated with glucocorticoids as first-line therapy were successful in discontinuing glucocorticoids completely at the time of the last visit. Only 51 (27%) of the 191 patients in whom this outcome was reported had been able to discontinue glucocorticoids entirely by the last visit. Three studies reported somewhat higher frequencies of glucocorticoid discontinuation (>50% of patients),[19,54,73] but the relapse rates were higher in these studies compared to those in which patients remained on glucocorticoids. In summary, successful discontinuation of glucocorticoids for prolonged periods of time appears to be the exception rather than the rule for most patients with IgG4-RD.
3.1.5. Outcome of untreated patients
In 14 studies, the authors detailed the outcome in 159 out of the 246 patients who did not receive treatment. Spontaneous improvement or resolution was reported in 68 (43%) cases, but long-term follow-up of the patients were seldom reported.
3.1.6. Mortality
Information on survival was detailed in only 7 studies,[16,18,19,24,50–52] which included a total of 294 patients. After a mean follow-up of 29.2 months (range 1–224 months), mortality was reported in 26 (8.8%) patients. The main causes of death included IgG4-RD progression (n = 7, including pulmonary disease in 4, aneurysm in 1, cholangitis in 1, and renal failure in 1) and cancer (n = 7). Other causes of death included cardiovascular disease (n = 4), infection (n = 3), and other/unknown (n = 5).
3.2. Association between study characteristics and outcomes
3.2.1. Classification criteria bias
Four studies[19,22,24,49] used classification criteria that include a positive response to glucocorticoids as one of the inclusion criteria (ICDC and HiSort criteria for autoimmune pancreatitis [AIP]); the efficacy of first-line glucocorticoid-based regimens was reported in 817/830 (98%) patients (no significant difference with respect to the remaining studies).
3.2.2. Geographical bias
Table 3 summarizes the differences in the main baseline variables between Asian, American, and European studies. Epidemiologically, Asian studies included patients with a higher mean age (62 years vs 56.9 in American studies and 57.9 in European studies; P = 0.034). With respect to first-line therapeutic regimens, European studies more-frequently used glucocorticoids, American studies more-frequently used drug-free regimens, and Asian studies had the highest frequency of untreated patients (P < 0.001). With respect to outcomes (Fig. 2), American studies reported a lower efficacy (OR 0.41, 95% CI 0.20–0.87). Both American and European studies a higher frequency of relapses (OR 1.59, 95% CI 1.01–2.48 and OR 3.18, 95% CI 2.02–5.01, respectively) compared with Asian studies.
Table 3.
Differences in main baseline variables between Asian, American, and European studies.
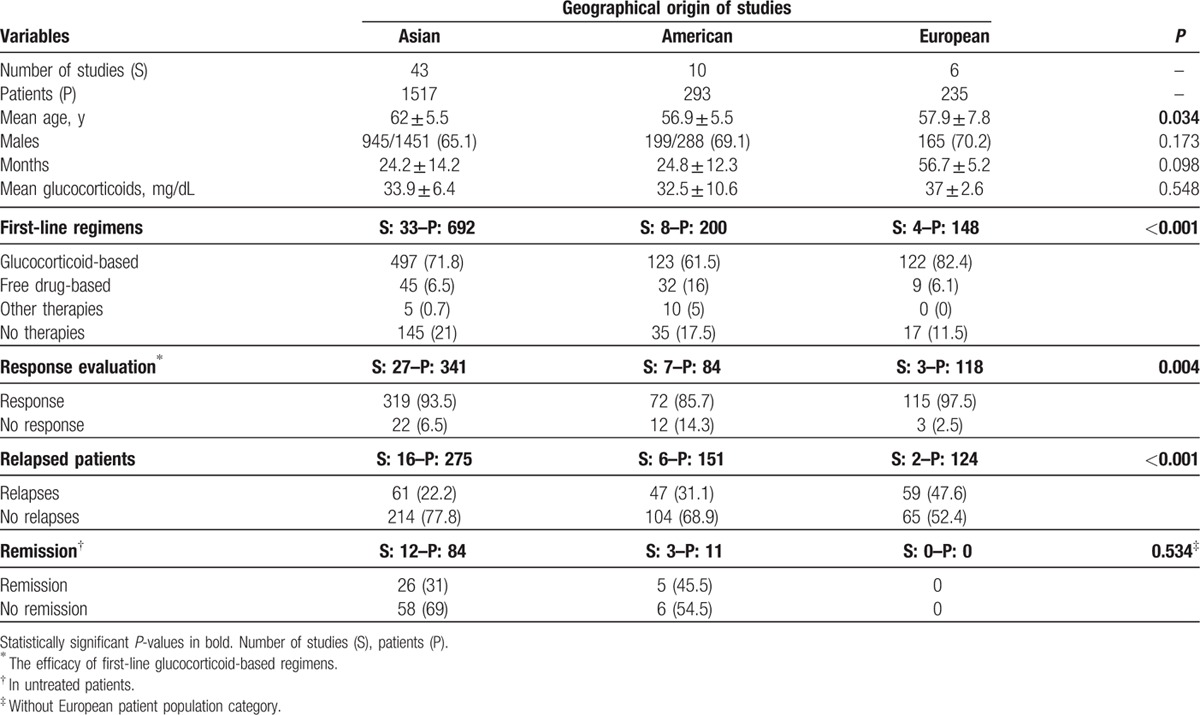
Figure 2.
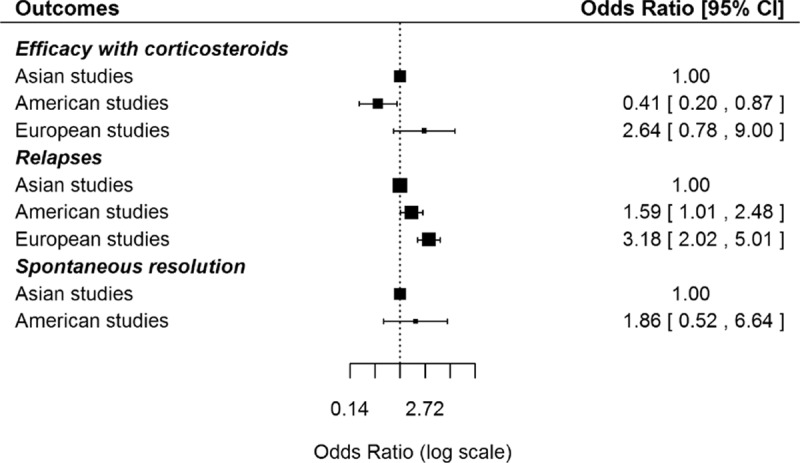
Differences in the main outcomes (efficacy of glucocorticoids, relapses, and spontaneous resolution) between Asian, American and European studies.
3.2.3. Organ-by-organ selection bias
Table 4 compares the main variables according to the predominant organ involvement selected for each study. With regard to epidemiological features, we found significant differences with respect to the geographical (P < 0.001) and gender distribution (P < 0.001). In addition, wide heterogeneity was observed with respect to the first-line therapeutic approaches used for the different organ-specific subsets of patients (Fig. 3) and the outcomes reported (Fig. 4), including differences in the mean dose of glucocorticoids used for the different types of organ involvement (Fig. 5).
Table 4.
Comparison of the main variables according to the predominant involved organ selected for each study: AIP/SC, lymphadenopathy or skin involvement, ocular involvement, other involvements, renal involvement, salivary involvement, and systemic/multiorgan involvement.
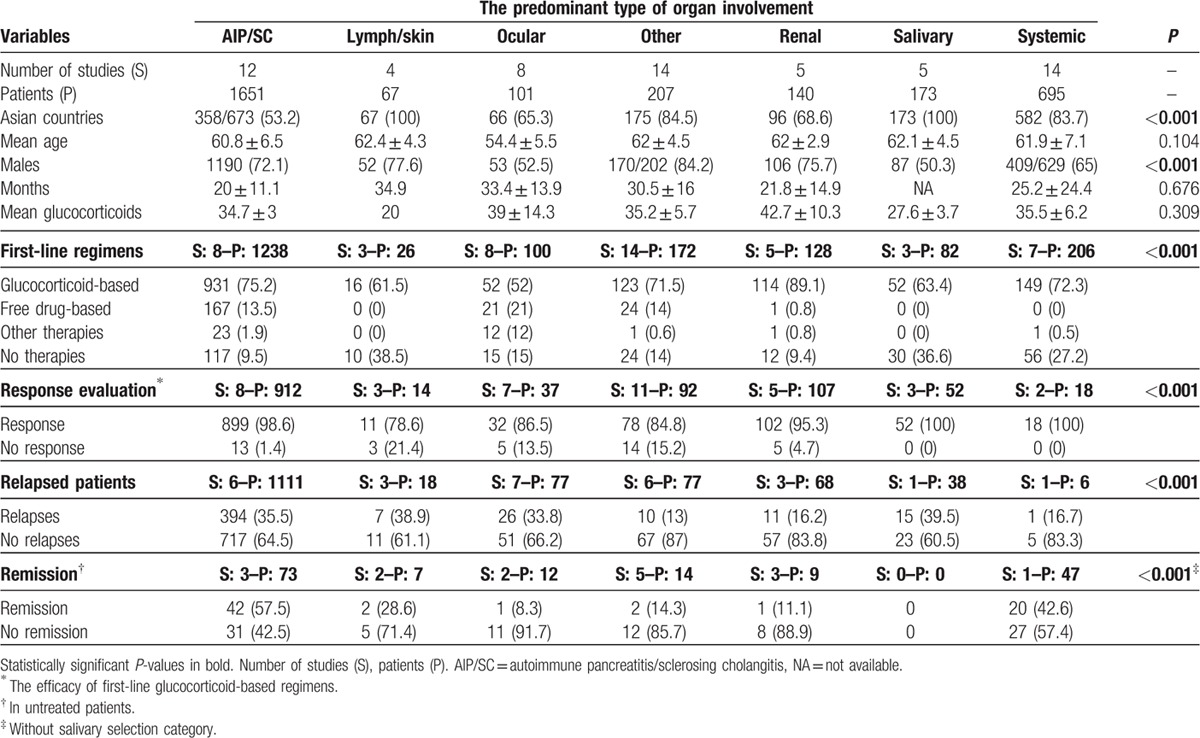
Figure 3.
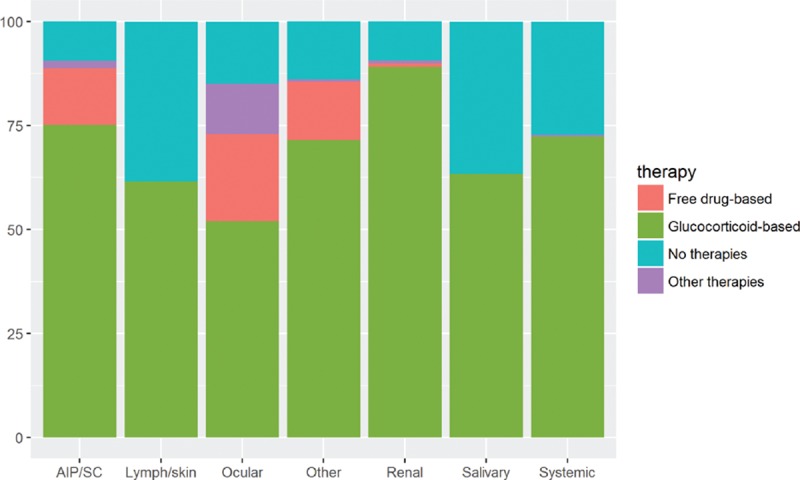
First-line therapeutic approaches used in the studies grouped according to the predominant organ involvement selected in each study (AIP = autoimmune pancreatitis, SC = sclerosing cholangitis).
Figure 4.
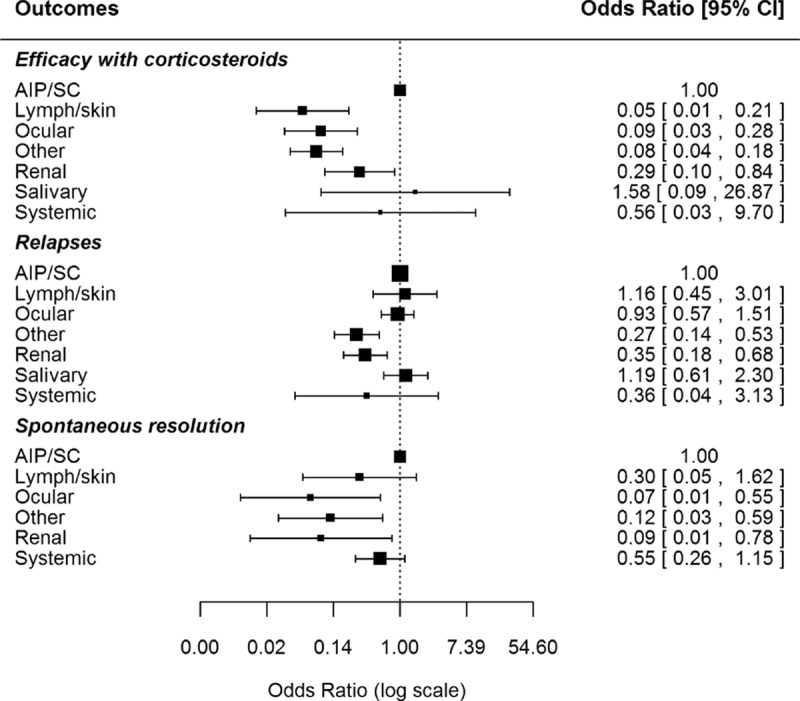
Differences in the main outcomes (efficacy of glucocorticoids, relapses, and spontaneous resolution) according to the predominant organ involvement selected in each study (AIP = autoimmune pancreatitis, SC = sclerosing cholangitis).
Figure 5.
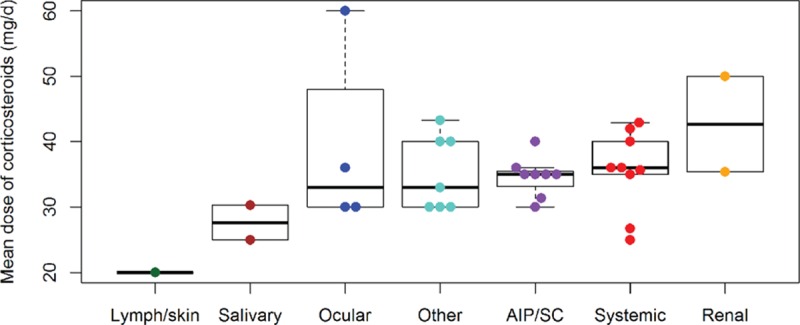
Differences in the mean dose of glucocorticoids used according to the predominant organ involvement selected in each study (AIP = autoimmune pancreatitis, SC = sclerosing cholangitis).
4. Discussion
IgG4-RD, first described some 15 years ago, remains an emerging disease. The rapid increase in number of studies has mainly been due to descriptive characterizations of the disease. Few studies have been designed specifically to evaluate the therapeutic response, and information on treatment has, to date, been presented almost exclusively in descriptive studies rather than formal clinical trials. As a result, the evidence base on which to predicate therapeutic recommendations remains slim. Consequently, an evidence-based systematic review of published uncontrolled studies may provide the best available understanding of current treatment approaches. Although several reviews on IgG4-RD have recently been published,[74,75] they did not specifically focus on the therapeutic management of the disease.
Since the first recognition of IgG4-RD as a discrete disease entity, glucocorticoid treatment has comprised the cornerstone of therapy for a majority of patients. Although such an approach is understandable given the affordability of this intervention and the substantial observational data that a majority of IgG4-RD patients respond to moderate to high doses of glucocorticoids, no controlled study has specifically evaluated their use. As a result, the optimal starting dose, the rate of glucocorticoid taper, the wisdom of discontinuing glucocorticoids altogether, patients’ long-term treatment response, and multiple other questions about this treatment approach remain uncertain.
The central place of glucocorticoids in the therapeutic armamentarium for IgG4-RD was extrapolated from observational studies in patients with type 1 AIP, in whom monotherapy with glucocorticoids has been largely uncontested as the first-line therapeutic approach. Our review confirms that glucocorticoid-based regimens were the therapeutic option of choice in the majority of patients (85%) in whom a specific therapeutic intervention was reported. A recent guideline recommends glucocorticoids as the first-line therapeutic agent, with 94% of interexpert agreement.[4] This is supported by our findings: the efficacy of glucocorticoid-based first-line regimens was 97%. However, 4 major areas of uncertainty concerning the use of glucocorticoids in IgG4-RD persist: the definition of the induction dose, how to taper glucocorticoids, the duration of therapy, and the balance between efficacy and side effects.
The recent consensus guidelines[4] recommend the use of prednisolone (0.6 mg/kg/d ) for 4 weeks as induction therapy. This mean dose was the most frequently used in our review (70% of studies), but varied widely according to geographical, epidemiological, and clinical features. Some studies[41,68,69] used higher doses in patients with severe complications (pancreatic, pulmonary, renal, and retroperitoneal involvement), while others used lower doses in patients with diabetes, those at high risk of developing steroid-related side effects,[24] or patients aged >80 years.[69] Although the optimal starting glucocorticoids dose remains undefined, it appears that doses equivalent to <10 mg/d of prednisone are associated with a lack of response.[55]
After the first 4 weeks of induction therapy, the glucocorticoid dose can be tapered gradually.[4] One approach, frequently employed in Japan, is to taper glucocorticoids over several months to a daily dose equivalent of somewhere between 2.5 and 10.0 mg/d of prednisone and then to maintain this dose for several years. This approach has been developed through consensus[76] rather than through rigorous, controlled studies, however, and its long-term efficacy has not been evaluated prospectively. A retrospective, multicenter study of 459 AIP patients in Japan reported that 82% of patients still received glucocorticoids as maintenance therapy at the last visit.[77] An alternative approach to the use of glucocorticoids, commonly employed in North America, is to taper prednisone to discontinuation after 2 to 3 months and to add a steroid-sparing agent if the disease relapses during or after the taper. The strategy acknowledges the facts that glucocorticoids do not cure IgG4-RD and the long-term morbidity of glucocorticoid use may be substantial in this patient population, which often has a number of comorbidities concomitantly with IgG4-RD.
Although the goal should probably be discontinuation of glucocorticoids after achieving a maintained clinical response, we found this only happened in 25% of patients in whom information on therapy at the last visit was detailed.
A key finding of this review is the very limited reported information on the side effects associated with widespread glucocorticoid use. Only one small series detailed the frequency of glucocorticoid-related side effects, which were found in two thirds of patients. DM is the most frequently reported side effect.[51] A detailed study by Ito et al evaluated the need for DM therapies after glucocorticoid treatment in 22 IgG4-RD patients without AIP and found that the percentage of untreated patients was reduced from 72% to 50% after starting glucocorticoids, while the rate of patients requiring insulin therapy increased from 4% to 27%; in addition, 83% of patients were aged >50 years and diagnosed with DM or impaired glucose tolerance.
The selection of a “steroid-sparing” agent is challenged by the paucity of data on the efficacy of conventional agents for IgG4-RD. Conventional immunomodulatory agents have been reported as first-line therapies in fewer than 10 patients. The recent therapeutic consensus[4] reported differing clinical practice according to country, with more Japanese physicians supporting glucocorticoid monotherapy as the first-line option compared with experts from other countries (North America, Europe, Korea, and China). However, in our geographical analysis, Asian studies had the highest frequency of untreated patients while glucocorticoids were more frequently used in Europeans and drug-free treatment in US patients: surprizingly, the rate of relapses was lower in the Asian population. This might be explained by taking into account the fact that Asian studies did not detail information on relapses (missing information would provide biased results) and because Asian studies included a lower frequency of patients affected by AIP (AIP is one of the IgG4-related involvements with the highest rate of relapses).
There is widespread consensus on the use of immunosuppressive agents as steroid-sparing agents in refractory/complicated cases. We found that immunosuppressants were used in nearly 40% of cases who relapsed but that these agents were employed overwhelmingly in combination with glucocorticoids, complicating any assessment of their efficacy alone. Azathioprine was used in 85% of cases, probably reflecting clinical practice in cases of AIP, followed by mycophenolate mofetil, methotrexate, tacrolimus, and cyclophosphamide. No study has compared the efficacy of different immunosuppressive agents. The efficacy of the azathioprine/glucocorticoid combination was estimated to be on the order of 80%. Some patients relapsed when treated with low doses of azathioprine (50 mg/d) or mycophenolate (1 g/d) and the disease was controlled by increasing the dose.[19] This was probably in fact azathioprine coupled simultaneously with an increase in prednisone dose, making it difficult to be certain about how much improvement was due to azathioprine and how much to increased steroids. Interpretation of the efficacy of all conventional agents in IgG4-RD is hampered by the dearth of prospective, controlled experiences with these medications. In addition, most data describing the use of conventional immunosuppressive medications are confounded by concomitant glucocorticoid use.
The emergence of biological therapies has increased the therapeutic armamentarium available for treating the most refractory/severe cases of IgG4-RD, but their use is limited by the lack of licensing. Rituximab was the most-reported biological option in our systematic review, being used in 9 studies: it was used less frequently as induction therapy (only 8 cases with an efficacy of 75%)[20,64] and more frequently as rescue therapy in patients who failed to achieve or sustain disease remission with glucocorticoid treatment.[11,18,22,31,32,49,66] Rituximab use was first reported in 2010[78] and was then used especially in patients with orbital involvement.[64,66] Carruthers et al[79] have recently published the results of an open-label prospective study including 30 IgG4-RD patients treated with 2 doses of rituximab (1000 mg each), with a disease response rate of 97%. The primary outcome was achieved by 23 participants (77%): 14 (47%) were in complete remission at 6 months, and 12 (40%) remained in complete remission at 12 months. These effects were achieved largely without glucocorticoid use (26 of the 30 patients were not treated with glucocorticoids) and without readministration of rituximab for the purpose of remission maintenance. Yamamoto et al[80] have also reported the successful use of rituximab without associated glucocorticoids. Further studies are needed to better define the place of rituximab in the therapeutic approach to IgG4-RD: although currently considered a rescue therapy, it might be considered as a first-line option in some cases. The apparent success of B cell depletion strategies has raised interest in other treatment strategies targeting the B cell lineage. A trial of Xmab5871, a homodimer that binds simultaneously to CD19 and Fc-gammaRIIIb – leading in theory to inhibition of the targeted cell – is now under study.
Fifteen percent of first-line therapeutic interventions in IgG4-RD were glucocorticoid-free regimens: these were mainly surgical (11% of the total) with radiotherapy used in only 13 cases. As a cardinal feature of IgG4-RD is single or multiple organ swelling that often raises concerns about malignancy, surgery is mainly reported to treat masses located in the pancreas, kidneys, lungs, biliary tract, and prostate. In these patients, IgG4-RD is diagnosed accidentally after surgical removal of a potential tumor. Most patients were retrospectively diagnosed with IgG4-RD based on histopathologic findings. The best examples are: presumed cholangiocarcinoma,[19] pancreatic cancer,[19] pulmonary or pleural tumors,[72] prostate adenocarcinoma,[13] or renal cancer.[50]
On the other hand, an IgG4-specific surgical approach has also been used in patients with specific involvements (mainly infiltrative masses that involve tubular anatomical structures and may lead to obstructive processes). The best examples are: dilatation/stenting of the biliary tract in patients with IgG4-related sclerosing cholangitis,[49,62] ureteral stents/ureterolysis,[32] transurethral resection of the prostate/suprapubic prostatectomy,[13] ocular surgical excision,[53,64] or surgical resection of submaxillary tumors.[34] In these cases, long-standing highly fibrotic lesions may respond poorly to drug-based therapies, and surgical debulking may be an appropriate and useful option.
A further key finding of our review was the lack of any therapeutic intervention in 13% of patients. In some specific organ involvements, this percentage was higher, reaching 71% in IgG4-related lymphadenopathy, 35% in IgG4-related salivary involvement, or 40% in IgG4-related skin involvement. Eight studies[18,25,29,41,46,54,67,72] detailed the reasons in 54 cases, which included mild/residual involvement or the patient refusing to be treated (n = 50), severe DM (n = 3), and active tuberculosis infection (n = 1). The outcome of these patients shows relapse or progression in nearly 60% of cases, a rate 2-fold higher than that found for patients treated with GC-therapeutic regimens. Wait and see management may be appropriate in asymptomatic patients with lymphadenopathy, cutaneous features, or mild salivary gland enlargement.
Another key finding of our review is the widespread lack of a standardized method of evaluating the therapeutic response. Of the 48 studies that evaluated the efficacy of first-line therapy, a specific definition of the type of therapeutic response was detailed in only 25. Only 2 studies[18,19] used an objective score; the remaining 23 used a subjective evaluation including clinical, laboratory, imaging, and/or organ-specific diagnostic tests (10 studies used ≥2 items and 13 only 1 item). Only 4 studies detailed reductions in serum IgG4 levels as a parameter of therapeutic efficacy. The development and validation of rigorous outcome measures are an important goal for the field of IgG4-RD investigation. An international effort to validate the IgG4-RD Responder Index[81] is now under way.
Recent studies have investigated novel cellular and molecular pathways involved in the pathogenesis of IgG4-RD that may be therapeutically targeted by biological therapies, including a potential role for basophil activation mediated by IgE or by Toll-like receptors.[82] Clayton et al[83] tested the use of omalizumab (biological therapy against IgE) in patients with IgG4-related eosinophilic esophagitis, while other studies[84,85] have found a raised expression of some cytokines, such as IL10 and Blys/BAFF, opening a possible therapeutic approach by targeting these molecules.
IgG4-RD is an increasingly recognized condition in adults, with a heterogeneous clinical presentation affecting a wide range of organ systems. The principal bases of therapeutic decision-making currently remain clinical experience and expert opinion, due to the low level of evidence with respect to therapeutic data. The body of evidence relies predominantly on descriptive series including a range of 5 to 50 patients (74% of studies). Our review found that although nearly 70% of reported IgG4-RD patients are treated with monotherapy with oral glucocorticoids, there is wide heterogeneity and therapeutic management is heavily influenced by geographical, epidemiological, and clinical factors, especially with respect to the predominant organ affected by the disease. International efforts are required to collect and characterize large multicenter and multidisciplinary cohorts of patients in order to develop consensual therapeutic approaches and endpoints.
Supplementary Material
Acknowledgments
The authors thank David Buss for his editorial assistance.
Footnotes
Abbreviations: AIP = autoimmune pancreatitis, DM = diabetes mellitus, IgG4-RD = IgG4-related disease.
Funding: Supported by Grants Fondo de Investigaciones Sanitarias (MRC, INT15/00085) and “Ajut per a la Recerca Josep Font” (PBZ, Hospital Clinic-Barcelona 2012).
The authors have no conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
References
- 1.Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med 2012; 366:539–551. [DOI] [PubMed] [Google Scholar]
- 2.Brito-Zeron P, Ramos-Casals M, Bosch X, et al. The clinical spectrum of IgG4-related disease. Autoimmun Rev 2014; 13:1203–1210. [DOI] [PubMed] [Google Scholar]
- 3.Stone JH, Brito-Zeron P, Bosch X, et al. Diagnostic approach to the complexity of IgG4-related disease. Mayo Clin Proc 2015; 90:927–939. [DOI] [PubMed] [Google Scholar]
- 4.Khosroshahi A, Wallace ZS, Crowe JL, et al. International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheumatol (Hoboken, NJ) 2015; 67:1688–1699. [DOI] [PubMed] [Google Scholar]
- 5.Shea BJ, Grimshaw JM, Wells GA, et al. Development of AMSTAR: a measurement tool to assess the methodological quality of systematic reviews. BMC Med Res Methodol 2007; 7:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Masaki Y, Shimizu H, Sato Nakamura T, et al. IgG4-related disease: diagnostic methods and therapeutic strategies in Japan. J Clin Exp Hematop 2014; 54:95–101. [DOI] [PubMed] [Google Scholar]
- 7.von Elm E, Altman DG, Egger M, et al. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) Statement: guidelines for reporting observational studies. Lancet 2007; 370:1453–1457. [DOI] [PubMed] [Google Scholar]
- 8.Manchikanti L, Datta S, Smith HS, et al. Evidence-based medicine, systematic reviews, and guidelines in interventional pain management: part 6. Systematic reviews and meta-analyses of observational studies. Pain Physician 2009; 12:819–850. [PubMed] [Google Scholar]
- 9.Shamliyan T, Kane RL, Jansen S. Quality of systematic reviews of observational nontherapeutic studies. Prev Chronic Dis 2010; 7:A133. [PMC free article] [PubMed] [Google Scholar]
- 10.Dreyer NA, Schneeweiss S, McNeil BJ, et al. GRACE principles: recognizing high-quality observational studies of comparative effectiveness. Am J Manag Care 2010; 16:467–471. [PubMed] [Google Scholar]
- 11.Alexander MP, Larsen CP, Gibson IW, et al. Membranous glomerulonephritis is a manifestation of IgG4-related disease. Kidney Int 2013; 83:455–462. [DOI] [PubMed] [Google Scholar]
- 12.Bosco JJ, Suan D, Varikatt W, Lin MW. Extra-pancreatic manifestations of IgG4-related systemic disease: a single-centre experience of treatment with combined immunosuppression. Intern Med J 2013; 43:417–423. [DOI] [PubMed] [Google Scholar]
- 13.Buijs J, Maillette de Buy Wenniger L, van Leenders G, et al. Immunoglobulin G4-related prostatitis: a case-control study focusing on clinical and pathologic characteristics. Urology 2014; 83:521–526. [DOI] [PubMed] [Google Scholar]
- 14.Carruthers MN, Khosroshahi A, Augustin T, et al. The diagnostic utility of serum IgG4 concentrations in IgG4-related disease. Ann Rheum Dis 2014:14–18. [DOI] [PubMed] [Google Scholar]
- 15.Chen H, Lin W, Wang Q, et al. IgG4-related disease in a Chinese cohort: a prospective study. Scand J Rheumatol 2014; 43:70–74. [DOI] [PubMed] [Google Scholar]
- 16.Cheuk W, Yuen HKL, Chu SYY, et al. Lymphadenopathy of IgG4-related sclerosing disease. Am J Surg Pathol 2008; 32:671–681. [DOI] [PubMed] [Google Scholar]
- 17.Detlefsen S, Zamboni G, Frulloni L, et al. Clinical features and relapse rates after surgery in type 1 autoimmune pancreatitis differ from type 2: a study of 114 surgically treated European patients. Pancreatology 2012; 12:276–283. [DOI] [PubMed] [Google Scholar]
- 18.Ebbo M, Daniel L, Pavic M, et al. IgG4-related systemic disease: features and treatment response in a French cohort: results of a multicenter registry. Medicine (Baltimore) 2012; 91:49–56. [DOI] [PubMed] [Google Scholar]
- 19.Ghazale A, Chari ST, Zhang L, et al. Immunoglobulin G4-associated cholangitis: clinical profile and response to therapy. Gastroenterology 2008; 134:706–715. [DOI] [PubMed] [Google Scholar]
- 20.Ginat DT, Freitag SK, Kieff D, et al. Radiographic patterns of orbital involvement in IgG4-related disease. Ophthal Plast Reconstr Surg 2013; 29:261–266. [DOI] [PubMed] [Google Scholar]
- 21.Go H, Kim JE, Kim Ya, et al. Ocular adnexal IgG4-related disease: comparative analysis with mucosa-associated lymphoid tissue lymphoma and other chronic inflammatory conditions. Histopathology 2012; 60:296–312. [DOI] [PubMed] [Google Scholar]
- 22.Hart PA, Kamisawa T, Brugge WR, et al. Long-term outcomes of autoimmune pancreatitis: a multicentre, international analysis. Gut 2013; 62:1771–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirano K, Tada M, Sasahira N, et al. Incidence of malignancies in patients with IgG4-related disease. Intern Med 2014; 53:171–176. [DOI] [PubMed] [Google Scholar]
- 24.Huggett MT, Culver EL, Kumar M, et al. Type 1 autoimmune pancreatitis and IgG4-related sclerosing cholangitis is associated with extrapancreatic organ failure, malignancy, and mortality in a prospective UK cohort. Am J Gastroenterol 2014; 109:1675–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inoue D, Zen Y, Abo H, et al. Immunoglobulin G4-related periaortitis and periarteritis: CT findings in 17 patients. Radiology 2011; 261:625–633. [DOI] [PubMed] [Google Scholar]
- 26.Inoue D, Zen Y, Sato Y, et al. IgG4-related perineural disease. Int J Rheumatol 2012; 2012:401890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ito N, Yagi K, Kawano M, et al. Analysis of pancreatic endocrine function in patients with IgG4-related diseases, in whom autoimmune pancreatitis was ruled out by diagnostic imaging. Endocr J 2014; 61:765–772. [DOI] [PubMed] [Google Scholar]
- 28.Kamisawa T, Takuma K, Tabata T, et al. Serum IgG4-negative autoimmune pancreatitis. J Gastroenterol 2011; 46:108–116. [DOI] [PubMed] [Google Scholar]
- 29.Katsura M, Mori H, Kunimatsu A, et al. Radiological features of IgG4-related disease in the head, neck, and brain. Neuroradiology 2012; 54:873–882. [DOI] [PubMed] [Google Scholar]
- 30.Kawano M, Saeki T, Nakashima H, et al. Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol 2011; 15:615–626. [DOI] [PubMed] [Google Scholar]
- 31.Khosroshahi A, Carruthers MN, Deshpande V, et al. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Medicine (Baltimore) 2012; 91:57–66. [DOI] [PubMed] [Google Scholar]
- 32.Khosroshahi A, Carruthers MN, Stone JH, et al. Rethinking Ormond's disease: “idiopathic” retroperitoneal fibrosis in the era of IgG4-related disease. Medicine (Baltimore) 2013; 92:82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim TY, Park KS, Choi JS, et al. Comparative clinical manifestations of IgG4- related and IgG4-negative primary tubulointerstitial nephritis. Clin Nephrol 2011; 76:440–446. [DOI] [PubMed] [Google Scholar]
- 34.Kitagawa S, Zen Y, Harada K, et al. Abundant IgG4-positive plasma cell infiltration characterizes chronic sclerosing sialadenitis (Küttner's tumor). Am J Surg Pathol 2005; 29:783–791. [DOI] [PubMed] [Google Scholar]
- 35.Kiyama K, Kawabata D, Hosono Y, et al. Serum BAFF and APRIL levels in patients with IgG4-related disease and their clinical significance. Arthritis Res Ther 2012; 14:R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koizumi S, Kamisawa T, Kuruma S, et al. Clinical features of IgG4-related dacryoadenitis. Graefes Arch Clin Exp Ophthalmol 2014; 252:491–497. [DOI] [PubMed] [Google Scholar]
- 37.Kubota T, Moritani S. Ocular adnexal IgG4-related lymphoplasmacytic infiltrative disorder. Arch Ophthalmol 2010; 128:577–584. [DOI] [PubMed] [Google Scholar]
- 38.Kuruma S, Kamisawa T, Tabata T, et al. Clinical characteristics of patients with autoimmune pancreatitis with or without Mikulicz's disease and Mikulicz's disease alone. Gut Liver 2013; 7:96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laco J, Podhola M, Kamarádová K, et al. Idiopathic vs. secondary retroperitoneal fibrosis: a clinicopathological study of 12 cases, with emphasis to possible relationship to IgG4-related disease. Virchows Arch 2013; 463:721–730. [DOI] [PubMed] [Google Scholar]
- 40.Lindstrom KM, Cousar JB, Lopes MBS. IgG4-related meningeal disease: clinico-pathological features and proposal for diagnostic criteria. Acta Neuropathol 2010; 120:765–776. [DOI] [PubMed] [Google Scholar]
- 41.Masaki Y, Dong L, Kurose N, et al. Proposal for a new clinical entity, IgG4-positive multiorgan lymphoproliferative syndrome: analysis of 64 cases of IgG4-related disorders. Ann Rheum Dis 2009; 68:1310–1315. [DOI] [PubMed] [Google Scholar]
- 42.Matsubayashi H, Sawai H, Kimura H, et al. Characteristics of autoimmune pancreatitis based on serum IgG4 level. Dig Liver Dis 2011; 43:731–735. [DOI] [PubMed] [Google Scholar]
- 43.Matsui S, Hebisawa A, Sakai F, et al. Immunoglobulin G4-related lung disease: clinicoradiological and pathological features. Respirology 2013; 18:480–487. [DOI] [PubMed] [Google Scholar]
- 44.Matsuo T, Ichimura K, Sato Y, et al. Immunoglobulin G4 (IgG4)-positive or -negative ocular adnexal benign lymphoid lesions in relation to systemic involvement. J Clin Exp Hematop 2010; 50:129–142. [DOI] [PubMed] [Google Scholar]
- 45.Mizushima I, Inoue D, Yamamoto M, et al. Clinical course after corticosteroid therapy in IgG4-related aortitis/periaortitis and periarteritis: a retrospective multicenter study. Arthritis Res Ther 2014; 16:R156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moteki H, Yasuo M, Hamano H, et al. IgG4-related chronic rhinosinusitis: a new clinical entity of nasal disease. Acta Otolaryngol 2011; 131:518–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oh H-C, Kim M-H, Lee KT, et al. Clinical clues to suspicion of IgG4-associated sclerosing cholangitis disguised as primary sclerosing cholangitis or hilar cholangiocarcinoma. J Gastroenterol Hepatol 2010; 25:1831–1837. [DOI] [PubMed] [Google Scholar]
- 48.Ohta N, Kurakami K, Ishida A, et al. Clinical and pathological characteristics of IgG4-related sclerosing sialadenitis. Laryngoscope 2012; 122:572–577. [DOI] [PubMed] [Google Scholar]
- 49.Patel H, Khalili K, Kyoung KT, et al. IgG4 related disease – a retrospective descriptive study highlighting Canadian experiences in diagnosis and management. BMC Gastroenterol 2013; 13:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raissian Y, Nasr SH, Larsen CP, et al. Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol 2011; 22:1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saeki T, Kawano M, Mizushima I, et al. The clinical course of patients with IgG4-related kidney disease. Kidney Int 2013; 84:826–833. [DOI] [PubMed] [Google Scholar]
- 52.Sakamoto A, Nagai R, Saito K, et al. Idiopathic retroperitoneal fibrosis, inflammatory aortic aneurysm, and inflammatory pericarditis-retrospective analysis of 11 case histories. J Cardiol 2012; 59:139–146. [DOI] [PubMed] [Google Scholar]
- 53.Sato Y, Ohshima KI, Ichimura K, et al. Ocular adnexal IgG4-related disease has uniform clinicopathology. Pathol Int 2008; 58:465–470. [DOI] [PubMed] [Google Scholar]
- 54.Sato Y, Inoue D, Asano N, et al. Association between IgG4-related disease and progressively transformed germinal centers of lymph nodes. Mod Pathol 2012; 25:956–967. [DOI] [PubMed] [Google Scholar]
- 55.Sato Y, Takeuchi M, Takata K, et al. Clinicopathologic analysis of IgG4-related skin disease. Mod Pathol 2013; 26:523–532. [DOI] [PubMed] [Google Scholar]
- 56.Suzuki M, Nakamaru Y, Akazawa S, et al. Nasal manifestations of immunoglobulin G4-related disease. Laryngoscope 2013; 123:829–834. [DOI] [PubMed] [Google Scholar]
- 57.Tabata T, Kamisawa T, Takuma K, et al. Serial changes of elevated serum IgG4 levels in IgG4-related systemic disease. Intern Med 2011; 50:69–75. [DOI] [PubMed] [Google Scholar]
- 58.Takahashi H, Yamashita H, Morooka M, et al. The utility of FDG-PET/CT and other imaging techniques in the evaluation of IgG4-related disease. Jt Bone Spine 2014; 81:331–336. [DOI] [PubMed] [Google Scholar]
- 59.Takagi Y, Nakamura H. IgG4-related Mikulicz’ s disease: ultrasonography of the salivary and lacrimal glands for monitoring the efficacy of corticosteroid therapy. Clin Exp Rheumatol 2013; 773–775. [PubMed] [Google Scholar]
- 60.Takagi D, Nakamaru Y, Fukuda S. Otologic manifestations of immunoglobulin G4-related disease. Ann Otol Rhinol Laryngol 2014; 123:420–424. [DOI] [PubMed] [Google Scholar]
- 61.Takuma K, Kamisawa T, Anjiki H, et al. Metachronous extrapancreatic lesions in autoimmune pancreatitis. Intern Med 2010; 49:529–533. [DOI] [PubMed] [Google Scholar]
- 62.Tanaka A, Tazuma S, Okazaki K, et al. Nationwide survey for primary sclerosing cholangitis and IgG4-related sclerosing cholangitis in Japan. J Hepatobiliary Pancreat Sci 2014; 21:43–50. [DOI] [PubMed] [Google Scholar]
- 63.Triantopoulou C, Malachias G, Maniatis P, et al. Renal lesions associated with autoimmune pancreatitis: CT findings. Acta Radiol 2010; 51:702–707. [DOI] [PubMed] [Google Scholar]
- 64.Wallace ZS, Deshpande V, Stone JH. Ophthalmic manifestations of IgG4-related disease: single-center experience and literature review. Semin Arthritis Rheum 2014; 43:806–817. [DOI] [PubMed] [Google Scholar]
- 65.Watanabe T, Maruyama M, Ito T, et al. Clinical features of a new disease concept, IgG4-related thyroiditis. Scand J Rheumatol 2013; 42:325–330. [DOI] [PubMed] [Google Scholar]
- 66.Wu A, Andrew NH, Tsirbas A, et al. Rituximab for the treatment of IgG4-related orbital disease: experience from five cases. Eye (Lond) 2015; 29:122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamada K, Hamaguchi Y, Saeki T, et al. Investigations of IgG4-related disease involving the skin. Mod Rheumatol 2013; 23:986–993. [DOI] [PubMed] [Google Scholar]
- 68.Yamamoto M, Takahashi H, Ishigami K, et al. Relapse patterns in IgG4-related disease. Ann Rheum Dis 2012; 71:1755–11755. [DOI] [PubMed] [Google Scholar]
- 69.Yamamoto M, Nojima M, Takahashi H, et al. Identification of relapse predictors in IgG4-related disease using multivariate analysis of clinical data at the first visit and initial treatment. Rheumatology (Oxford) 2015; 54:45–49. [DOI] [PubMed] [Google Scholar]
- 70.Yoo JJ, Park JJ, Kang EH, et al. Risk factors for the recurrence of IgG 4-related sclerosing disease without autoimmune pancreatitis. J Clin Rheumatol 2011; 17:392–394. [DOI] [PubMed] [Google Scholar]
- 71.You M-W, Kim JH, Byun JH, et al. Relapse of IgG4-related sclerosing cholangitis after steroid therapy: image findings and risk factors. Eur Radiol 2014; 24:1039–1048. [DOI] [PubMed] [Google Scholar]
- 72.Zen Y, Inoue D, Kitao A, et al. IgG4-related lung and pleural disease: a clinicopathologic study of 21 cases. Am J Surg Pathol 2009; 33:1886–1893. [DOI] [PubMed] [Google Scholar]
- 73.Khosroshahi A, Carruthers MN, Deshpande V, et al. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Medicine (Baltimore) 2012; 91:57–66. [DOI] [PubMed] [Google Scholar]
- 74.Karim AF, Verdijk RM, Guenoun J, et al. An inflammatory condition with different faces: immunoglobulin G4-related disease. Neth J Med 2016; 74:110–115. [PubMed] [Google Scholar]
- 75.Mulholland GB, Jeffery CC, Satija P, et al. Immunoglobulin G4-related diseases in the head and neck: a systematic review. J Otolaryngol Head Neck Surg 2015; 44:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kamisawa T, Okazaki K, Kawa S, et al. Japanese consensus guidelines for management of autoimmune pancreatitis: III. Treatment and prognosis of AIP. J Gastroenterol 2010; 45:471–477. [DOI] [PubMed] [Google Scholar]
- 77.Kamisawa T, Shimosegawa T, Okazaki K, et al. Standard steroid treatment for autoimmune pancreatitis. Gut 2009; 58:1504–1507. [DOI] [PubMed] [Google Scholar]
- 78.Khosroshahi A, Bloch DB, Deshpande V, et al. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum 2010; 62:1755–1762. [DOI] [PubMed] [Google Scholar]
- 79.Carruthers MN, Topazian MD, Khosroshahi A, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis 2015; 74:1171–1177. [DOI] [PubMed] [Google Scholar]
- 80.Yamamoto M, Awakawa T, Takahashi H. Is rituximab effective for IgG4-related disease in the long term? Experience of cases treated with rituximab for 4 years. Ann Rheum Dis 2015; 74:e46. [DOI] [PubMed] [Google Scholar]
- 81.Carruthers MN, Stone JH, Deshpande V, et al. Development of an IgG4-RD responder index. Int J Rheumatol 2012; 2012:259408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sharma M, Bayry J. Autoimmunity: basophils in autoimmune and inflammatory diseases. Nat Rev Rheumatol 2015; 11:129–131. [DOI] [PubMed] [Google Scholar]
- 83.Clayton F, Fang JC, Gleich GJ, et al. Eosinophilic esophagitis in adults is associated with IgG4 and not mediated by IgE. Gastroenterology 2014; 147:602–609. [DOI] [PubMed] [Google Scholar]
- 84.Furukawa S, Moriyama M, Tanaka A, et al. Preferential M2 macrophages contribute to fibrosis in IgG4-related dacryoadenitis and sialoadenitis, so-called Mikulicz's disease. Clin Immunol 2015; 156:9–18. [DOI] [PubMed] [Google Scholar]
- 85.Lin W, Jin L, Chen H, et al. B cell subsets and dysfunction of regulatory B cells in IgG4-related diseases and primary Sjogren's syndrome: the similarities and differences. Arthritis Res Ther 2014; 16:R118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.