

Abstract
Acute pancreatitis (AP) is a leading cause of hospitalization among non-malignant gastrointestinal disorders. The mortality of severe AP can reach 30–50%, which is most probably owing to the lack of specific treatment. Therefore, AP is a major healthcare problem, which urges researchers to identify novel drug targets. Studies from the last decades highlighted that the toxic cellular Ca2+ overload and mitochondrial damage are key pathogenic steps in the disease development affecting both acinar and ductal cell functions. Moreover, recent observations showed that modifying the cellular Ca2+ signalling might be beneficial in AP. The inhibition of Ca2+ release from the endoplasmic reticulum or the activity of plasma membrane Ca2+ influx channels decreased the severity of AP in experimental models. Similarly, inhibition of mitochondrial permeability transition pore (MPTP) opening also seems to improve the outcome of AP in in vivo animal models. At the moment MPTP blockers are under detailed clinical investigation to test whether interventions in MPTP openings and/or Ca2+ homeostasis of the cells can be specific targets in prevention or treatment of cell damage in AP.
This article is part of the themed issue ‘Evolution brings Ca2+ and ATP together to control life and death’.
Keywords: acute pancreatitis, mitochondrial damage, Ca2+ overload
1. Ca2+ is controlling secretory events in pancreatic acinar and ductal cells
Intracellular Ca2+ signalling plays central role in the regulation of the secretory processes of the exocrine pancreas. It is a well-known fact that in the exocrine pancreas acinar cells secrete digestive enzymes and pancreatic ductal epithelial cells secrete 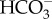 rich alkaline fluid that washes the digestive enzymes out from the pancreas. The prompt coordination of the secretory events of the two cell types is essential and Ca2+ has a central role in both pancreatic physiology and pathophysiology. Recent studies suggest that these two cell types cannot be handled separately since they are more likely integrated into a functional unit [1]. This is further amplified by the neurohormonal regulation of exocrine pancreatic secretion. It has been demonstrated that acetylcholine (the main stimulatory neurotransmitter of the pancreas) is released from the parasympathic nerve endings, releasing digestive enzymes from the acinar cells [2], whereas at the same time enhances the pancreatic ductal fluid and
rich alkaline fluid that washes the digestive enzymes out from the pancreas. The prompt coordination of the secretory events of the two cell types is essential and Ca2+ has a central role in both pancreatic physiology and pathophysiology. Recent studies suggest that these two cell types cannot be handled separately since they are more likely integrated into a functional unit [1]. This is further amplified by the neurohormonal regulation of exocrine pancreatic secretion. It has been demonstrated that acetylcholine (the main stimulatory neurotransmitter of the pancreas) is released from the parasympathic nerve endings, releasing digestive enzymes from the acinar cells [2], whereas at the same time enhances the pancreatic ductal fluid and 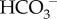 secretion via M3 metabotropic cholinerg receptor (M3R) mediated Ca2+ release [3]. In addition, the circulating hormone cholecystokinin (CCK) regulates pancreatic secretion via oscillatory Ca2+ signals [4]. In pancreatic ductal epithelial cells (PDECs), the role of CCK stimulation differs between species, in humans it has negligible direct effects, but remarkably potentiates the stimulatory effect of secretin on the
secretion via M3 metabotropic cholinerg receptor (M3R) mediated Ca2+ release [3]. In addition, the circulating hormone cholecystokinin (CCK) regulates pancreatic secretion via oscillatory Ca2+ signals [4]. In pancreatic ductal epithelial cells (PDECs), the role of CCK stimulation differs between species, in humans it has negligible direct effects, but remarkably potentiates the stimulatory effect of secretin on the 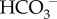 secretion [5]. The proper control of secretion is further potentiated by the strong synergy between Ca2+ and cAMP signalling [6]. The physiological roles of Ca2+ signalling in epithelial secretion have been outlined in more detail in excellent reviews [7–10].
secretion [5]. The proper control of secretion is further potentiated by the strong synergy between Ca2+ and cAMP signalling [6]. The physiological roles of Ca2+ signalling in epithelial secretion have been outlined in more detail in excellent reviews [7–10].
2. The price of versatility: Ca2+ toxicity in acute pancreatitis
Although it is well established that physiological Ca2+ signalling controls the normal pancreatic functions on multiple levels, it is also well documented that uncontrolled cellular Ca2+ overload leads to cellular damage and pathogenesis of acute pancreatitis (AP; figure 1). In this chapter, we will summarize the effects of the common stress factors that cause AP.
Figure 1.

Hypothetical sequence of events in the pathogenesis of AP. Pancreatitis inducing toxic stress factors can release the intracellular Ca2+ from the stores, such as the endoplasmic reticulum (ER), or acidic organelles. However, the constant presence of toxins will lead to the elongation of the Ca2+ signals via multiple mechanisms. First, the ER Ca2+ depletion activates the influx of extracellular (EC) Ca2+. Second, the direct mitochondrial toxicity of the stress factors (such as bile acids or non-oxidative ethanol metabolites), increases reactive oxygen species production and the sustained Ca2+ increase will lead to the opening of the MPTP that will damage the mitochondria. The lack of intracellular ATP impairs the function of Ca2+ extrusion and reuptake pumps such as PMCA or SERCA. These changes together will generate a vicious cycle leading to inhibited secretion and intracellular activation of digestive enzymes in acinar cells and impaired ductal fluid and 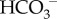 secretion. Altogether, these changes will trigger cell necrosis and AP.
secretion. Altogether, these changes will trigger cell necrosis and AP.
(a). Bile acids
Biliary pancreatitis is one of the most common forms of AP, although the exact pathogenesis is not known in detail. One possible explanation is the ‘common channel’ theory, which suggests that an impacted gallstone creates communication behind the stone connecting the common bile duct to the pancreatic duct. This would theoretically allow bile acids (BAs) to reach the pancreatic ductal lumen or even the acinar cells [11]. However, this hypothesis was questioned by several studies suggesting that instead of the reflux mechanism, increased luminal pressure would cause the pancreatic damage [12–15]. Whether or not BAs reach the pancreatic tissue directly from the luminal side, several observations suggest that BA reaching the ductals cells from either basolateral or luminal sides can trigger multiple cellular responses in acinar and ductal cells that might contribute to the development of AP.
Earlier, our group showed that the hydrophobic, non-conjugated BA, chenodeoxycholate (CDCA) dose-dependently affects 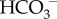 secretion of pancreatic ductal epithelia [16]. We found that lower concentration of CDCA (100 µM) stimulated and high concentration (1 mM) severely inhibited the ion transport activities including the ductal
secretion of pancreatic ductal epithelia [16]. We found that lower concentration of CDCA (100 µM) stimulated and high concentration (1 mM) severely inhibited the ion transport activities including the ductal 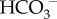 secretion. The explanation for this dual effect might be the type of Ca2+ signals triggered by CDCA. Luminal administration of 100 µM CDCA evoked short oscillatory Ca2+ signals, which were fully abolished by IP3 receptor inhibition. On the other hand, challenging the pancreatic ductal cells with 1 mM CDCA caused a sustained Ca2+ elevation [16] and severe damage of the mitochondrial morphology and function [17]. Interestingly, in our hands N,N′-[1,2-ethanediylbis(oxy-2,1-phenylene)]bis[N-[2-[(acetyloxy)methoxy]-2-oxoethyl]]-,bis[(acetyloxy)methyl]ester (BAPTA-AM) failed to prevent the mitochondrial damage and therefore the inhibitory effect of CDCA on the
secretion. The explanation for this dual effect might be the type of Ca2+ signals triggered by CDCA. Luminal administration of 100 µM CDCA evoked short oscillatory Ca2+ signals, which were fully abolished by IP3 receptor inhibition. On the other hand, challenging the pancreatic ductal cells with 1 mM CDCA caused a sustained Ca2+ elevation [16] and severe damage of the mitochondrial morphology and function [17]. Interestingly, in our hands N,N′-[1,2-ethanediylbis(oxy-2,1-phenylene)]bis[N-[2-[(acetyloxy)methoxy]-2-oxoethyl]]-,bis[(acetyloxy)methyl]ester (BAPTA-AM) failed to prevent the mitochondrial damage and therefore the inhibitory effect of CDCA on the 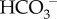 secretion [16], which might be explained by the existence of a Ca2+-independent direct mitochondrial toxicity of bile acids [18]. Similarly to ductal cells, pancreatic acinar cells respond with intracellular Ca2+ elevation to BA challenge [19] due to IP3R and ryanodine receptor activation. It is also well documented that taurolithocholicacid 3-sulfate diminishs cellular ATP production [20] and dissipate the mitochondrial membrane potential (ΔΨm), which was not affected by BAPTA-AM treatment [21]. Although BA directly affects the acinar cells, the observations of Perides et al. actually suggest that biliary pancreatitis is a receptor mediated disease [22]. They showed that the G-protein-coupled cell surface bile acid receptor (Gpbar1, or TGR5) is expressed at the apical membrane of pancreatic acinar cells and its activation is associated with pathological Ca2+ signals, intracellular activation of digestive enzymes and cell injury, i.e. the hallmarks of AP. Whereas the genetic deletion of Gpbar1 markedly reduced the severity of taurolithocholic acid 3-sulfate (TLCS)-induced, but not caerulein-induced AP. Very recently, Katona et al. provided solid evidence that specific BA might be used as treatment option against biliary pancreatitis [23]. They showed that pre-treatment of pancreatic ducts with ursodeoxycholate (UDCA) remarkably ameliorated the toxic effects of UDCA. Chenodeoxycholate-induced intracellular ATP depletion, mitochondrial injury, and as a consequence, cell death were completely prevented by UDCA, whereas the activity of the epithelial acid–base transporters was preserved in in vitro experiments. In addition, in vivo experiments showed that oral administration of UDCA significantly reduced the severity of CDCA-induced AP. Interestingly, UDCA had no effect on the sustained Ca2+ elevation triggered by CDCA, raising the possibility of a direct mitochondrial protective effect, which is yet to be determined. These observations nicely supplement the previous results of Seyhun et al., who showed that the endoplasmic reticulum (ER) chaperone tauroursodeoxycholic acid inhibits the unfolded protein response (UPR) in vitro [24] and in vivo [25]. This effect reduced the activation of UPR components and reduced intracellular trypsin activation, oedema formation and cell damage in pancreatic acinar cells.
secretion [16], which might be explained by the existence of a Ca2+-independent direct mitochondrial toxicity of bile acids [18]. Similarly to ductal cells, pancreatic acinar cells respond with intracellular Ca2+ elevation to BA challenge [19] due to IP3R and ryanodine receptor activation. It is also well documented that taurolithocholicacid 3-sulfate diminishs cellular ATP production [20] and dissipate the mitochondrial membrane potential (ΔΨm), which was not affected by BAPTA-AM treatment [21]. Although BA directly affects the acinar cells, the observations of Perides et al. actually suggest that biliary pancreatitis is a receptor mediated disease [22]. They showed that the G-protein-coupled cell surface bile acid receptor (Gpbar1, or TGR5) is expressed at the apical membrane of pancreatic acinar cells and its activation is associated with pathological Ca2+ signals, intracellular activation of digestive enzymes and cell injury, i.e. the hallmarks of AP. Whereas the genetic deletion of Gpbar1 markedly reduced the severity of taurolithocholic acid 3-sulfate (TLCS)-induced, but not caerulein-induced AP. Very recently, Katona et al. provided solid evidence that specific BA might be used as treatment option against biliary pancreatitis [23]. They showed that pre-treatment of pancreatic ducts with ursodeoxycholate (UDCA) remarkably ameliorated the toxic effects of UDCA. Chenodeoxycholate-induced intracellular ATP depletion, mitochondrial injury, and as a consequence, cell death were completely prevented by UDCA, whereas the activity of the epithelial acid–base transporters was preserved in in vitro experiments. In addition, in vivo experiments showed that oral administration of UDCA significantly reduced the severity of CDCA-induced AP. Interestingly, UDCA had no effect on the sustained Ca2+ elevation triggered by CDCA, raising the possibility of a direct mitochondrial protective effect, which is yet to be determined. These observations nicely supplement the previous results of Seyhun et al., who showed that the endoplasmic reticulum (ER) chaperone tauroursodeoxycholic acid inhibits the unfolded protein response (UPR) in vitro [24] and in vivo [25]. This effect reduced the activation of UPR components and reduced intracellular trypsin activation, oedema formation and cell damage in pancreatic acinar cells.
(b). Ethanol and non-oxidative ethanol metabolites
The second most frequent form of pancreatitis is alcohol-induced AP [26]. Whereas genetic factors seem to be involved in the disease development [27], several studies investigated the direct effects of ethanol and different ethanol metabolites on the exocrine pancreas. Ethanol and its oxidative metabolite acetaldehyde have moderate effects on the [Ca2+]i in pancreatic acinar cells even in extremely high concentrations [28]. Whereas the non-oxidative ethanol metabolites (fatty acid ethyl esters, FAEE) induced sustained [Ca2+]i elevation and a drop of cellular ATP leading to necrosis [28–30]. Importantly, the breakdown of FAEE to fatty acids (FA) by intracellular hydrolases significantly contribute to the toxic effects of non-oxidative ethanol metabolites [30]. This fact has been further emphasized in a recent elegant study by Huang et al. [31]. They showed that the inhibition of oxidative ethanol metabolism significantly enhance, whereas inhibition of non-oxidative ethanol metabolism augment pancreatic damage in an in vivo model of ethanol-fatty acid induced AP. On the other hand pancreatic ductal cells respond to low to high concentrations of alcohol, likewise to BA. Yamamoto et al. showed that 1 mM ethanol induces [Ca2+]i, elevation and augments fluid secretion, whereas high concentration moderately inhibits the stimulated fluid secretion in secretin-stimulated guinea pig pancreatic ducts [32]. Our group recently investigated the effects of ethanol and ethanol metabolites in more detail [33]. We showed that alcohol and fatty acids inhibit fluid and 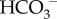 secretion, as well as cystic fibrosis transmembrane conductance regulator (CFTR) activity, in pancreatic ductal cells. Interestingly, in the case of FAEE only the inhibition of the CFTR channel was observed in high concentrations [34]; however, the inhibition of
secretion, as well as cystic fibrosis transmembrane conductance regulator (CFTR) activity, in pancreatic ductal cells. Interestingly, in the case of FAEE only the inhibition of the CFTR channel was observed in high concentrations [34]; however, the inhibition of 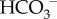 secretion was not observed [33]. The remarkable inhibitory effects of alcohol and fatty acids were mediated by sustained increase of intracellular Ca2+, inhibited adenosine 3’,5’-cyclic monophosphate and ATP production and depolarization of ΔΨm. We also showed that ethanol reduced expression of CFTR via multiple pathways, which in turn augmented the severity of experimental alcohol-induced AP in mice.
secretion was not observed [33]. The remarkable inhibitory effects of alcohol and fatty acids were mediated by sustained increase of intracellular Ca2+, inhibited adenosine 3’,5’-cyclic monophosphate and ATP production and depolarization of ΔΨm. We also showed that ethanol reduced expression of CFTR via multiple pathways, which in turn augmented the severity of experimental alcohol-induced AP in mice.
(c). Other stress factors
As demonstrated above, the two most common pathogenic factors of AP—BA and ethanol—damage the exocrine pancreas via Ca2+ toxicity and mitochondrial injury. Notably, these cellular changes seem to be the key of AP pathogenesis since a considerable number of studies showed that other stress factors provoke the same alterations in Ca2+ signalling and energy metabolism. Intrapancreatic trypsinogen activation is a hallmark of AP pathogenesis and we showed earlier that trypsin acting via PAR2 on the luminal membrane induces intracellular Ca2+ elevation and inhibits the luminal acid/base transporters in PDEC [35]. Moreover, the inhibitory effect was abolished by BAPTA-AM preincubation, similarly to the inhibitory effects of ethanol and fatty acids. Very recently, Jin et al. investigated the pathomechanism of an iatrogen form of AP, the post-ERCP pancreatitis [36]. Using sophisticated in vitro and in vivo models, they showed that exposure of pancreatic acinar cells to iohexol (a radiocontrast agent) triggered sustained intracellular Ca2+ elevation. The downstream activation of NF-κB and NFAT is completely abolished by the suppression of the Ca2+ signals. Moreover, they proved that the downstream effects of Ca2+ were mediated by calcineurin since genetic, or pharmacological inhibition of calcineurin prevented the radiocontrast-induced damage. This interesting study further underlines the central role of pathophysiological Ca2+ signalling in the pathogenesis of AP regardless of the etiological factor.
3. Sources of Ca2+ in pancreatic acinar and ductal cells
(a). Ca2+ release from the endoplasmic reticulum
Agonist binding (Ach, ATP) to G-protein-coupled receptors activate phospholipase C β (PLCβ) in pancreatic acinar and ductal cells. The activated PLCβ releases inositol trisphosphate (IP3) by hydrolysing phosphatidylinositol 4,5-bisphosphate (PIP2) [37]. Under physiological conditions, the intracellular Ca2+ signals have a strict spatio-temporal localization [38,39], mostly limiting Ca2+ signals to the apical pole of the cells. As in other non-excitable cell types, this is ensured by two ATP-dependent pumps that clear the cytosol from the free Ca2+. The sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pumps and the plasma membrane Ca2+-ATPase (PMCA) pumps move Ca2+ from the cytosol to the ER and the extracellular space, respectively. This activity restores basal intracellular Ca2+ levels and refills the ER Ca2+ stores. In PDEC, the Ca2+ signalling is not characterized in such detail; however, the overall polarity of the ductal cells including the ion channels and transporters, IP3 receptors and mitochondria [17], suggest a very similarly regulated Ca2+ signalling, like in acinar cells. Further studies are required for the clarification of these questions.
(b). Extracellular Ca2+ influx
The complex role of extracellular Ca2+ influx to orchestrate non-excitable cell functions has been established several decades ago [40]; however, the molecular components participating in the process remained unknown until 2005. Hoth et al. found that agonist-mediated depletion of the intracellular Ca2+ stores induced a Ca2+ selective sustained inwardly rectifying current, which was termed ICRAC (calcium release-activated calcium current) [41]. The real revolution of the field began by the discovery of the ER Ca2+ sensor stromal interaction molecule 1 (Stim1) [42] and the plasma membrane Ca2+ channel Orai1 [43,44]. Briefly, the process of store operated Ca2+ entry (SOCE) consist of the following elements. In resting conditions the ER Ca2+ stores are refilled and Stim1 distributes in the ER membrane. However during physiological stimulation the ER Ca2+ stores are quickly depleted, which induces the dissociation of the bound Ca2+ from the EF hand of Stim1. This is followed by a conformational change and translocation of Stim1 to defined ER-PM junctions, termed as puncta formation [45]. This translocation is required for the activation of the plasma membrane Ca2+ influx channel Orai1, where the Stim Orai1-activating region (SOAR) and polybasic domains of Stim1 interact with different binding sites of Orai1 that results in clustering and activation of the channel [46]. In addition to Orai1, other possible Ca2+ entry channels that seem to play a role in Stim1-mediated SOCE are the TRPC channels [47,48]. These channels function as Ca2+-permeable non-selective cation channels mediating receptor evoked Ca2+ influx in many cells [49]. SOCE have been investigated mostly in acinar cells of various exocrine glands, as models of polarized epithelial cells [50–52]. Interestingly, the role of SOCE in the physiological functions of PDEC, especially in 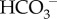 secretion remained elusive. Kim et al. found that intracellular Ca2+ elevation, caused by the activation of SOCE might play a role in exocytosis in pancreatic ductal cells isolated from dog main pancreatic duct [53,54]; however, they did not investigate
secretion remained elusive. Kim et al. found that intracellular Ca2+ elevation, caused by the activation of SOCE might play a role in exocytosis in pancreatic ductal cells isolated from dog main pancreatic duct [53,54]; however, they did not investigate 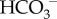 secretion of PDEC, which therefore needs further investigation.
secretion of PDEC, which therefore needs further investigation.
4. Mitochondrial Ca2+ handling and Ca2+ overload of mitochondria
During physiological Ca2+ signalling, mitochondria takes up Ca2+, which has been shown to directly increase energy output by enhancing the activity of tricarboxylic acid cycle dehydrogenases and the ATP synthase [55]. The pioneer work of Rizzuto et al. highlighted that the cytosolic Ca2+ signals propagate to the mitochondria [56] and a couple of years later Csordas et al. found that ER membrane and the outer mitochondrial membrane form a quasi-synaptic connection [57] that is the structural bases of the Ca2+ hotspots [58]. Despite the functional characterization of the mitochondrial Ca2+ signalling the molecular background of the process was not known. In 2011, two groups independently identified the mitochondrial Ca2+ uniporter (MCU), an inner mitochondrial membrane protein that is responsible for the mitochondrial Ca2+ uptake [59,60]. The Ca2+ efflux from the mitochondria is mediated by the mitochondrial Na+/Ca2+ exchanger (NCLX) [61], thus the mitochondrial Ca2+ level is tightly regulated under physiological conditions. However, pathophysiological signals can lead mitochondrial injury, which can activate both apoptosis and necrosis. The classical mitochondrial apoptotic pathway involves the outer membrane permeabilization by Bax and Bak (two members of the pro-death Bcl-2 family) that will allow apoptotic factors like cytochrome c, Smac/DIABLO and apoptosis inducing factor to be released from the intermembrane space into the cytosol, leading to cell death by apoptosis [62]. On the other hand, Ca2+ overload or increased reactive oxygen species (ROS) production can cause the opening of mitochondrial permeability transition pore (MPTP) that results in the loss of mitochondrial inner membrane potential, uncoupling of the respiratory chain with a consequent drop of mitochondrial ATP synthesis, and increased permeability of the inner mitochondrial membrane that eventually leads to mitochondrial swelling, rupture and necrotic cell death [63,64]. Notably, recent studies lead to the reconsideration of the role of MPTP in cellular physiology, since it has been proved to be important in several physiological processes such as energy metabolism [65], mitochondrial Ca2+ efflux [66] and ROS signalling [67] as well. The molecular identity of MPTP is still a matter of investigation [68]. The historical model of MPTP included the voltage-dependent anion channel (VDAC) in the outer mitochondrial membrane, the adenine nucleotide translocator (ANT) in the inner mitochondrial membrane, and CypD as its regulator in the matrix of the mitochondria [69]. However recent intensive efforts revealed new molecules that might contribute to the MPTP formation (reviewed in detail [68,70]). A growing number of evidence suggest that VDAC is not very likely to contribute to the MPTP formation. On the other hand, studies on ANT suggest that it is not required for MPTP formation, but it regulates MPTP activity [71]. CypD is an important regulator of MPTP as supported by genetically modified mice [72] and pharmacologic inhibition of CypD by cyclosporine A [73]. On the other hand, several studies suggested that the activity of F1F0 ATP synthase or the proapoptotic Bax/Bak proteins [74] are required for proper MPTP function [75], whereas other proteins, such as mitochondrial phosphate carrier, might impact the pore opening indirectly [76]. At the moment the role of MPTP in the pathogenesis of AP is supported by limited, but still solid evidence. Mukherjee et al. demonstrated that both genetic and pharmacologic inhibition of MPTP opening (using the Cyclophilin D-deficient Ppif gene knockout mice, or in vivo treatment with cyclosporine A derivates, respectively) significantly ameliorated pancreatic damage in different experimental AP models in mice [77]. Importantly MPTP blockade protected the pancreatic acinar cells from necrosis whereas apoptosis was not affected, which is in strong agreement of earlier studies [72].
5. Novel therapeutic targets in acute pancreatitis
In pancreatic acinar cells, IP3-mediated Ca2+ release from the ER is an essential component of the physiological response to agonist stimulation, but it could also contribute to the pathological Ca2+ overload of the cells evoked by toxic factors that induce AP (cerluien hyperstimulation, bile acids, or ethanol and ethanol metabolites) [39]. Caffeine is a known inhibitor of IP3Rs due to multiple actions that include the inhibition of phospholipase C-mediated production of IP3 [78], antagonism of IP3Rs [79] and direct binding to IP3Rs that reduce the channels open-state probability [80]. Interestingly, coffee consumption moderately reduces the risk of alcohol-associated pancreatitis suggesting that the inhibitory effect of caffeine on IP3-mediated Ca2+ signalling may be protective in AP [81]. Based on these considerations Huang et al. recently studied the effects of caffeine and its xanthine metabolites on pancreatic acinar IP3R-mediated Ca2+ signalling and experimental AP [82]. They found that caffeine and dimethylxanthines (but not monomethylxanthines) blocks IP3-mediated Ca2+ oscillations in response to uncaged IP3 or toxins, prevented mitochondrial depolarization and necrotic cell death in vitro and significantly impaired the severity of experimental AP in three different models. These observations suggest that caffeine, or its metabolites might be suitable starting points to develop therapy for AP (figure 2).
Figure 2.
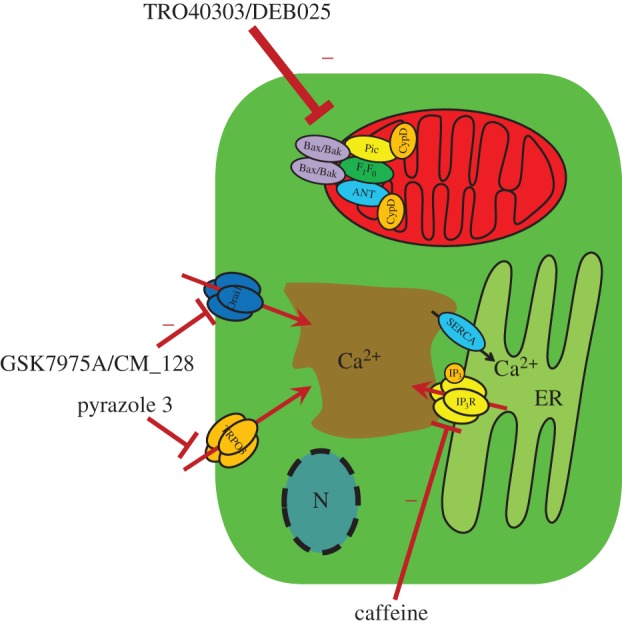
Novel therapeutic targets in AP. Experimental studies from recent years identified several proteins in cellular Ca2+ signaling machinery that might be potential target molecules in AP treatment. Caffeine and dimethylxanthines were shown to block IP3-mediated Ca2+ release from the ER that decreased the severity of AP in experimental models. Similarly, the inhibition of the plasma membrane Ca2+ influx channels Orai1 and TRPC3 reduced the severity of AP in animal models. Another treatment possibility might be the inhibition of the MPTP opening, which improved the disease outcome in rodents.
As discussed above, store operated Ca2+ entry could be a key component in the development of cellular Ca2+ overload. Earlier Kim et al. showed that genetic [83] or pharmacological inhibition (using the TRPC3-specific inhibitor pyrazole 3) [84] of TRPC3 significantly reduce the sustained Ca2+ elevation in pancreatic acinar cells evoked by cell stressors (bile acid or fatty acid ethyl ester). In addition, it prevented the pathological inhibition of digestive enzyme secretion and markedly reduced intracellular trypsin activation and excessive actin depolymerization in vitro and the severity of pancreatitis in vivo. Recently, Gerasimenko et al. demonstrated the pharmacological inhibition of another Ca2+ entry channel Orai1 by a specific inhibitor called GSK-7975A which prevents acinar cell necrosis in vitro [85]. This important observation was supported by Wen et al., who tested the effects of two specific Orai1 inhibitors (GSK-7975A and CM_128) in isolated human and rodent pancreatic acinar cells and in different experimental AP models [86]. They showed that both Orai1 inhibitors prevented the sustained Ca2+ elevation in vitro and significantly impaired signs of pancreatic injury including pancreatic oedema, inflammation and necrosis in all tested experimental models.
Mitochondrial permeability transition is a key feature of cellular damage in many cell types and diseases (see above); therefore, MPTP blockers are under detailed clinical investigation in different studies. In a recent clinical study, the efficacy and safety of TRO40303 (an MPTP inhibitor) have been evaluated for the reduction of reperfusion injury in patients undergoing revascularization for ST-elevation myocardial infarction (MITOCARE study) [87]. This study did not show any effect of TRO40303 in limiting reperfusion injury of the ischaemic myocardium. In another recently completed CIRCUS trial, the effects of i.v. administrated cyclosporine have been evaluated on the clinical outcome of patients with anterior STEMI [88]. Similarly to the MITOCARE study, CIRCUS trial did not report any improvement in the cyclosporine-treated patients. The reasons for the failure of the studies might be explained by pharamological limitations of the administrated compounds [89] that include low tissue penetration due to the lack of collateral blood flow and high metabolism of the compound in the blood. In addition, MPTP blockers have been suggested to be beneficial in hepatitis C therapy, since they inhibited hepatitis C virus (HCV) replication by preventing a cyclophilin-A induced cis–trans isomerization in domain II of NS5A [90]. However, it was not investigated in clinical trials further. Very recently, Mukherjee et al. tested the effect of MPTP inhibition on the severity of AP in rodent experimental AP models [77]. They have shown that the inhibition of MPTP with pharmacological compounds (two cyclosporine A derivate: DEB025 or TRO40303), or genetic deletion of the Ppif gene (that encodes cyclophylin D, a component of MPTP) significantly decreases the severity of AP in different independent models. These observations suggest that the MPTP inhibition might be potentially beneficial in the AP therapy. Other indirect evidence for this hypothesis has been provided by Judak et al., who showed that the supplementation of cellular ATP in vitro diminished the inhibitory effect of ethanol metabolites on the ion transport activities in isolated guinea pig pancreatic ductal cells [34]. These results suggest that the restoration of the cellular energy level can be beneficial in AP, which can prevent the cellular dysfunction and cell damage.
6. Closing remarks
Although there are several promising results and potential drug targets that play a role in the pathogenesis of AP, it remains a great challenge for researchers and clinicians. A number of unanswered questions are waiting for answers. Moreover, it will take several years to test the experimental results on clinical patients as well. To be able take up these challenges, clinicians and researchers should work closely together in the future.
Competing interests
We declare we have no competing interests.
Funding
Our research was supported by the Hungarian Scientific Research Fund (PD115974 (J.M.), K116634 (P.H.)) and the Momentum grant of the Hungarian Academy of Sciences (LP2014-10/2014 (P.H.)).
References
- 1.Maleth J, Hegyi P. 2014. Calcium signaling in pancreatic ductal epithelial cells: an old friend and a nasty enemy. Cell Calcium 55, 337–345. ( 10.1016/j.ceca.2014.02.004) [DOI] [PubMed] [Google Scholar]
- 2.Matthews EK, Petersen OH, Williams JA. 1973. Pancreatic acinar cells: acetylcholine-induced membrane depolarization, calcium efflux and amylase release. J. Physiol. 234, 689–701. ( 10.1113/jphysiol.1973.sp010367) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hootman SR, Zukerman J, Kovalcik SA. 1993. Muscarinic receptors in isolated guinea pig pancreatic ducts. Biochem. Pharmacol. 46, 291–296. ( 10.1016/0006-2952(93)90417-U) [DOI] [PubMed] [Google Scholar]
- 4.Murphy JA, et al. 2008. Direct activation of cytosolic Ca2+ signaling and enzyme secretion by cholecystokinin in human pancreatic acinar cells. Gastroenterology 135, 632–641. ( 10.1053/j.gastro.2008.05.026) [DOI] [PubMed] [Google Scholar]
- 5.You CH, Rominger JM, Chey WY. 1983. Potentiation effect of cholecystokinin-octapeptide on pancreatic bicarbonate secretion stimulated by a physiologic dose of secretin in humans. Gastroenterology 85, 40–45. [PubMed] [Google Scholar]
- 6.Ahuja M, Jha A, Maleth J, Park S, Muallem S. 2014. cAMP and Ca(2)(+) signaling in secretory epithelia: crosstalk and synergism. Cell Calcium 55, 385–393. ( 10.1016/j.ceca.2014.01.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petersen OH. 2014. Calcium signalling and secretory epithelia. Cell Calcium 55, 282–289. ( 10.1016/j.ceca.2014.01.003) [DOI] [PubMed] [Google Scholar]
- 8.Jung J, Lee MG. 2014. Role of calcium signaling in epithelial bicarbonate secretion. Cell Calcium 55, 376–384. ( 10.1016/j.ceca.2014.02.002) [DOI] [PubMed] [Google Scholar]
- 9.Gerasimenko J, Peng S, Gerasimenko O. 2014. Role of acidic stores in secretory epithelia. Cell Calcium 55, 346–354. ( 10.1016/j.ceca.2014.04.002) [DOI] [PubMed] [Google Scholar]
- 10.Lee MG, Ohana E, Park HW, Yang D, Muallem S. 2012. Molecular mechanism of pancreatic and salivary gland fluid and HCO3 secretion. Physiol. Rev. 92, 39–74. ( 10.1152/physrev.00011.2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lerch MM, Aghdassi AA. 2010. The role of bile acids in gallstone-induced pancreatitis. Gastroenterology 138, 429–433. ( 10.1053/j.gastro.2009.12.012) [DOI] [PubMed] [Google Scholar]
- 12.Lerch MM, Saluja AK, Runzi M, Dawra R, Saluja M, Steer ML. 1993. Pancreatic duct obstruction triggers acute necrotizing pancreatitis in the opossum. Gastroenterology 104, 853–861. [DOI] [PubMed] [Google Scholar]
- 13.Menguy RB, Hallenbeck GA, Bollman JL, Grindlay JH. 1958. Intraductal pressures and sphincteric resistance in canine pancreatic and biliary ducts after various stimuli. Surg. Gynecol. Obstet. 106, 306–320. [PubMed] [Google Scholar]
- 14.DiMagno EP, Shorter RG, Taylor WF, Go VL. 1982. Relationships between pancreaticobiliary ductal anatomy and pancreatic ductal and parenchymal histology. Cancer 49, 361–368. ( 10.1002/1097-0142(19820115)49:2%3C361::AID-CNCR2820490225%3E3.0.CO;2-O) [DOI] [PubMed] [Google Scholar]
- 15.Lerch MM, Weidenbach H, Hernandez CA, Preclik G, Adler G. 1994. Pancreatic outflow obstruction as the critical event for human gall stone induced pancreatitis. Gut 35, 1501–1503. ( 10.1136/gut.35.10.1501) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Venglovecz V, Rakonczay Z Jr, Ozsvari B, Takacs T, Lonovics J, Varro A, Gray MA, Argent BE, Hegyi P. 2008. Effects of bile acids on pancreatic ductal bicarbonate secretion in guinea pig. Gut 57, 1102–1112. ( 10.1136/gut.2007.134361) [DOI] [PubMed] [Google Scholar]
- 17.Maleth J, Venglovecz V, Razga Z, Tiszlavicz L, Rakonczay Z Jr, Hegyi P. 2011. Non-conjugated chenodeoxycholate induces severe mitochondrial damage and inhibits bicarbonate transport in pancreatic duct cells. Gut 60, 136–138. ( 10.1136/gut.2009.192153) [DOI] [PubMed] [Google Scholar]
- 18.Schulz S, et al. 2013. Progressive stages of mitochondrial destruction caused by cell toxic bile salts. Biochim. Biophys. Acta 1828, 2121–2133. ( 10.1016/j.bbamem.2013.05.007) [DOI] [PubMed] [Google Scholar]
- 19.Gerasimenko JV, Flowerdew SE, Voronina SG, Sukhomlin TK, Tepikin AV, Petersen OH, Gerasimenko OV. 2006. Bile acids induce Ca2+ release from both the endoplasmic reticulum and acidic intracellular calcium stores through activation of inositol trisphosphate receptors and ryanodine receptors. J. Biol. Chem. 281, 40 154–40 163. ( 10.1074/jbc.M606402200) [DOI] [PubMed] [Google Scholar]
- 20.Voronina SG, Barrow SL, Simpson AW, Gerasimenko OV, da Silva Xavier G, Rutter GA, Petersen OH, Tepikin AV. 2010. Dynamic changes in cytosolic and mitochondrial ATP levels in pancreatic acinar cells. Gastroenterology 138, 1976–1987. ( 10.1053/j.gastro.2010.01.037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Voronina SG, Barrow SL, Gerasimenko OV, Petersen OH, Tepikin AV. 2004. Effects of secretagogues and bile acids on mitochondrial membrane potential of pancreatic acinar cells: comparison of different modes of evaluating DeltaPsim. J. Biol. Chem. 279, 27 327–27 338. ( 10.1074/jbc.M311698200) [DOI] [PubMed] [Google Scholar]
- 22.Perides G, Laukkarinen JM, Vassileva G, Steer ML. 2010. Biliary acute pancreatitis in mice is mediated by the G-protein-coupled cell surface bile acid receptor Gpbar1. Gastroenterology 138, 715–725. ( 10.1053/j.gastro.2009.10.052) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katona M, Hegyi P, Kui B, Balla Z, Rakonczay Z Jr, Razga Z, Tiszlavicz L, Maleth J, Venglovecz V. 2016. A novel, protective role of ursodeoxycholate in bile-induced pancreatic ductal injury. Am. J. Physiol. Gastrointest. Liver Physiol. 310, G193–G204. [DOI] [PubMed] [Google Scholar]
- 24.Malo A, Kruger B, Seyhun E, Schafer C, Hoffmann RT, Goke B, Kubisch CH. 2010. Tauroursodeoxycholic acid reduces endoplasmic reticulum stress, trypsin activation, and acinar cell apoptosis while increasing secretion in rat pancreatic acini. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G877–G886. ( 10.1152/ajpgi.00423.2009) [DOI] [PubMed] [Google Scholar]
- 25.Seyhun E, Malo A, Schafer C, Moskaluk CA, Hoffmann RT, Goke B, Kubisch CH. 2011. Tauroursodeoxycholic acid reduces endoplasmic reticulum stress, acinar cell damage, and systemic inflammation in acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 301, G773–G782. ( 10.1152/ajpgi.00483.2010) [DOI] [PubMed] [Google Scholar]
- 26.Yadav D, Lowenfels AB. 2013. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 144, 1252–1261. ( 10.1053/j.gastro.2013.01.068) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whitcomb DC. 2012. Genetics of alcoholic and nonalcoholic pancreatitis. Curr. Opin. Gastroenterol. 28, 501–506. ( 10.1097/MOG.0b013e328356e7f3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Criddle DN, Raraty MG, Neoptolemos JP, Tepikin AV, Petersen OH, Sutton R. 2004. Ethanol toxicity in pancreatic acinar cells: mediation by nonoxidative fatty acid metabolites. Proc. Natl Acad. Sci. USA 101, 10 738–10 743. ( 10.1073/pnas.0403431101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Criddle DN, McLaughlin E, Murphy JA, Petersen OH, Sutton R. 2007. The pancreas misled: signals to pancreatitis. Pancreatology 7, 436–446. ( 10.1159/000108960) [DOI] [PubMed] [Google Scholar]
- 30.Criddle DN, Murphy J, Fistetto G, Barrow S, Tepikin AV, Neoptolemos JP, Sutton R, Petersen OH. 2006. Fatty acid ethyl esters cause pancreatic calcium toxicity via inositol trisphosphate receptors and loss of ATP synthesis. Gastroenterology 130, 781–793. ( 10.1053/j.gastro.2005.12.031) [DOI] [PubMed] [Google Scholar]
- 31.Huang W, et al. 2014. Fatty acid ethyl ester synthase inhibition ameliorates ethanol-induced Ca2+-dependent mitochondrial dysfunction and acute pancreatitis. Gut 63, 1313–1324. ( 10.1136/gutjnl-2012-304058) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamamoto A, et al. 2003. Ethanol induces fluid hypersecretion from guinea-pig pancreatic duct cells. J. Physiol. 551, 917–926. ( 10.1113/jphysiol.2003.048827) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maleth J, et al. 2015. Alcohol disrupts levels and function of the cystic fibrosis transmembrane conductance regulator to promote development of pancreatitis. Gastroenterology 148, 427–439. ( 10.1053/j.gastro.2014.11.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Judak L, Hegyi P, Rakonczay Z Jr, Maleth J, Gray MA, Venglovecz V. 2014. Ethanol and its non-oxidative metabolites profoundly inhibit CFTR function in pancreatic epithelial cells which is prevented by ATP supplementation. Pflugers Arch. 466, 549–562. ( 10.1007/s00424-013-1333-x) [DOI] [PubMed] [Google Scholar]
- 35.Pallagi P, et al. 2011. Trypsin reduces pancreatic ductal bicarbonate secretion by inhibiting CFTR Cl− channels and luminal anion exchangers. Gastroenterology 141, 2228–2239. ( 10.1053/j.gastro.2011.08.039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin S, Orabi AI, Le T, Javed TA, Sah S, Eisses JF, Bottino R, Molkentin JD, Husain SZ. 2015. Exposure to radiocontrast agents induces pancreatic inflammation by activation of nuclear factor-kappaB, calcium signaling, and calcineurin. Gastroenterology 149, 753–764. ( 10.1053/j.gastro.2015.05.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berridge MJ. 1993. Inositol trisphosphate and calcium signalling. Nature 361, 315–325. ( 10.1038/361315a0) [DOI] [PubMed] [Google Scholar]
- 38.Thorn P, Lawrie AM, Smith PM, Gallacher DV, Petersen OH. 1993. Ca2+ oscillations in pancreatic acinar cells: spatiotemporal relationships and functional implications. Cell Calcium 14, 746–757. ( 10.1016/0143-4160(93)90100-K) [DOI] [PubMed] [Google Scholar]
- 39.Petersen OH, Tepikin AV. 2008. Polarized calcium signaling in exocrine gland cells. Annu. Rev. Physiol. 70, 273–299. ( 10.1146/annurev.physiol.70.113006.100618) [DOI] [PubMed] [Google Scholar]
- 40.Putney JW., Jr 1978. Stimulus-permeability coupling: role of calcium in the receptor regulation of membrane permeability. Pharmacol. Rev. 30, 209–245. [PubMed] [Google Scholar]
- 41.Hoth M, Penner R. 1992. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 355, 353–356. ( 10.1038/355353a0) [DOI] [PubMed] [Google Scholar]
- 42.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr, Meyer T. 2005. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15, 1235–1241. ( 10.1016/j.cub.2005.05.055) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feske S, et al. 2006. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179–185. ( 10.1038/nature04702) [DOI] [PubMed] [Google Scholar]
- 44.Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. 2006. Orai1 is an essential pore subunit of the CRAC channel. Nature 443, 230–233. ( 10.1038/nature05122) [DOI] [PubMed] [Google Scholar]
- 45.Lee KP, Yuan JP, Hong JH, So I, Worley PF, Muallem S. 2010. An endoplasmic reticulum/plasma membrane junction: STIM1/Orai1/TRPCs. FEBS Lett. 584, 2022–2027. ( 10.1016/j.febslet.2009.11.078) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF, Muallem S. 2009. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 11, 337–343. ( 10.1038/ncb1842) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S. 2007. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat. Cell Biol. 9, 636–645. ( 10.1038/ncb1590) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim JY, Zeng W, Kiselyov K, Yuan JP, Dehoff MH, Mikoshiba K, Worley PF, Muallem S. 2006. Homer 1 mediates store- and inositol 1,4,5-trisphosphate receptor-dependent translocation and retrieval of TRPC3 to the plasma membrane. J. Biol. Chem. 281, 32 540–32 549. ( 10.1074/jbc.M602496200) [DOI] [PubMed] [Google Scholar]
- 49.Nilius B, Owsianik G, Voets T, Peters JA. 2007. Transient receptor potential cation channels in disease. Physiol. Rev. 87, 165–217. ( 10.1152/physrev.00021.2006) [DOI] [PubMed] [Google Scholar]
- 50.Hong JH, Li Q, Kim MS, Shin DM, Feske S, Birnbaumer L, Cheng KT, Ambudkar IS, Muallem S. 2011. Polarized but differential localization and recruitment of STIM1, Orai1 and TRPC channels in secretory cells. Traffic 12, 232–245. ( 10.1111/j.1600-0854.2010.01138.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lur G, Haynes LP, Prior IA, Gerasimenko OV, Feske S, Petersen OH, Burgoyne RD, Tepikin AV. 2009. Ribosome-free terminals of rough ER allow formation of STIM1 puncta and segregation of STIM1 from IP(3) receptors. Curr. Biol. 19, 1648–1653. ( 10.1016/j.cub.2009.07.072) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ambudkar IS. 2012. Polarization of calcium signaling and fluid secretion in salivary gland cells. Curr. Med. Chem. 19, 5774–5781. ( 10.2174/092986712804143321) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim MH, Seo JB, Burnett LA, Hille B, Koh DS. 2013. Characterization of store-operated Ca2+ channels in pancreatic duct epithelia. Cell Calcium 54, 266–275. ( 10.1016/j.ceca.2013.07.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koh DS, Moody MW, Nguyen TD, Hille B. 2000. Regulation of exocytosis by protein kinases and Ca2+ in pancreatic duct epithelial cells. J. Gen. Physiol. 116, 507–520. ( 10.1085/jgp.116.4.507) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hansford RG, Zorov D. 1998. Role of mitochondrial calcium transport in the control of substrate oxidation. Mol. Cell. Biochem. 184, 359–369. ( 10.1023/A:1006893903113) [DOI] [PubMed] [Google Scholar]
- 56.Rizzuto R, Simpson AW, Brini M, Pozzan T. 1992. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 358, 325–327. ( 10.1038/358325a0) [DOI] [PubMed] [Google Scholar]
- 57.Csordas G, Thomas AP, Hajnoczky G. 1999. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J. 18, 96–108. ( 10.1093/emboj/18.1.96) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rizzuto R, Duchen MR, Pozzan T. 2004. Flirting in little space: the ER/mitochondria Ca2+ liaison. Sci. STKE 2004, re1. ( 10.1126/stke.2152004re1) [DOI] [PubMed] [Google Scholar]
- 59.De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. 2011. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. ( 10.1038/nature10230) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baughman JM, et al. 2011. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. ( 10.1038/nature10234) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Palty R, et al. 2010. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl Acad. Sci. USA 107, 436–441. ( 10.1073/pnas.0908099107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tait SW, Green DR. 2010. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 11, 621–632. ( 10.1038/nrm2952) [DOI] [PubMed] [Google Scholar]
- 63.Halestrap AP. 2009. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 46, 821–831. ( 10.1016/j.yjmcc.2009.02.021) [DOI] [PubMed] [Google Scholar]
- 64.Golstein P, Kroemer G. 2007. Cell death by necrosis: towards a molecular definition. Trends Biochem. Sci. 32, 37–43. ( 10.1016/j.tibs.2006.11.001) [DOI] [PubMed] [Google Scholar]
- 65.Elrod JW, et al. 2010. Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Invest. 120, 3680–3687. ( 10.1172/JCI43171) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.De Marchi E, Bonora M, Giorgi C, Pinton P. 2014. The mitochondrial permeability transition pore is a dispensable element for mitochondrial calcium efflux. Cell Calcium 56, 1–13. ( 10.1016/j.ceca.2014.03.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. 2000. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 192, 1001–1014. ( 10.1084/jem.192.7.1001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kwong JQ, Molkentin JD. 2015. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 21, 206–214. ( 10.1016/j.cmet.2014.12.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crompton M, Virji S, Ward JM. 1998. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur. J. Biochem. 258, 729–735. ( 10.1046/j.1432-1327.1998.2580729.x) [DOI] [PubMed] [Google Scholar]
- 70.Halestrap AP, Richardson AP. 2015. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 78, 129–141. ( 10.1016/j.yjmcc.2014.08.018) [DOI] [PubMed] [Google Scholar]
- 71.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. 2004. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427, 461–465. ( 10.1038/nature02229) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Baines CP, et al. 2005. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662. ( 10.1038/nature03434) [DOI] [PubMed] [Google Scholar]
- 73.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. 2005. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434, 652–658. ( 10.1038/nature03317) [DOI] [PubMed] [Google Scholar]
- 74.Karch J, et al. 2013. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. Elife 2, e00772 ( 10.7554/eLife.00772) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Giorgio V, Bisetto E, Soriano ME, Dabbeni-Sala F, Basso E, Petronilli V, Forte MA, Bernardi P, Lippe G. 2009. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J. Biol. Chem. 284, 33 982–33 988. ( 10.1074/jbc.M109.020115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gutierrez-Aguilar M, Douglas DL, Gibson AK, Domeier TL, Molkentin JD, Baines CP. 2014. Genetic manipulation of the cardiac mitochondrial phosphate carrier does not affect permeability transition. J. Mol. Cell. Cardiol. 72, 316–325. ( 10.1016/j.yjmcc.2014.04.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mukherjee R, et al. In press. Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: inhibition prevents acute pancreatitis by protecting production of ATP. Gut ( 10.1136/gutjnl-2014-308553) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Toescu EC, O'Neill SC, Petersen OH, Eisner DA. 1992. Caffeine inhibits the agonist-evoked cytosolic Ca2+ signal in mouse pancreatic acinar cells by blocking inositol trisphosphate production. J. Biol. Chem. 267, 23 467–23 470. [PubMed] [Google Scholar]
- 79.Wakui M, Osipchuk YV, Petersen OH. 1990. Receptor-activated cytoplasmic Ca2+ spiking mediated by inositol trisphosphate is due to Ca2(+)-induced Ca2+ release. Cell 63, 1025–1032. ( 10.1016/0092-8674(90)90505-9) [DOI] [PubMed] [Google Scholar]
- 80.Saleem H, Tovey SC, Molinski TF, Taylor CW. 2014. Interactions of antagonists with subtypes of inositol 1,4,5-trisphosphate (IP3) receptor. Br. J. Pharmacol. 171, 3298–3312. ( 10.1111/bph.12685) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Morton C, Klatsky AL, Udaltsova N. 2004. Smoking, coffee, and pancreatitis. Am. J. Gastroenterol. 99, 731–738. ( 10.1111/j.1572-0241.2004.04143.x) [DOI] [PubMed] [Google Scholar]
- 82.Huang W, et al. In press. Caffeine protects against experimental acute pancreatitis by inhibition of inositol 1,4,5-trisphosphate receptor-mediated Ca2+ release. Gut ( 10.1136/gutjnl-2015-309363) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim MS, Hong JH, Li Q, Shin DM, Abramowitz J, Birnbaumer L, Muallem S. 2009. Deletion of TRPC3 in mice reduces store-operated Ca2+ influx and the severity of acute pancreatitis. Gastroenterology 137, 1509–1517. ( 10.1053/j.gastro.2009.07.042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim MS, Lee KP, Yang D, Shin DM, Abramowitz J, Kiyonaka S, Birnbaumer L, Mori Y, Muallem S. 2011. Genetic and pharmacologic inhibition of the Ca2+ influx channel TRPC3 protects secretory epithelia from Ca2+-dependent toxicity. Gastroenterology 140, 2107–2115. ( 10.1053/j.gastro.2011.02.052) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gerasimenko JV, et al. 2013. Ca2+ release-activated Ca2+ channel blockade as a potential tool in antipancreatitis therapy. Proc. Natl Acad. Sci. USA 110, 13 186–13 191. ( 10.1073/pnas.1300910110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wen L, et al. 2015. Inhibitors of ORAI1 prevent cytosolic calcium-associated injury of human pancreatic acinar cells and acute pancreatitis in 3 mouse models. Gastroenterology 149, 481–492. ( 10.1053/j.gastro.2015.04.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Atar D, et al. 2015. Effect of intravenous TRO40303 as an adjunct to primary percutaneous coronary intervention for acute ST-elevation myocardial infarction: MITOCARE study results. Eur. Heart J. 36, 112–119. ( 10.1093/eurheartj/ehu331) [DOI] [PubMed] [Google Scholar]
- 88.Cung TT, et al. 2015. Cyclosporine before PCI in patients with acute myocardial infarction. N. Engl. J. Med. 373, 1021–1031. ( 10.1056/NEJMoa1505489) [DOI] [PubMed] [Google Scholar]
- 89.Monassier L, Ayme-Dietrich E, Aubertin-Kirch G, Pathak A. 2015. Targeting myocardial reperfusion injuries with cyclosporine in the CIRCUS trial: pharmacological reasons for failure. Fundam. Clin. Pharmacol. 2, 191–193. ( 10.1111/fcp.12177) [DOI] [PubMed] [Google Scholar]
- 90.Coelmont L, et al. 2010. DEB025 (Alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PLoS ONE 5, e13687 ( 10.1371/journal.pone.0013687) [DOI] [PMC free article] [PubMed] [Google Scholar]