

Abstract
Vitamin D is a hormone that maintains healthy cells. It functions by regulating the low resting levels of cell signalling components such as Ca2+ and reactive oxygen species (ROS). Its role in maintaining phenotypic stability of these signalling pathways depends on the ability of vitamin D to control the expression of those components that act to reduce the levels of both Ca2+ and ROS. This regulatory role of vitamin D is supported by both Klotho and Nrf2. A decline in the vitamin D/Klotho/Nrf2 regulatory network may enhance the ageing process, and this is well illustrated by the age-related decline in cognition in rats that can be reversed by administering vitamin D. A deficiency in vitamin D has also been linked to two of the major diseases in man: heart disease and Alzheimer's disease (AD). In cardiac cells, this deficiency alters the Ca2+ transients to activate the gene transcriptional events leading to cardiac hypertrophy and the failing heart. In the case of AD, it is argued that vitamin D deficiency results in the Ca2+ landscape that initiates amyloid formation, which then elevates the resting level of Ca2+ to drive the memory loss that progresses to neuronal cell death and dementia.
This article is part of the themed issue ‘Evolution brings Ca2+ and ATP together to control life and death’.
Keywords: vitamin D, calcium, cardiac disease, Alzheimer's disease, klotho, inositol trisphosphate
1. Introduction
A large number of cellular processes are regulated by calcium (Ca2+). An important component of Ca2+ signalling is the InsP3/Ca2+ signalling pathway, which has two main operational modes. It functions either as a primary signalling pathway or it can operate as a modulatory signal. Its primary role is evident mainly in non-excitable cells where it generates the Ca2+ signals to control processes as diverse as fertilization, proliferation, metabolism, secretion and smooth muscle contraction. In excitable cells, the primary Ca2+ signal depends on the entry of Ca2+ through voltage-operated channels and the release of Ca2+ by ryanodine receptors (RYRs) on the internal stores. This primary Ca2+ pathway regulates processes such as contraction in the heart or memory formation in neurons. The InsP3/Ca2+ signalling pathway provides a modulatory signal that can induce subtle changes in the generation and function of this primary Ca2+ signal. In this review, I will argue that subtle changes in the nature of this modulatory role of the InsP3/Ca2+ signalling pathway may be responsible for the onset of two major human diseases: Alzheimer's disease (AD) and cardiovascular disease.
Both cardiac disease and AD are age related and what is remarkable is their very slow progression. Most individuals who develop these diseases are completely unaware that the disease is developing, and it is this aspect that may be explained by the subtle modulatory activity of the InsP3/Ca2+ signalling pathway. It will be argued that one of the main causes of the alteration in this modulatory pathway is vitamin D deficiency that causes the small alterations in the Ca2+ signalling pathway responsible for the onset of these two diseases [1–3]. All this evidence raises a major question concerning what it is about vitamin D that makes it such an important component of a healthy life. Any hypothesis as to how vitamin D deficiency might contribute to disease has to take into account a possible relationship between ageing and vitamin D deficiency. There is increasing evidence that vitamin D acts by maintaining the integrity of cell signalling pathways such as those regulated by Ca2+ and reactive oxygen species (ROS) [1–3]. It will be argued that low vitamin D levels result in an increase in the activity of these two signalling pathways that not only act to accelerate the ageing process but may also set the stage for the onset of a large number of diseases.
2. Integrated calcium and redox signalling pathways
A large number of cellular processes are regulated by Ca2+ signalling pathways often operating in conjunction with the redox signalling pathway [1,2]. What is remarkable about these two signalling systems is the way they interact with each other [4] (figure 1). When Ca2+ builds up within the mitochondrion, it increases mitochondrial metabolism resulting in an increased formation of superoxide 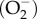 . Another action of Ca2+ is to stimulate nitric oxide synthase (NOS) to increase the formation of NO that contributes to the generation of peroxynitrite (ONOO−). Similarly, ROS can enhance Ca2+ signalling. For example, ROS sensitizes both the RYRs [5–7] and the InsP3Rs [8] to increase the release of Ca2+ from the internal store. The expression of Bcl-2, which regulates Ca2+ signalling by controlling Ca2+ release by the InsP3 receptors [9] and RYRs [10,11], is reduced by ROS [12]. ROS can activate a number of TRP channels that gate Ca2+ (e.g. TRPM2, TRPA1 and TRPV1) [13].
. Another action of Ca2+ is to stimulate nitric oxide synthase (NOS) to increase the formation of NO that contributes to the generation of peroxynitrite (ONOO−). Similarly, ROS can enhance Ca2+ signalling. For example, ROS sensitizes both the RYRs [5–7] and the InsP3Rs [8] to increase the release of Ca2+ from the internal store. The expression of Bcl-2, which regulates Ca2+ signalling by controlling Ca2+ release by the InsP3 receptors [9] and RYRs [10,11], is reduced by ROS [12]. ROS can activate a number of TRP channels that gate Ca2+ (e.g. TRPM2, TRPA1 and TRPV1) [13].
Figure 1.

The main reactive oxygen species (ROS) in cells are superoxide 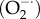 , hydrogen peroxide (H2O2) and peroxynitrite (ONOO−). The dashed lines represent the many interactions that operate between the Ca2+ and redox signalling pathways. An increase in Ca2+ can promote ROS formation by entering the mitochondria to form
, hydrogen peroxide (H2O2) and peroxynitrite (ONOO−). The dashed lines represent the many interactions that operate between the Ca2+ and redox signalling pathways. An increase in Ca2+ can promote ROS formation by entering the mitochondria to form 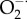 that is converted into H2O2 by SOD2. Ca2+ can also stimulate the nitric oxide synthase (NOS) that forms NO that interacts with
that is converted into H2O2 by SOD2. Ca2+ can also stimulate the nitric oxide synthase (NOS) that forms NO that interacts with 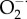 to form ONOO−. In a reciprocal way, an increase in cytosolic ROS can markedly enhance Ca2+ signalling by either increasing the activity of various channels such as the InsP3Rs and RYRs or by inhibiting the PMCA pump.
to form ONOO−. In a reciprocal way, an increase in cytosolic ROS can markedly enhance Ca2+ signalling by either increasing the activity of various channels such as the InsP3Rs and RYRs or by inhibiting the PMCA pump.
The reason for concentrating on these two pathways is because the expression of many of the genes responsible for regulating them is controlled by vitamin D [1,2]. Any deficiency in vitamin D will result in an alteration in how they operate, and this can have profound consequences for many different cellular processes and may be responsible for triggering a number of the diseases that have been linked to vitamin D deficiency.
3. Vitamin D regulation of the Ca2+ and redox signalling pathways
The active component of vitamin D is 1α,25-dihydroxyvitamin D3 [1α,25(OH)2D3] that is formed by a series of reactions that begin in the skin where sunlight converts 7-dehydrocholesterol to vitamin D3 (cholecalciferol) (figure 2). The latter is transferred to the liver where a hydroxyl group is added to the C-25 position by a vitamin D-25 hydroxylase (encoded by the CYP27A1 gene) to form 25-hydroxyvitamin D3 [25(OH)D3] that is the immediate precursor for active vitamin D. This 25(OH)D3 is carried in the blood to enter multiple cell types where a 25(OH)D3-1α-hydroxylase (encoded by the CYP27B1 gene) adds another hydroxyl group to the 1 position to form the active hormone 1α,25(OH)2D3, which will be referred to hereafter as vitamin D, that functions to regulate many different cellular processes [14].
Figure 2.

The vitamin D hormone 1,25-dihydroxyvitamin D3 [1α,25(OH)2D3] binds to the vitamin D receptor (VDR) that interacts with the retinoid X receptor (RXR) to form the VDR/RXR heterodimer that binds to the vitamin D response element (VDRE). Once in place, the VDR initiates the expression of a large number of genes located in many different cell types to express proteins that function in a number of cellular processes. Many of its actions also depend on its ability to increase the expression of both Klotho and Nrf2 that carry out many of its homeostatic functions.
Vitamin D can act in two ways. Firstly, it has non-genomic actions where it alters the activity of various signalling pathways. Secondly, it has a genomic action that is mediated by its binding to the vitamin D receptor (VDR), which interacts with the retinoid X receptor (RXR) before binding to the vitamin D response element (VDRE) located on a large number of vitamin D-sensitive target genes (figure 2). Two of the important genes that are activated by vitamin D are Nrf2 and the anti-ageing gene Klotho, both of which have multiple roles in maintaining the integrity of cellular signalling systems (figure 2). Many of the genes that are controlled by the vitamin D/Klotho/Nrf2 regulatory network function to maintain Ca2+ and redox homeostasis. For example, vitamin D increases the expression of Ca2+ pumps, exchangers and buffers to maintain low levels of Ca2+. In addition, vitamin D together with Klotho and Nrf2 all increase cellular antioxidants to maintain the normal reducing environment within the cell [15,16].
Vitamin D may also play a significant role in regulating the balance between autophagy and apoptosis [17,18]. This ability of vitamin D to promote autophagy over apoptosis may depend on its ability to regulate Ca2+ signalling that plays a significant role in controlling autophagy [19,20]. Subsequent studies revealed that the InsP3/Ca2+ signalling pathway plays a prominent role in regulating autophagy [21–23]. Elevation in various pathological aggregates such as amyloid, tau, α-synucleins and mutant Huntington fragments, which contribute to neurodegenerative disease such as AD, Parkinson's disease and Huntington's disease, may accumulate because of a decline in autophagy due to an alteration in Ca2+ signalling that occurs when vitamin D is deficient.
Another important action of vitamin D is to control the epigenetic landscape of multiple gene promoters to maintain the transcription activity of all the genes that function in its regulatory network [24]. Vitamin D influences the epigenetic landscape by controlling both the acetylation and methylation states of multiple gene promotor regions. The VDR/RXR dimer recruits histone acetyltransferases (HATs) such as p300/CBP and steroid receptor coactivators 1 and 2 (SRC1 and SRC2) that carry out the acetylation reactions that open up the chromatin structure to facilitate transcription so as to maintain phenotypic stability (figure 2).
Vitamin D can also regulate phenotypic stability by regulating demethylation. Many of the genes regulated by vitamin D are silenced by methylation of CpG islands located in their promotor regions [25]. For example, the decline in SERCA2a activity in cardiovascular disease may be caused by hypermethylation of its promotor region [26]. Expression of the Klotho gene, which acts together with vitamin D to regulate phenotypic stability, is silenced by methylation [27,28]. Such hypermethylation of promotor regions increases during ageing and is evident in many of the diseases such as cancer, cardiovascular and neurodegenerative diseases [29]. For example, hypermethylation of promotors in GABAergic neurons may contribute to the phenotypic remodelling responsible for schizophrenia and bipolar disorder [30]. Vitamin D modulates methylation by inducing the expression of a number of key DNA demethylases such as Jumonji domain-containing protein 1A and 3 (JMJD1A, JMJD3) and lysine-specific demethylase 1 and 2 (LSD1, LSD2) that contributes to its ability to maintain phenotypic stability [31].
This ability of vitamin D to modulate the epigenetic landscape is in keeping with its proposed role in maintaining phenotypic stability, and this may explain why vitamin D deficiency has been linked to both ageing and so many of the age-related diseases.
4. Vitamin D and ageing
There is increasing evidence that vitamin D may play an important role in the process of ageing. For example, the decline in cognition that occurs normally in older adults has been linked to vitamin D deficiency [32–35]. The ability of human skin to synthesize vitamin D declines with age [36], and this may account for the decline in the level of vitamin D and Klotho during ageing. Vitamin D and Klotho deficiency may contribute to the ageing process through dysregulation of the Ca2+ and redox cell signalling pathways. Nrf2 may also act to regulate longevity [37]. Dysregulation of Ca2+ signalling, which is closely linked to mitochondrial dysfunction and ROS formation, has been implicated in ageing [38,39]. In ageing striatal neurons, there is a marked decline in the expression of Bcl2 [38], which would contribute to the dysregulation of Ca2+, because one of its functions is to inhibit the InsP3Rs [9] (figure 1).
There has long been an interest in the possibility that alterations in the cellular redox balance [40,41] and Ca2+ signalling [42] might be responsible for ageing [43]. The way in which vitamin D deficiency and a concomitant decline in both Klotho and Nrf2 function contributes to many diseases may be explained through the ability of these custodial systems to maintain the stability of the redox and Ca2+ signalling systems described earlier [2]. For example, during ageing, there is a decline in the capacity of cells to maintain NAD(P)H levels in neurons [44,45], and this accounts for a decline in the levels of glutathione (GSH), which is essential to maintain low redox levels [46]. Such a decline in GSH results in a selective decline in the activity of GABAergic neurons in the hippocampus and could contribute to schizophrenia [47]. Vitamin D acts to maintain the expression of the Nrf2 antioxidant pathway [48]. There is a marked decline in the level of Nrf2 in the AD brain compared with age-matched controls [49]. Genetic ablation of the VDR results in premature ageing in mice suggesting that vitamin D can maintain normal physiological ageing [50].
Some of the most convincing evidence that vitamin D deficiency contributes to the ageing process has emerged from studies on the decline of memory in ageing rats. When considering memory mechanisms, it is important to point out that the ageing process does not affect long-term memories, but it induces a slow and progressive deterioration in the formation and retention of new memories [51]. This initial age-related decline in working memory is very subtle and has been linked to small changes in both the Ca2+ and redox signalling pathways [51–53]. An alteration in Ca2+ signalling has been linked to ageing in the brain [54–58]. The early loss of memory is caused by a number of subtle changes such as an elevation in the resting level of Ca2+ [56] and an increase in the expression of the CaV1.2 L-type Ca2+ channel [58], which is one of the proteins that is normally down-regulated by vitamin D (figure 2). Such changes may also depend on a decrease in the neuronal Ca2+ buffers and a decline in the mechanisms responsible for extruding Ca2+ from the cytoplasm [59]. Enhancing the intracellular buffering capacity markedly enhanced the learning capacity of aged rats [60].
At the electrophysiological level, the loss of memory during ageing has been linked to the progressive increase in the amplitude of the slow after hyperpolarization (sAHP) [52,61]. This sAHP is caused by increased fluxes of Ca2+ through CaV1.2 L-type voltage-gated channels, which are known to be elevated during ageing [58], and the RYRs resulting in abnormally high Ca2+ transients that activate SK potassium channels to hyperpolarize the membrane [51,55,57]. This sAHP reduces working memory in two ways. Firstly, the hyperpolarization reduces the spiking activity necessary for memory formation through long-term potentiation (LTP). Secondly, the increase in Ca2+ stimulates calcineurin to induce the long-term depolarization (LTD) that erases memories [52].
One of the interesting aspects of this dysregulation is that the relatively subtle elevation in the sensitivity of the Ca2+ signalling pathway appears to be driven by an increase in the oxidative state of the neurons [62]. In ageing mice, there is a marked increase in oxidative stress that contributes to a reduction in memory formation [63]. The enhanced ROS levels may increase sAHP by sensitizing the RYRs (figure 1). This would seem to be the case because the sAHP can be reversed by treating neurons with dithiothreitol (DTT) [64]. Similarly, a decrease in ROS could also contribute to the increase in cognition observed in ageing rats following treatment with the anti-inflammatory drug montelukast that is used normally to treat asthma [65]. The dysregulation of both Ca2+ and ROS signalling that is responsible for development of the sAHP during ageing seems to depend on vitamin D deficiency. The vitamin D/Klotho/NRF2 regulatory system can prevent the dysregulation of the Ca2+ and ROS signalling responsible for the sAHP through multiple mechanisms. For example, vitamin D suppresses the expression of the CaV1.2 L-type Ca2+ channel [66] that initiates the Ca2+ signal that induces the sAHP, and it also maintains the expression of PMCA and NCX1, which extrude Ca2+ from the cell. Klotho acts to stimulate the Na+/K+-ATPase responsible for maintaining the Na+ gradient necessary for Ca2+ extrusion by NCX1. Finally, NRF2 increases the expression of many antioxidants that ensure that ROS levels are kept low, which will prevent the sensitization of the RYRs that are triggering the sAHP and memory erasure.
Such a conclusion is strongly supported by the observation that vitamin D can reverse the Ca2+-dependent processes responsible for the age-related decline in memory [67]. What is more significant is that vitamin D can enhance hippocampal synaptic function, and more significantly, it could prevent the decline in cognition [68].
The fact that vitamin D deficiency brings about a dysregulation of both the Ca2+ and redox signalling pathways during ageing has raised an interesting possibility that it could also contribute to the initiation of age-related diseases [2].
5. The vitamin D/Klotho/Nrf2 regulatory network and disease
While most attention has been focused on establishing a link between vitamin D deficiency and disease, there is little information as to what the mechanism might be. To answer this question, I have developed a phenotypic stability hypothesis that is based on the idea that vitamin D may play an essential role through its ability to maintain both the redox and Ca2+ signalling pathways as described earlier [1,2]. A decline in the activity of the vitamin D/klotho/Nrf2 regulatory network has been linked to many diseases. Roselli & Caroni [69] have emphasized the importance of studying the early preclinical phases of neurodegenerative diseases. AD is a case in point in that the preclinical phase can last for many years before the disease is diagnosed. The following conceptual framework attempts to explain what might drive the early preclinical disease development and how this may be related to the ageing process. The basic idea is that there is a slow but progressive dysregulation of the Ca2+ and redox signalling pathways resulting from a deficiency in vitamin D [1,2]. It will be argued that this dysregulation results in an alteration in the modulatory activity of the InsP3/Ca2+ signalling pathway, and this creates subtle alterations in the normal cellular signalling pathway resulting in the onset of disease. To understand why such subtle alterations occur can lead to various disease states, it is important to consider the way the Ca2+ signalling system is organized in each specific cell type to provide either primary or modulatory signals.
(a). Cardiovascular disease
Vitamin D deficiency has been linked to hypertension and cardiovascular disease [70–76]. The ability of vitamin D to protect the cardiovascular system may depend on its ability to maintain the stability of the ROS and Ca2+ signalling systems, which are known to be dysregulated in hypertension, cardiac hypertrophy, congestive heart failure (CHF) and atrial arrhythmias.
One of the main causes of cardiac hypertrophy and CHF is hypertension. The renin–angiotensin system (RAS) plays a major role in regulating blood pressure. One of the primary actions of vitamin D is to curb RAS to prevent the hypertension that is a major risk factor for heart disease [77]. Vitamin D regulates the secretion of renin by renin-producing granular cells, which is controlled by the cyclic AMP signalling pathway. Vitamin D acts by preventing the cyclic AMP response element-binding protein (CREB) from binding to the renin gene promotor [78]. In mice, deletion of either the enzyme 25(OH)D 1α-hydroxylase or the VDR resulted in an increase in the renin–angiotensin system, hypertension and the onset of cardiac hypertrophy [79–81]
In patients with type 2 diabetes, the associated hypertension was improved following vitamin D supplementation [82]. The excessive release of renin and the resulting increase in angiotensin II can have multiple effects on some of the key components of the cardiovascular system. One of the actions of angiotensin II is to increase the formation of endothelin-1 (ET-1), which is a potent vasoconstrictor and thus contributes to angiotensin II-induced hypertension [83,84].
The changes in Ca2+ signalling in ventricular cardiac cells, which result in hypertrophy and CHF, are relatively minor. There is a small increase in the amplitude of the Ca2+ transient that occurs during each heartbeat. This amplification of each transient is caused by an increase in the activity of InsP3/Ca2+ modulatory signalling pathway, which is driven by the increased levels of angiotensin II and ET-1 [85]. In the presence of these two hormones, there are subtle changes in the spatial properties of the individual Ca2+ transients. It was proposed that the increase in InsP3 acts on perinuclear InsP3R2s to create a nuclear Ca2+ signal responsible for driving the transcriptional processes that initiate hypertrophy [86] (figure 3). There is now considerable experimental evidence to show that activation of InsP3Rs can indeed function to induce the nuclear Ca2+ transients that activate the transcriptional events responsible for the onset of hypertrophy [87–93]. One of the genes that is activated is ITPR2 that codes for the InsP3R2 that is responsible for the nuclear Ca2+ signal that drives hypertrophy [94]. In cardiomyoctes, miR-133a acts to inhibit the expression of InsP3R2 [95]. Down-regulation of miR-133a accounts for an increase in the level of the InsP3R2s, and this is a major contributory factor for the onset of cardiac hypertrophy.
Figure 3.

The role of enhanced Ca2+ and ROS levels in cardiac hypertrophy. A number of signalling pathways have been implicated in the activation of hypertrophy. A major pathway is induced by angiotensin II and endothelin that stimulate the formation of InsP3 that triggers a nuclear Ca2+ signal that activates the HDAC and NFAT shuttles to stimulate the transcription factors responsible for switching on the fetal genes that induce hypertrophy. These hormones also activate NOX to form reactive oxygen species (ROS) that contributes to hypertrophy by enhancing the sensitivity of InsP3R (I) and ryanodine receptor 2 (RYR2).
Vitamin D deficiency contributes to the onset of hypertrophy by increasing the Ca2+ and redox signalling pathways. For example, there is a decrease in the expression of both SERCA and phospholamban (PLN) that contributes to an increased Ca2+ transient amplitude and a decline in the recovery phase [96]. Vitamin D deficiency will also result in an increase in ROS levels that then enhances the Ca2+ signalling events that initiate the processes of hypertrophy that results in CHF [97,98]. The angiotensin II and ET-1 not only act to increase Ca2+, but they also increase ROS levels by stimulating NOX at the plasma membrane [99–101] (figure 3). ROS acts by increasing the activity of the ion channels (NaV1.5 sodium channel, CaV1.2 channels and RYR2) and pumps (SERCA) that contribute to the Ca2+cycling events that occur during each heartbeat. In addition, ROS can also act indirectly by increasing the activity of protein kinases such as PKA and CaMKIIδc that act normally to regulate cardiac activity [101]. These increased ROS and Ca2+ signalling processes contribute to the alterations in gene transcription that result in hypertrophy [98]. The cardiac hypertrophy in spontaneously hypertensive rats is reduced by vitamin D [102], and vitamin D supplementation can also markedly improve the outcome of patients suffering from heart failure [74,103].
(b). Alzheimer's disease
AD is another example of a major human disease where the initial change is so subtle that it can go undiagnosed for long periods. The initial symptoms are a decline in working memory, which closely resemble those that occur in ageing as described earlier. The onset of AD depends on the accumulation of extracellular β-amyloid (Aβ) deposits that disrupt neuronal signalling pathways to reduce cognition. The Ca2+ hypothesis considers that the loss of memory depends on an up-regulation of neuronal Ca2+ signalling [104–109]. When Ca2+ is measured in the spines and dendrites of cortical pyramidal neurons of transgenic mice, there was a higher than normal resting level in those neurons located close to amyloid deposits [110]. Similarly, the resting level of Ca2+ in the cortical neurons of 3xTg-AD animals was 247 nmol l−1, which was twice that found in the non-Tg controls (110 nmol l−1) [111]. Such evidence of a persistent elevation in the resting level of Ca2+ led to the suggestion that it may continuously activate LTD to explain why memories are erased shortly after they are formed [112,113].
The relatively small elevation in the resting level of Ca2+ does not alter the overall function of the brain. Information from the sensory organs can still be processed, new memories can be formed, but they are not retained because the persistent elevation in Ca2+ erases them shortly after they are formed. A number of mechanisms have been proposed to explain the elevation of intracellular Ca2+ levels by the Aβ protein [114,115]. Many of these mechanisms depend on the InsP3/Ca2+ signalling pathway [116–119]. Aβ can bind to the cellular prion protein (PrPC), which is coupled to mGluR5 that increases InsP3 formation and Ca2+ release [117] (figure 4). The formation of InsP3 is also increased by Aβ acting on the calcium-sensing receptor (CaSR) [118]. Phospholipase Cη1 (PLCη1), which is activated by Ca2+, may contribute to the dysregulation of Ca2+ signalling by amplifying these Aβ-dependent elevations in Ca2+ [119]. Activation of the mGluR5 receptor by the Aβ protein has been shown to enhance the process of LTD responsible for memory loss [120]. The significance of InsP3R activation in the pathogenesis of AD has also emerged from studies on the effects of presenilin mutations. In familial Alzheimer's disease (FAD), presenilin mutations enhance the activity of InsP3Rs resulting in an increase in Ca2+ signalling in both human cells and mouse neurons [121,122]. In a mouse model of AD, which had mutations in presenilin, the AD symptoms were reversed following a reduction in the expression of the InsP3R, thus supporting the notion that the InsP3/Ca2+ signalling pathway plays a significant role in disease pathogenesis [122].
Figure 4.

Dysregulation of Ca2+ and redox signalling in Alzheimer's disease (AD). The calcium hypothesis of AD suggests that the formation of Aβ oligomers brings about an overall increase in Ca2+ signalling that results in a permanent elevation in the resting level of Ca2+ to 300 nM that then erases memories soon after they are formed by activating calcineurin (CaN) inducing long-term depression (LTD). An elevation of Ca2+ sets up a positive feedback loop by acting to stimulate the hydrolysis of the amyloid precursor protein (APP) to generate the Aβ oligomers that bind to the cellular prion protein (PrPC) that then activates the InsP3/Ca2+ signalling pathway. Vitamin D reduces the risk of AD by acting to maintain Ca2+ and redox signalling at their normal low resting levels.
There are an increasing number of studies indicating that a deficiency in vitamin D may contribute to the onset of AD [123–128]. The level of vitamin D in AD patients is lower than that in controls [129]. Enhanced dietary vitamin D intake lowered the risk of developing AD in a study of older women [130]. VDR polymorphisms have been associated with age-related decline in cognition and are also a risk factor for AD [126,131,132]. Since AD seems to be caused by abnormal elevations in Ca2+, it is reasonable to propose that the deleterious effect of vitamin D deficiency may be explained by a decrease in its normal role as a custodian of Ca2+ and ROS homeostasis. Similarly, a decrease in ROS could also contribute to the increase in cognition observed in ageing rats following treatment with the anti-inflammatory drug montelukast that is used normally to treat asthma [65].
Vitamin D may prevent the onset of AD by regulating a number of processes. Firstly, vitamin D can increase the expression of the multidrug resistance protein 1 (MDR1) gene that codes for the P-glycoprotein (P-gp), which is an efflux transporter that acts to reduce the accumulation of Aβ [133]. Secondly, vitamin D may act to control the expression of those toolkit components responsible for maintaining low ROS and Ca2+ levels. For example, vitamin D stimulates the expression of Ca2+ pumps and exchangers (PMCA and NCX) and Ca2+ buffers such as calbindin (CB) and parvalbumin (figure 4). The expression of neuronal CB is known to be reduced in AD [134]. Mice expressing mutant APP also display a decline in the level of CB especially in the dentate gyrus region of the hippocampus, which functions in learning and memory [135]. Vitamin D can curb the influx of external Ca2+ by reducing the expression of L-type voltage-sensitive channels, which are markedly elevated in rat hippocampal neurons [66].
Many of the deleterious effects of vitamin D deficiency in AD may depend on a decline in the expression of its two collaborators Nrf2 and klotho. Nrf2 levels are markedly reduced in the brain of patient with AD [49]. Vector-mediated expression of Nrf2 in the hippocampus of AD transgenic mice resulted in a marked improvement in cognition [135]. One of the main functions of Nrf2 is to maintain the cellular level of the redox buffer GSH [136], which is a critical factor in preventing AD [44]. The level of Bcl-2, which inhibits the ability of InsP3 to activate the InsP3 receptors [9] and the RYRs [10,11] (figure 1), is maintained by Nrf2 and Klotho, thereby reducing the level of Ca2+. The ability of Bcl-2 to reduce the symptoms of AD in transgenic mice [137,138] may be explained by this reduction in the activity of both the InsP3R and RYRs. Klotho may also play a role in AD, because its levels in the CSF of patients with AD are lower than those in age-matched controls [139]. In the senescence-accelerated mouse prone-8 (SAMP8) mouse, a decline in the expression of klotho has been linked to symptoms of AD, including a decline in cognition and an accumulation of amyloid-β1–42 [140].
It is clear that dysregulation of the vitamin D/klotho/Nrf2 regulatory network results in a decline in cell signalling stability that results in the elevated neuronal Ca2+ and ROS levels that seem to responsible for the onset of AD. Such a mechanism suggests an interesting explanation for the sporadic nature of AD. Despite it being referred to as an age-related disease, not everyone who ages develops AD. So what is it that triggers the onset of sporadic AD? One possibility is that it is induced in those individuals who are deficient in vitamin D and thus have abnormally elevated levels of Ca2+ that may initiate the formation of the pathological Aβ oligomers [2,3]. This possibility is supported by the fact that Ca2+ acts to stimulate the formation of Aβ (figure 4) [57,107,141–145]. Inhibiting the RYR2 with dantrolene that reduces their release of Ca2+ was found to markedly reduce the formation of Aβ [145]. Such Ca2+-induced increases in amyloid formation then initiates a positive feedback loop, because it is followed by Aβ-induced Ca2+ signalling and it is this Aβ/Ca2+ positive feedback loop that may be responsible for the onset of AD [146]. Such a scenario is entirely consistent with the fact the vitamin D deficiency is such a strong risk factor for AD.
6. Conclusion
The phenotypic stability of the interacting Ca2+ and ROS signalling pathways is maintained by vitamin D. It is argued that a deficiency in vitamin D results in an elevation in both the ROS and Ca2+ signalling pathways that may contribute to the process of ageing. An example of this is the age-related decline in the cognition of rats that can be reversed by administering vitamin D. Such deficiencies in vitamin D may also set the stage for the onset of both heart disease and AD.
Competing interests
I have no competing interests.
Funding
I received no funding for this study.
References
- 1.Berridge MJ. 2015. Vitamin D: a custodian of cell signalling stability in health and disease. Biochem. Soc. Trans. 43, 349–358. ( 10.1042/BST20140279) [DOI] [PubMed] [Google Scholar]
- 2.Berridge MJ. 2015. Vitamin D cell signalling in health and disease. Bioch. Biophys. Res. Commun. 460, 53–71. ( 10.1016/j.bbrc.2015.01.008) [DOI] [PubMed] [Google Scholar]
- 3.Berridge MJ. 2015. Vitamin D, cell signalling phenotypic stability and Alzheimer's disease. Austin J. Clin. Neurol. 2, 1033–1036. [Google Scholar]
- 4.Hidalgo C, Donoso P. 2008. Crosstalk between calcium and redox signaling: from molecular mechanisms to health implications. Antioxid. Redox Signal. 10, 1275–1312. ( 10.1089/ars.2007.1886) [DOI] [PubMed] [Google Scholar]
- 5.Donoso P, Sanchez G, Bull R, Hidalgo C. 2011. Modulation of cardiac ryanodine receptor activity by ROS and RNS. Front. Biosci. 16, 553–567. ( 10.2741/3705) [DOI] [PubMed] [Google Scholar]
- 6.Prosser BL, Khairallah RJ, Ziman AP, Ward CW, Lederer WJ. 2013. X-ROS signaling in the heart and skeletal muscle: stretch-dependent local ROS regulates [Ca2+](i). J. Mol. Cell Cardiol. 58, 172–181. ( 10.1016/j.yjmcc.2012.11.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hidalgo C. 2005. Cross talk between Ca2+ and redox signalling cascades in muscle and neurons through the combined activation of ryanodine receptors/Ca2+ release channels. Phil. Trans. R. Soc. B 360, 2237–2246. ( 10.1098/rstb.2005.1759) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bánsághi S, Golenár T, Madesh M, Csordás G, RamachandraRao S, Sharma K, Yule DI, Joseph SK, Hajnóczky G. 2014. Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J. Biol. Chem. 289, 8170–8181. ( 10.1074/jbc.M113.504159) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rong YP, Distelhorst CW. 2008. Bcl-2 protein family: versatile regulators of calcium signaling in cell survival and apoptosis. Annu. Rev. Physiol. 70, 73–91. ( 10.1146/annurev.physiol.70.021507.105852) [DOI] [PubMed] [Google Scholar]
- 10.Vervliet T, et al. 2014. Bcl-2 binds to and inhibits ryanodine receptors. J. Cell Sci. 127, 2782–2792. ( 10.1242/jcs.150011) [DOI] [PubMed] [Google Scholar]
- 11.Vervliet T, Parys JB, Bultynck G. 2015. Bcl-2 and FKBP12 bind to IP3 and ryanodine receptors at overlapping sites: the complexity of protein-protein interactions for channel regulation. Biochem. Soc. Trans. 43, 396–404. ( 10.1042/BST20140298) [DOI] [PubMed] [Google Scholar]
- 12.Hildeman DA, Mitchell T, Aronow B, Wojciechowski S, Kappler J, Marrack P. 2003. Control of Bcl-2 expression by reactive oxygen species. Proc. Natl Acad. Sci. USA 100, 15 035–15 040. ( 10.1073/pnas.1936213100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kozai D, Ogawa N, Mori Y. 2014. Redox regulation of transient receptor potential channels. Antioxid. Redox Signal. 21, 971–986. ( 10.1089/ars.2013.5616) [DOI] [PubMed] [Google Scholar]
- 14.Dusso AS. 2014. Update on the biologic role of vitamin D on the endocrine system. Curr. Vasc. Pharmacol. 12, 272–277. ( 10.2174/15701611113119990026) [DOI] [PubMed] [Google Scholar]
- 15.Kaspar JW, Niture SK, Jaiswal AK. 2009. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 47, 1304–1309. ( 10.1016/j.freeradbiomed.2009.07.035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayes JD, Dinkova-Kostova AT. 2014. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 39, 199–218. ( 10.1016/j.tibs.2014.02.002) [DOI] [PubMed] [Google Scholar]
- 17.Høyer-Hansen M, Nordbrandt SP, Jäättelä M. 2010. Autophagy as a basis for the health-promoting effects of vitamin D. Trends Mol. Med. 16, 295–302. ( 10.1016/j.molmed.2010.04.005) [DOI] [PubMed] [Google Scholar]
- 18.Uberti F, Lattuada D, Morsanuto V, Nava U, Bolis G, Vacca G, Squarzanti DF, Cisari C, Molinari C. 2014. Vitamin D protects human endothelial cells from oxidative stress through the autophagic and survival pathways. J. Clin. Endocrinol. Metab. 99, 1367–1374. ( 10.1210/jc.2013-2103) [DOI] [PubMed] [Google Scholar]
- 19.Galluzzi L, Pietrocola F, Levine B, Kroemer G. 2014. Metabolic control of autophagy. Cell 159, 1263–1276. ( 10.1016/j.cell.2014.11.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Høyer-Hansen M, et al. 2007. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell. 25, 193–205. ( 10.1016/j.molcel.2006.12.009) [DOI] [PubMed] [Google Scholar]
- 21.Cárdenas C. 2010. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283. ( 10.1016/j.cell.2010.06.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harr MW, Distelhorst CW. 2010. Apoptosis and autophagy: decoding calcium signals that mediate life or death. Cold Spring Harb. Perspect. Biol. 2, a005579 ( 10.1101/cshperspect.a005579) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Decuypere JP, Bultynck G, Parys JB. 2011. A dual role for Ca2+ in autophagy regulation. Cell Calcium 50, 242–250. ( 10.1016/j.ceca.2011.04.001) [DOI] [PubMed] [Google Scholar]
- 24.Fetahu IS, Höbaus J, Kállay E. 2014. Vitamin D and the epigenome. Front. Physiol. 5, 164 ( 10.3389/fphys.2014.00164) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saccone D, Asani F, Bornman L. 2015. Regulation of the vitamin D receptor gene by environment, genetics and epigenetics. Gene 561, 171–180. ( 10.1016/j.gene.2015.02.024) [DOI] [PubMed] [Google Scholar]
- 26.Kao YH, Cheng CC, Chen YC, Chung CC, Lee TI, Chen SA, Chen YJ. 2011. Hydralazine-induced promoter demethylation enhances sarcoplasmic reticulum Ca2+-ATPase and calcium homeostasis in cardiac myocytes. Lab. Invest. 91, 1291–1297. ( 10.1038/labinvest.2011.92) [DOI] [PubMed] [Google Scholar]
- 27.King GD, Rosene DL, Abraham CR. 2011. Promoter methylation and age-related downregulation of Klotho in rhesus monkey. Age 34, 1405–1419. ( 10.1007/s11357-011-9315-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee J, et al. 2010. Theranti-aging gene KLOTHO is a novel target for epigenetic silencing in human cervical carcinoma. Mol. Cancer 9, 109–119. ( 10.1186/1476-4598-9-109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Otterdijk SD, Mathers LC, Strathdee G. 2013. Do age-related changes in DNA methylation play a role in the development of age-related diseases? Biochem. Soc. Trans. 41, 803–807. ( 10.1042/BST20120358) [DOI] [PubMed] [Google Scholar]
- 30.Guidotti AJ, et al. 2011. Epigenetic GABAergic targets in schizophrenia and bipolar disorder. Neuropharmacology 60, 1007–1016. ( 10.1016/j.neuropharm.2010.10.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pereira F, Barbáchano A, Singh PK, Campbell MJ, Muñoz A, Larriba MJ. 2012. Vitamin D has wide regulatory effects on histone demethylase genes. Cell Cycle 11, 1081–1089. ( 10.4161/cc.11.6.19508) [DOI] [PubMed] [Google Scholar]
- 32.Przybelski RJ, Binkley NC. 2007. Is vitamin D important for preserving cognition? A positive correlation of serum 25-hydroxyvitamin D concentration with cognitive function. Arch. Biochem. Biophys. 460, 202–205. ( 10.1016/j.abb.2006.12.018) [DOI] [PubMed] [Google Scholar]
- 33.Kuningas M, Mooijaart SP, Jolles J, Slagboom PE, Westendorp RG, van Heemst D. 2009. VDR gene variants associate with cognitive function and depressive symptoms in old age. Neurobiol. Aging. 30, 466–473. ( 10.1016/j.neurobiolaging.2007.07.001) [DOI] [PubMed] [Google Scholar]
- 34.Beydoun MA, Ding EL, Beydoun HA, Tanaka T, Ferrucci L, Zonderman AB. 2012. Vitamin D receptor and megalin gene polymorphisms and their associations with longitudinal cognitive change in US adults. Am. J. Clin. Nutr. 95, 163–178. ( 10.3945/ajcn.111.017137) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schlögl M, Holick MF. 2014. Vitamin D and neurocognitive function. Clin. Interv. Aging 9, 559–568. ( 10.2147/CIA.S51785) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacLaughlin J, Holick MF. 1985. Aging decreases the capacity of human skin to produce vitamin D3. J. Clin. Invest. 76, 1536–1538. ( 10.1172/JCI112134) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewis KN, Mele J, Hayes JD, Buffenstein R. 2010. Nrf2, a guardian of healthspan and gatekeeper of species longevity. Integr. Comp. Biol. 50, 829–843. ( 10.1093/icb/icq034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ureshino RP, Bertoncini CR, Fernandes MJ, Abdalla FM, Porto CS, Hsu YT, Lopes GS, Smaili SS. 2010. Alterations in calcium signaling and a decrease in Bcl-2 expression: possible correlation with apoptosis in aged striatum. J. Neurosci. Res. 88, 438–447. ( 10.1002/jnr.22214) [DOI] [PubMed] [Google Scholar]
- 39.Decuypere J-P, Monaco G, Missiaen L, De Smedt H, Parys JB, Bultynck G. 2011. IP3 receptors, mitochondria, and Ca2+ signaling: implications for aging. J. Aging Res. 2011, 920178 ( 10.4061/2011/920178) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harman D. 1956. Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300. ( 10.1093/geronj/11.3.298) [DOI] [PubMed] [Google Scholar]
- 41.Finkel T, Holbrook NJ. 2000. Oxidants, oxidative stress and the biology of ageing. Nature 408, 239–247. ( 10.1038/35041687) [DOI] [PubMed] [Google Scholar]
- 42.Khachaturian ZS. 1989. The role of calcium regulation in brain aging: reexamination of a hypothesis. Aging (Milano) 1, 17–34. ( 10.1007/BF03323872) [DOI] [PubMed] [Google Scholar]
- 43.Ureshino RP, Rocha KK, Lopes GS, Trindade CB, Smaili SS. 2014. Calcium signaling alterations, oxidative stress, and autophagy in aging. Antioxid. Redox Signal. 21, 123–137. ( 10.1089/ars.2013.5777) [DOI] [PubMed] [Google Scholar]
- 44.Ghosh D, LeVault KR, Brewer GJ. 2014. Dual-energy precursor and nuclear erythroid-related factor 2 activator treatment additively improve redox glutathione levels and neuron survival in aging and Alzheimer mouse neurons upstream of reactive oxygen species. Neurobiol. Aging 35, 179–190. ( 10.1016/j.neurobiolaging.2013.06.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghosh D, LeVault KR, Brewer GJ. 2014. Relative importance of redox buffers GSH and NAD(P)H in agerelated neurodegeneration and Alzheimer disease-like mouse neurons. Aging Cell 13, 631–640. ( 10.1111/acel.12216) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghosh D, LeVault KR, Barnett AJ, Brewer GJ. 2012. A reversible early oxidized redox state that precedes macromolecular ROS damage in aging nontransgenic and 3xTg-AD mouse neurons. J. Neurosci. 32, 5821–5832. ( 10.1523/JNEUROSCI.6192-11.2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steullet P, Cabungcal JH, Kulak A, Kraftsik R, Chen Y, Dalton TP, Cuenod M, Do KQ. 2010. Redox dysregulation affects the ventral but not dorsal hippocampus: impairment of parvalbumin neurons, gamma oscillations, and related behaviors. J. Neurosci. 30, 2547–2558. ( 10.1523/JNEUROSCI.3857-09.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakai K, et al. 2014. Vitamin D activates the Nrf2-Keap1 antioxidant pathway and ameliorates nephropathy in diabetic rats. Am. J. Hypertens. 27, 586–595. ( 10.1093/ajh/hpt160) [DOI] [PubMed] [Google Scholar]
- 49.Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA, Hamilton RL, Chu CT, Jordan-Sciutto KL. 2007. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 66, 75–85. ( 10.1097/nen.0b013e31802d6da9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keisala TMA, Lou YR, Zou J, Kalueff AV, Pyykkö I, Tuohimaa P. 2009. Premature aging in vitamin D receptor mutant mice. J. Steroid Biochem. Mol. Biol. 115, 91–97. ( 10.1016/j.jsbmb.2009.03.007) [DOI] [PubMed] [Google Scholar]
- 51.Toescu EC, Verkhratsky A. 2007. The importance of being subtle: small changes in calcium homeostasis control cognitive decline in normal aging. Aging Cell 6, 267–273. ( 10.1111/j.1474-9726.2007.00296) [DOI] [PubMed] [Google Scholar]
- 52.Foster TC. 2007. Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell 6, 319–325. ( 10.1111/j.1474-9726.2007.00283.x) [DOI] [PubMed] [Google Scholar]
- 53.Mattson MP. 2007. Calcium and neurodegeneration. Aging Cell 6, 337–350. ( 10.1111/j.1474-9726.2007.00275) [DOI] [PubMed] [Google Scholar]
- 54.Gibson GE, Peterson C. 1987. Calcium and the aging nervous system. Neurobiol. Aging 8, 329–343. ( 10.1016/0197-4580(87)90072-8) [DOI] [PubMed] [Google Scholar]
- 55.Landfield PW. 1987. ‘Increased calcium-current’ hypothesis of brain aging. Neurobiol. Aging 8, 346–347. ( 10.1016/0197-4580(87)90074-1) [DOI] [PubMed] [Google Scholar]
- 56.Kirischuk S, Verkhratsky A. 1996. Calcium homeostasis in aged neurones. Life Sci. 59, 451–459. ( 10.1016/0024-3205(96)00324-4) [DOI] [PubMed] [Google Scholar]
- 57.Thibault O, Gant JC, Landfield PW. 2007. Expansion of the calcium hypothesis of brain aging and Alzheimer's disease: minding the store. Aging Cell 6, 307–317. ( 10.1111/j.1474-9726.2007.00295.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zanos P, Bhat S, Terrillion CE, Smith RJ, Tonelli LH, Gould TD. 2015. Sex-dependent modulation of age-related cognitive decline by the L-type calcium channel gene Cacna1c (Cav 1.2). Eur. J. Neurosci. 42, 2499–2507. ( 10.1111/ejn.12952) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zaidi A, Gao J, Squier TC, Michaelis ML. 1998. Age-related decrease in brain synaptic membrane Ca2+-ATPase in F344/BNF1 rats. Neurobiol. Aging 19, 487–495. ( 10.1016/S0197-4580(98)00078-5) [DOI] [PubMed] [Google Scholar]
- 60.Tonkikh A, Janus C, El-Beheiry H, Pennefather PS, Samoilova M, McDonald P, Ouanounou A, Carlen PL. 2006. Calcium chelation improves spatial learning and synaptic plasticity in aged rats. Exp. Neurol. 197, 291–300. ( 10.1016/j.expneurol.2005.06.014) [DOI] [PubMed] [Google Scholar]
- 61.Disterhoft J, Oh M. 2007. Alterations in intrinsic neuronal excitability during normal aging. Aging Cell 6, 327–336. ( 10.1111/j.1474-9726.2007.00297.x) [DOI] [PubMed] [Google Scholar]
- 62.Serrano F, Klann E. 2004. Reactive oxygen species and synaptic plasticity in the aging hippocampus. Ageing Res. Rev. 3, 431–443. ( 10.1016/j.arr.2004.05.002) [DOI] [PubMed] [Google Scholar]
- 63.Watson JB, Arnold MM, Ho YS, O'Dell TJ. 2006. Age-dependent modulation of hippocampal long-term potentiation by antioxidant enzymes. J. Neurosci. Res. 84, 1564–1574. ( 10.1002/jnr.21040) [DOI] [PubMed] [Google Scholar]
- 64.Bodhinathan K, Kumar A, Foster TC. 2010. Redox sensitive calcium stores underlie enhanced after hyperpolarization of aged neurons: role for ryanodine receptor mediated calcium signaling. J. Neurophysiol. 104, 2586–2593. ( 10.1152/jn.00577.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marschallinger J, et al. 2015. Structural and functional rejuvenation of the aged brain by an approved anti-asthmatic drug. Nat. Commun. 6, 8466 ( 10.1038/ncomms9466) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brewer LD, Thibault V, Chen KC, Langub MC, Landfield PW, Porter NM. 2001. Vitamin D hormone confers neuroprotection in parallel with downregulation of L-type Ca2+ channel expression in hippocampal neurons. J. Neurosci. 21, 98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brewer LD, Porter NM, Kerr DS, Landfield PW, Thibault O. 2006. Chronic 1α,25-(OH)2vitamin D3 treatment reduces Ca2+-mediated hippocampal biomarkers of aging. Cell Calcium 40, 277–286. ( 10.1016/j.ceca.2006.04.001) [DOI] [PubMed] [Google Scholar]
- 68.Latimer CS, et al. 2014. Vitamin D prevents cognitive decline and enhances hippocampal synaptic function in aging rats. Proc. Natl Acad. Sci. USA 111, E4359–E4366. ( 10.1073/pnas.1404477111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roselli F, Caroni P. 2015. From intrinsic firing properties to selective neuronal vulnerability in neurodegenerative diseases. Neuron 85, 901–910. ( 10.1016/j.neuron.2014.12.063) [DOI] [PubMed] [Google Scholar]
- 70.Scragg R, Jackso R, Holdaway IM, Lim T, Beaglehole R. 1990. Myocardial infarction is inversely associated with plasma 25-hydroxyvitamin D3 levels: a community-based study. Int. J. Epidemiol. 19, 559–563. ( 10.1093/ije/19.3.559) [DOI] [PubMed] [Google Scholar]
- 71.Heaney RH. 2008. Vitamin D in Health and Disease. Clin. J. Am. Soc. Nephrol. 3, 1535–1541. ( 10.2215/CJN.01160308) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim DH, Sabour S, Sagar UN, Adams S, Whellan DJ. 2008. Prevalence of hypovitaminosis D in cardiovascular diseases (from the National Health and Nutrition Examination Survey 2001 to 2004). Am. J. Cardiol. 102, 1540–1544. ( 10.1016/j.amjcard.2008.06.067) [DOI] [PubMed] [Google Scholar]
- 73.Anderson JL, May HT, Horne BD, Bair TL, Hall NL, Carlquist JF, Lappé DL, Muhlestein JB. 2010. Relation of vitamin D deficiency to cardiovascular risk factors, disease status, and incident events in a general healthcare population. Am. J. Cardiol. 106, 963–968. ( 10.1016/j.amjcard.2010.05.027) [DOI] [PubMed] [Google Scholar]
- 74.Vacek JL, Vanga SR, Good M, Lai SM, Lakkireddy D, Howard PA. 2012. Vitamin D deficiency and supplementation and relation to cardiovascular health. Am. J. Cardiol. 109, 359–363. ( 10.1016/j.amjcard.2011.09.020) [DOI] [PubMed] [Google Scholar]
- 75.Hossein-nezhad A, Holick MF. 2013. Vitamin D for health: a global perspective. Mayo Clin. Proc. 88, 720–755. ( 10.1016/j.mayocp.2013.05.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dong J, Lau CW, Wong SL, Huang Y. 2014. Cardiovascular benefits of vitamin D. Acta Physiologica Sinica 66, 30–36. ( 10.13294/j.aps.2014.0005) [DOI] [PubMed] [Google Scholar]
- 77.Weng S, Sprague JE, Oh J, Riek AE, Chin K, Garcia M, Bernal-Mizrachi C. 2013. Vitamin D deficiency induces high blood pressure and accelerates atherosclerosis in mice. PLoS ONE 8, e54625 ( 10.1371/journal.pone.0054625) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yuan W, et al. 2007. 1,25-Dihydroxyvitamin D3 suppresses renin gene transcription by blocking the activity of the cyclic AMP response element in the renin gene promoter. J. Biol. Chem. 282, 29 821–29 830. ( 10.1074/jbc.M705495200) [DOI] [PubMed] [Google Scholar]
- 79.Li YC, Kong J, Wei M, Chen Z-F, Liu SQ, Cao L-P. 2002. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin–angiotensin system. J. Clin. Invest. 110, 229–238. ( 10.1172/JCI15219) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xiang W, et al. 2005. Cardiac hypertrophy in vitamin D receptor knockout mice: role of the systemic and cardiac renin–angiotensin systems. Am. J. Physiol. Endocrinol. Metab. 288, E125–E132. ( 10.1152/ajpendo.00224.2004) [DOI] [PubMed] [Google Scholar]
- 81.Zhou C, Lu F, Cao K, Xu D, Goltzman D, Miao D. 2008. Calcium-independent and 1,25(OH)2D3-dependent regulation of the renin-angiotensin system in 1alpha-hydroxylase knockout mice. Kidney Int. 74, 170–179. ( 10.1038/ki.2008.101) [DOI] [PubMed] [Google Scholar]
- 82.Nasri H, Behradmanesh S, Ahmadi A, Rafieian-Kopaei M. 2014. Impact of oral vitamin D (cholecalciferol) replacement therapy on blood pressure in type 2 diabetes patients; a randomized, double-blind, placebo controlled clinical trial. J. Nephropathol. 3, 29–33. ( 10.12860/jnp.2014.07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Romero JC, Reckelhoff JF. 1999. Role of angiotensin and oxidative stress in essential hypertension. Hypertension 34, 943–949. ( 10.1161/01.HYP.34.4.943) [DOI] [PubMed] [Google Scholar]
- 84.Ortiz MC, Manriquez MC, Romero JC, Juncos JA. 2001. Antioxidants block angiotensin II-induced increases in blood pressure and endothelin. Hypertension 38, 655–659. ( 10.1161/01.HYP.38.3.655) [DOI] [PubMed] [Google Scholar]
- 85.Berridge MJ. 2012. Calcium signalling remodelling and disease. Biochem. Soc. Trans. 40, 297–309. ( 10.1042/BST20110766) [DOI] [PubMed] [Google Scholar]
- 86.Berridge MJ. 2006. Remodelling Ca2+ signalling systems and cardiac hypertrophy. Biochem. Soc. Trans. 34, 228–231. ( 10.1042/BST0340228) [DOI] [PubMed] [Google Scholar]
- 87.Lipp P, Laine M, Tovey SC, Burrell KM, Berridge MJ, Li W, Bootman MD. 2000. Functional InsP3 receptors that may modulate excitation-contraction coupling in the heart. Curr. Biol. 10, 939–942. ( 10.1016/S0960-9822(00)00624-2) [DOI] [PubMed] [Google Scholar]
- 88.Wu X, et al. 2006. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation–transcription coupling. J. Clin. Invest. 116, 675–682. ( 10.1172/JCI27374) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Molkentin JD. 2006. Dichotomy of Ca2+ in the heart: contraction versus intracellular signaling. J. Clin. Invest. 116, 623–626. ( 10.1172/JCI27824) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Luo D, et al. 2008. Nuclear Ca2+ sparks and waves mediated by inositol 1,4,5-trisphosphate receptors in neonatal rat cardiomyocytes. Cell Calcium 43, 165–174. ( 10.1016/j.ceca.2007.04.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Harzheim D, Movassagh M, Foo RS, Ritter O, Tashfeen A, Conway SJ, Bootman MD, Roderick HL. 2009. Increased InsP3Rs in the junctional sarcoplasmic reticulum augment Ca2+ transients and arrhythmias associated with cardiac hypertrophy. Proc. Natl Acad. Sci. USA 106, 11 406–11 411. ( 10.1073/pnas.0905485106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Higazi DR, et al. 2009. Endothelin-1-stimulated InsP3-induced Ca2+ release is a nexus for hypertrophic signalling in cardiac myocytes. Mol. Cell 33, 472–482. ( 10.1016/j.molcel.2009.02.005) [DOI] [PubMed] [Google Scholar]
- 93.Nakayama H, et al. 2010. The IP3 receptor regulates cardiac hypertrophy in response to select stimuli. Circ. Res. 107, 659–666. ( 10.1161/CIRCRESAHA.110.220038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sankar N, deTombe PP, Mignery GA. 2014. Calcineurin-NFATc regulates type 2 inositol 1,4,5-trisphosphate receptor (InsP3R2) expression during cardiac remodeling. J. Biol. Chem. 289, 188–198. ( 10.1074/jbc.M113.495242) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Drawnel FM, et al. 2012. Mutual antagonism between IP3RII and miRNA-133a regulates calcium signals and cardiac hypertrophy. J. Cell Biol. 199, 783–798. ( 10.1083/jcb.201111095) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Choudhury S, Bae S, Ke QJY, Lee JY, Singh SS, St-Arnaud R, del Monte F, Kang PM. 2014. Abnormal calcium handling and exaggerated cardiac dysfunction in mice with defective Vitamin D signaling. PLoS ONE 9, e108382 ( 10.1371/journal.pone.0108382) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sag CM, Santos CX, Shah AM. 2014. Redox regulation of cardiac hypertrophy. J. Mol. Cell Cardiol. 73, 103–111. ( 10.1016/j.yjmcc.2014.02.002) [DOI] [PubMed] [Google Scholar]
- 98.Köhler AC, Sag CM, Maier LS. 2014. Reactive oxygen species and excitation-contraction coupling in the context of cardiac pathology. J. Mol. Cell Cardiol. 73, 92–102. ( 10.1016/j.yjmcc.2014.03.001) [DOI] [PubMed] [Google Scholar]
- 99.Bendall JK, Cave AC, Heymes C, Gall N, Shah AM. 2002. Pivotal role of a gp91(phox)-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation. 105, 293–296. ( 10.1161/hc0302.103712) [DOI] [PubMed] [Google Scholar]
- 100.Li JM, Gall NP, Grieve DJ, Chen M, Shah AM. 2002. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension 40, 477–484. ( 10.1161/01.HYP.0000032031.30374.32) [DOI] [PubMed] [Google Scholar]
- 101.Burgoyne JR, Mongue-Din H, Eaton P, Shah AJ. 2012. Redox signalling in cardiac physiology and pathology. Circ. Res. 111, 1091–1106. ( 10.1161/CIRCRESAHA.111.255216) [DOI] [PubMed] [Google Scholar]
- 102.Mancuso P, Rahman A, Hershey SD, Dandu L, Nibbelink KA, Simpson RU. 2008. 1,25-dihydroxyvitamin-D3treatment reduces cardiac hypertrophy and left ventricular diameter in spontaneously hypertensive heart failure-prone (cp/+) rats independent of changes in serum leptin. J. Cardiovasc. Pharm. 51, 559–564. ( 10.1097/FJC.0b013e3181761906) [DOI] [PubMed] [Google Scholar]
- 103.Gotsman I, Shauer A, Zwas DR, Hellman Y, Keren A, Lotan C, Admon D. 2012. Vitamin D deficiency is a predictor of reduced survival in patients with heart failure; vitamin D supplementation improves outcome. Eur. J. Heart Fail. 14, 357–366. ( 10.1093/eurjhf/hfr175) [DOI] [PubMed] [Google Scholar]
- 104.Khachaturian ZS. 1989. Calcium, membranes, aging, and Alzheimer's disease. Introduction and overview. Ann. N Y Acad. Sci. 568, 1–4. ( 10.1111/j.1749-6632.1989.tb12485.x) [DOI] [PubMed] [Google Scholar]
- 105.LaFerla FM. 2002. Calcium dyshomeostasis and intracellular signaling in Alzheimer's disease. Nat. Rev. Neurosci. 3, 862–872. ( 10.1038/nrn960) [DOI] [PubMed] [Google Scholar]
- 106.Stutzmann GE. 2007. The pathogenesis of Alzheimer's disease is it a lifelong ‘calciumopathy’. Neuroscientist 13, 546–559. ( 10.1177/1073858407299730) [DOI] [PubMed] [Google Scholar]
- 107.Bezprozvanny I, Mattson MP. 2008. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 31, 454–463. ( 10.1016/j.tins.2008.06.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bojarski L, Herms J, Kuznicki J. 2008. Calcium dysregulation in Alzheimer's disease. Neurochem. Int. 52, 621–633. ( 10.1016/j.neuint.2007.10.002) [DOI] [PubMed] [Google Scholar]
- 109.Stutzmann GE, Mattson MP. 2011. Endoplasmic reticulum Ca2+ handling in cells in health and disease. Pharmacol. Rev. 63, 700–727. ( 10.1124/pr.110.003814) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu H-Y, Hyman BT, Bacskai BJA. 2008. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 59, 214–225. ( 10.1016/j.neuron.2008.06.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lopez JR, Lyckman A, Oddo S, Laferla FM, Querfurth HW, Shtifman A. 2008. Increased intraneuronal resting [Ca2+] in adult Alzheimer's disease mice. J. Neurochem. 105, 262–271. ( 10.1111/j.1471-4159.2007.05135.x) [DOI] [PubMed] [Google Scholar]
- 112.Berridge MJ. 2012. Dysregulation of neural calcium signalling in Alzheimer disease, bipolar disorder and schizophrenia. Prion 6, 1–12. ( 10.4161/pri.21767) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Berridge MJ. 2014. Calcium regulation of neural rhythms, memory and Alzheimer's disease. J. Physiol. 592, 281–293. ( 10.1113/jphysiol.2013.257527) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jensen LE, Bultynck G, Luyten T, Amijee H, Bootman MD, Roderick HL. 2013. Alzheimer's disease-associated peptide Aβ42 mobilizes ER Ca2+ via InsP3R-dependent and -independent mechanisms. Front. Mol. Neurosci 6, 36 ( 10.3389/fnmol.2013.00036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Brawek B, Garaschuk O. 2014. Network-wide dysregulation of calcium homeostasis in Alzheimer's disease. Cell Tissue Res. 357, 427–438. ( 10.1007/s00441-014-1798-8) [DOI] [PubMed] [Google Scholar]
- 116.Demoro A, Parker I. 2013. Cytotoxicity of intracellular Aβ42 amyloid oligomers Involves Ca2+ release from the endoplasmic reticulum by stimulated production of inositol trisphosphate. J. Neurosci. 33, 3824–3833. ( 10.1523/JNEUROSCI.4367-12.2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Um JW, et al. 2013. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 79, 887–902. ( 10.1016/j.neuron.2013.06.036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Armato U, Bonafini C, Chakravarthy B, Pacchiana R, Chiarini A, Whitfield JF, Dal Prà I. 2012. The calcium-sensing receptor: a novel Alzheimer's disease crucial target? J. Neurol. Sci. 322, 137–140. ( 10.1016/j.jns.2012.07.031) [DOI] [PubMed] [Google Scholar]
- 119.Popovics P, Stewart AL. 2012. Phospholipase C-η activity may contribute to Alzheimer's disease-associated calciumopathy. J. Alzheimer's Dis. 30, 737–744. ( 10.3233/JAD-2012-120241) [DOI] [PubMed] [Google Scholar]
- 120.Chen X, Lin R, Chang L, Xu S, Wei X, Zhang J, Wang C, Anwyl C, Wang Q. 2013. Enhancement of long-term depression by soluble amyloid β protein in rat hippocampus is mediated by metabotropic glutamate receptor and involves activation of p38MAPK, STEP and Caspase 3. Neuroscience 253, 435–443. ( 10.1016/j.neuroscience.2013.08.054) [DOI] [PubMed] [Google Scholar]
- 121.Cheung KH, Mei L, Mak DO, Hayashi I, Iwatsubo T, Kang DE, Foskett JK. 2010. Gain-of-function enhancement of IP3receptor modal gating by familial Alzheimer's disease-linked presenilin mutants in human cells and mouse neurons. Sci. Signal. 3, ra22. ( 10.1126/scisignal.2000818) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shilling D, Müller M, Takano H, Mak DO, Abel T, Coulter DA, Foskett JK. 2014. Suppression of InsP3 receptor-mediated Ca2+ signaling alleviates mutant presenilin-linked familial Alzheimer's disease pathogenesis. J Neurosci. 34, 6910–6923. ( 10.1523/JNEUROSCI.5441-13.2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tuohimaa P, Keisala T, Minasyan A, Cachat J, Kalueff A. 2009. Vitamin D nervous system and aging. Psychoneuroendocrinology 34(Suppl 1), S278–S286. ( 10.1016/j.psyneuen.2009.07.003) [DOI] [PubMed] [Google Scholar]
- 124.Annweiler C, Schott AM, Allali G, Bridenbaugh S, Kressig RW, Allain P, Herrmann FR, Beauchet O. 2010. Association of vitamin D deficiency with cognitive impairment in older women: cross-sectional study. Neurology 74, 27–32. ( 10.1212/WNL.0b013e3181beecd3) [DOI] [PubMed] [Google Scholar]
- 125.Annweiler C, Fantino B, Le Gall D, Schott AM, Berrut G, Beauchet O. 2011. Severe vitamin D deficiency is associated with advanced-stage dementia in geriatric inpatients. J. Am. Geriatr. Soc. 59, 169–171. ( 10.1111/j.1532-5415.2010.03166.x) [DOI] [PubMed] [Google Scholar]
- 126.Wang L, et al. 2012. Vitamin D receptor and Alzheimer's disease: a genetic and functional study. Neurobiol. Aging 33, 1844e1–1844e9. ( 10.1016/j.neurobiolaging.2011.12.038) [DOI] [PubMed] [Google Scholar]
- 127.DeLuca GC, Kimball SM, Kolasinski J, Ramagopalan SV, Ebers GC. 2013. Review: the role of vitamin D in nervous system health and disease. Neuropath. Appl. Neurobiol 39, 458–484. ( 10.1111/nan.12020) [DOI] [PubMed] [Google Scholar]
- 128.Gezen-Ak AD, Yilmazer S, Dursun E. 2014. Why vitamin D in Alzheimer's disease? The hypothesis. J. Alzheimer's Dis. 40, 257–269. ( 10.3233/JAD-131970) [DOI] [PubMed] [Google Scholar]
- 129.Annweiler C, Rolland Y, Schott AM, Blain H, Vellas B, Herrmann FR, Beauchet O. 2012. Higher vitamin D dietary intake is associated with lower risk of Alzheimer's disease: a 7-year follow-up. J. Gerontol. A Biol. Sci. Med. Sci. 67, 1205–1211. ( 10.1093/gerona/gls107) [DOI] [PubMed] [Google Scholar]
- 130.Annweiler C, Llewellyn DJ, Beauchet O. 2013. Low serum vitamin D concentrations in Alzheimer's disease: a systematic review and meta-analysis. J. Alzheimers Dis. 33, 659–674. ( 10.3233/JAD-2012-121432) [DOI] [PubMed] [Google Scholar]
- 131.Lehmann DJ, Refsum H, Warden DR, Medway C, Wilcock GK, Smith AD. 2011. The vitamin D receptor gene is associated with Alzheimer's disease. Neurosci. Lett. 504, 79–82. ( 10.1016/j.neulet.2011.08.057) [DOI] [PubMed] [Google Scholar]
- 132.Gezen-Ak DE, et al. 2007. Association between vitamin D receptor gene polymorphism and Alzheimer's disease. Tohoku J. Exp. Med. 212, 275–282. ( 10.1620/tjem.212.275) [DOI] [PubMed] [Google Scholar]
- 133.Durk MR, Han K, Chow EC, Ahrens R, Henderson JT, Fraser PE, Pang KS. 2014. 1α,25-Dihydroxyvitamin D3 reduces cerebral amyloid-β accumulation and improves cognition in mouse models of Alzheimer's disease. J Neurosci. 34, 7091–7101. ( 10.1523/JNEUROSCI.2711-13.2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sutherland MK, Somerville MJ, Yoong LKK, Bergeron C, Haussler MR, Mclachlan DRC. 1992. Reduction of vitamin D hormone receptor mRNA levels in Alzheimer as compared to Huntington hippocampus: correlation with calbindin-28 k mRNA levels. Mol. Brain Res. 13, 239–250. ( 10.1016/0169-328X(92)90032-7) [DOI] [PubMed] [Google Scholar]
- 135.Palop JJ, Jones B, Kekonius L, Chin J, Yu G-Q, Raber J, Masliah E, Mucke L. 2003. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc. Natl Acad. Sci. USA 100, 9572–9577. ( 10.1073/pnas.1133381100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Niture SK, Jaiswal AK. 2012. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 287, 9873–9886. ( 10.1074/jbc.M111.312694) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rohn TT, Vyas V, Hernandez-Estrada T, Nichol KE, Christie L-A, Head E. 2008. Lack of pathology in a triple transgenic mouse model of Alzheimer's disease after overexpression of the anti-apoptotic protein Bcl-2. J. Neurosci. 28, 3051–3059. ( 10.1523/JNEUROSCI.5620-07.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kanninen K, et al. 2009. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer's disease. Proc. Natl Acad. Sci. USA 106, 16 505–16 510. ( 10.1073/pnas.0908397106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Semba RD, Moghekar AR, Hu J, Sun K, Turner R, Ferrucci L, O'Brien R. 2014. Klotho in the cerebrospinal fluid of adults with and without Alzheimer's disease. Neurosci. Lett. 558, 37–40. ( 10.1016/j.neulet.2013.10.058) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kuang K, et al. 2014. Klotho upregulation contributes to the neuroprotection of ligustilide in an Alzheimer's disease mouse model. Neurobiol. Aging 35, 169–178. ( 10.1016/j.neurobiolaging.2013.07.019) [DOI] [PubMed] [Google Scholar]
- 141.Querfurth HK, Selkoe DJ. 1994. Calcium ionophore increases amyloid beta peptide production by cultured cells. Biochemistry 33, 4550–4561. ( 10.1021/bi00181a016) [DOI] [PubMed] [Google Scholar]
- 142.Itkin A, Dupres V, Dufrêne YF, Bechinger B, Ruysschaert JM, Raussens V. 2011. Calcium ions promote formation of amyloid β-peptide (1–40) oligomers causally implicated in neuronal toxicity of Alzheimer's disease. PLoS ONE 6, e18250 ( 10.1371/journal.pone.0018250) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pierrot N, Santos SF, Feyt C, Morel M, Brion JP, Octave JN. 2006. Calcium-mediated transient phosphorylation of tau and amyloid precursor protein followed by intraneuronal amyloid-beta accumulation. J. Biol. Chem. 281, 39 907–39 914. ( 10.1074/jbc.M606015200) [DOI] [PubMed] [Google Scholar]
- 144.Green KN, LaFerla FM. 2008. Linking calcium to Aβ and Alzheimer's disease. Neuron 59, 190–194. ( 10.1016/j.neuron.2008.07.013) [DOI] [PubMed] [Google Scholar]
- 145.Del Prete D, Checler F, Chami M. 2014. Ryanodine receptors: physiological function and deregulation in Alzheimer disease. Mol. Neurodegener. 9, 21–36. ( 10.1186/1750-1326-9-21) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.DeCaluwé J, Dupont G. 2013. The progression towards Alzheimer's disease described as a bistable switch arising from the positive loop between amyloids and Ca2+. J. Theor. Biol. 331, 12–18. ( 10.1016/j.jtbi.2013.04.015) [DOI] [PubMed] [Google Scholar]