

Abstract
Mathematical models have provided important insights into acute viral dynamics within individual patients. In this paper, we study the simplest target cell-limited models to investigate the within-host dynamics of influenza A virus infection in humans. Despite the biological simplicity of the models, we show how these can be used to understand the severity of the infection and the key attributes of possible immunotherapy and antiviral drugs for the treatment of infection at different times post infection. Through an analytic approach, we derive and estimate simple summary biological quantities that can provide novel insights into the infection dynamics and the definition of clinical endpoints. We focus on nine quantities, including the area under the viral load curve, peak viral load, the time to peak viral load and the level of cell death due to infection. Using Markov chain Monte Carlo methods, we fitted the models to data collected from 12 untreated volunteers who participated in two clinical studies that tested the antiviral drugs oseltamivir and zanamivir. Based on the results, we also discuss various difficulties in deriving precise estimates of the parameters, even in the very simple models considered, when experimental data are limited to viral load measures and/or there is a limited number of viral load measurements post infection.
Keywords: viral kinetics, immune control, acute viral infection, within-host modelling, influenza A
1. Introduction
Influenza continues to be a significant cause of morbidity and mortality worldwide [1]. Seasonal epidemics of influenza cause more than 300 000 deaths annually around the world. In the USA alone, a typical seasonal influenza A epidemic results in over 200 000 hospitalizations [2] and 36 000 deaths [3].
Influenza is a short-lived infection with an incubation period of approximately 2 days [4]. The standard pattern of virus kinetics is characterized by rapid exponential growth, with a peak in viral load occurring 1–3 days post infection, followed by a decline over the subsequent 3–5 days. In patients with immunodeficiency, the duration of infection may be prolonged [5,6].
Experimental studies on the typical course of influenza A in a patient have provided useful insights into the processes controlling viral dynamics, especially the associated immune response. Mathematical models have been used to improve understanding of the infection dynamics. Influenza A virus kinetics in the human body has been examined in a number of previous studies [7–10]. Various models that describe the infection dynamics in animals (for example in mice [10–12] and horses [13,14]) have also facilitated the investigation of the immunological mechanisms involved in controlling influenza A replication. Models have been developed to incorporate the innate immune response, the adaptive response or both types of responses against influenza A [7–9,11–18]. Mathematical modelling has also helped in assessing the efficacy of influenza antiviral treatments [19–22] such as neuraminidase inhibitors (oseltamivir and zanamivir) [7,8,17,23,24] and adamantanes (amantadine and rimantadine) [17,25]. The models developed typically consist of systems of ordinary differential equations (ODEs), based on the classic viral dynamic model describing uninfected and infected cells and free virus in the host [7,26–31]. Stochastic effects become important when the viral load is at low levels [8] (for reviews of the mathematical model development, see [32–34]).
Despite the availability of a range of mathematical and statistical tools, limitations in data availability and measurement issues in clinical studies of infected patients hinder the development of complex models that will accurately predict the infection dynamics and provide insight into the exact mechanisms responsible for its control. In this paper, we consider the simplest within-host models of viral dynamics. We assess the validity of these models by fitting to data collected from 12 untreated volunteers experimentally inoculated with human influenza A, but left untreated by drugs or immunotherapeutic agents. Despite the biological simplicity of these models, they can facilitate the assessment of infection-related morbidity and the efficacies of immunotherapies or antiviral treatments and help in the design of control and prevention strategies. We derive and estimate simple quantities that reflect the severity of the infection at different times post acquisition and can be considered as potential endpoints in clinical trials. Such quantities include the area under the viral load curve, the peak viral load, the time to peak viral load and the level of cell death due to infection. The analytical results derived shed light on important parameters that influence patterns in within-patient viral kinetics. Based on the results, we also discuss problems arising in predicting the viral dynamics and evaluating therapeutic interventions when the quality of clinical data on viral load is suboptimal. We provide guidance on what to measure, and when and how frequently, in order to accurately describe the infection dynamics and facilitate the accurate assessment of therapies that restrict viral growth.
2. Material and methods
2.1. Simple mathematical models of viral dynamics
The main effects of the immune response on viral populations can be simply classified as one of the following: (i) decreasing the infection of susceptible cells, (ii) reducing the production of virus by infected cells, (iii) killing infected cells, and (iv) increasing the clearance rate of free virus particles. The simplest model of influenza A virus infection that incorporates implicitly or explicitly these four mechanisms is the TIV model [7]. This model assumes that susceptible cells (primarily epithelial cells), T, become infected when in contact with free infectious virus particles, V, via a mass-action process with rate β. Virus is produced in direct proportion to the number of (productively) infected cells I at rate p per infected cell and is lost at rate c per virion due to non-specific mechanisms that include immune response and natural virus decomposition. An infected cell dies or is killed by immune cells and other non-specific mechanisms with rate δI. Hence, infected cells produce an average p/δI virions during their lifetime. The ODEs that describe this dynamical system are as follows:
| 2.1 |
| 2.2 |
| 2.3 |
with initial conditions
| 2.4 |
The TIV model, (2.1)–(2.4), is shown schematically (figure 1).
Figure 1.

A schematic diagram of the TIV model (2.1)–(2.4) of viral dynamics. (Online version in colour.)
Owing to the paucity of quantitative information on the clearance rate of the virus and the death rate of infected cells, we focus on a simpler version of the TIV model which assumes that the viral dynamics is much faster than the infected cell dynamics and that a quasi-stationary state at which V = pI/c is attained very quickly [35–37]. In this case, the TIV model is reduced to the following pair of equations:
| 2.5 |
and
| 2.6 |
with initial conditions
| 2.7 |
The parameter γ is the death rate of infected cells δI in the TIV model and r can be interpreted as p/c. However, as the parameters p and c cannot be estimated independently, we will consider r as a single parameter. Model (2.5)–(2.7) will be referred to as the TV model. The TV model has the same structure as the susceptible–infectious–recovered (SIR) model in infectious disease epidemiology of viral spread in a population of hosts [38–40] and thus most of the results derived for the SIR model in host populations can be applied in the context of within-host viral dynamics.
The TV model can be extended to the so-called TVA model to include a representation of the overall action strength of the immune response against influenza A (see electronic supplementary material, S1). All three models, TIV, TV and TVA, predict changes in viral load over time post infection accurately. However, the TV and TVA models are ‘better’ models; the fit of the TIV model to observed viral load data is almost equivalent to that of the TV and TVA models (data are not shown) and there are no data to support the estimation of the extra parameters in the more complex model. As there is no independent information about the dynamics of infected cells, the quasi-stationary state assumption in the TV and TVA models is therefore reasonable. We focus on the TV model, but in the electronic supplementary material, S3, we add to previous published analytical results for the TIV model and in the electronic supplementary material, S1, we discuss the TVA model in more detail.
Table 1 summarizes the variables and parameters of all the models considered in this paper.
Table 1.
Notation of the models' variables and parameters.
| notation | meaning | units |
|---|---|---|
| model variables | ||
| T | cells susceptible to infection | cell |
| I | infected cells | cell |
| V | viral load | TCID50 ml−1 |
| A | immune response | |
| model parameters | ||
| β | infection rate | (TCID50/ml)−1 × day−1 |
| p | viral production rate per infected cell | TCID50 ml−1 × day−1 |
| c | viral clearance rate | day−1 |
| δI | death rate of an infected cell | day−1 |
| γ | viral decay rate | day−1 |
| r | rate at which target cells that become infected produce virus in the TV and TVA models | TCID50 ml−1 |
| w | clearance rate of virus by immune response | day−1 |
The above models encapsulate a number of biological assumptions. For example, the regeneration and natural death of target cells have been neglected due to the long time scales of these processes compared with the time scale of influenza infection [41,42]. Therefore, even in cases of severe infection the virus will eventually decline due to the depletion of susceptible target cells. Including the regeneration of target cells does not improve the model fit in most cases [7,10–14,25,43]. An additional assumption is that there is no delay between cell infection and production of virus (a latent phase). Moreover, it should be noted that if no death of infected cells occurs during the latent phase, then the delay simply postpones the infection dynamics (delays the onset of the infection, reduces the peak of the viral load and increases the duration of the infection) without reducing the amount of viral shedding significantly (if regrowth of susceptible cells is considered then increasing the delay of virus production will result in a reduction in the amount of viral shedding). Finally, the loss of virions through cell entry is also considered negligible and absorbed into the loss term −cV, given that any one cell has the potential to produce between 103 and 104 virions [44], which is much more than the number of virions needed to infect a cell [45]. This loss might be important in in vitro models [25].
2.2. Parameter estimation
2.2.1. Datasets used
We use viral load data from two different datasets: the first consists of six volunteers from the placebo group of the oseltamivir trial conducted by Roche [46]. All the participants were healthy adults and screened for haemagglutination inhibition titre. Intranasal inoculation with 106 (50% tissue culture) of a safety-tested pool (TCID50) of human A/Texas/36/91 H1N1 influenza virus was performed on day 0. Nasal lavage fluids were collected for virus isolation and titration by standard methods on days 2–8. The second dataset also consists of six volunteers who were part of the placebo group of the zanamivir trial conducted by GlaxoSmithKline (GSK). The volunteers in this trial were also inoculated with human H1N1 influenza virus (A/Texas/91 H1N1) following a similar procedure.
For the model fitting and the estimation of the parameters, when two or more (sequential) viral load data points fall below the detection limit we exclude them all but the first one. In this case, the first undetectable viral load data point is set to be equal to the value of the detection limit. In the data analysed in this study, the value of the detection limit was not known and it was set to be equal to 0.7 TCID50 ml−1, which is 0.05 TCID50 ml−1 below the smallest measurement value in the two datasets considered.
2.2.2. Fitting procedure
In the TV model the parameters r, I0 and T0 cannot be estimated independently. We fixed T0 at 4 × 108 cells [7] and I0 at 0. A structural identifiability analysis of the TV model [47] shows that the estimation of the identifiable parameters (β, γ, r or T0) requires at least five measurements of viral load at distinct time points. However, examining the pairwise relationships between the parameters (see §3.2), it is observed that all the parameters are correlated and it is thus not possible to produce good estimates of each individually. Owing to the correlation between β and r, we reparametrized the model by replacing rβ in equation (2.6) by l and we estimated l and β (note: β and l are also correlated).
We fitted the TV model to data and estimated the parameters for each patient independently using a random-walk Metropolis–Hastings algorithm. Independent prior distributions were chosen for each of the unknown parameters V0, β, l and γ. An exponential distribution with mean 100 was chosen for V0 and γ. A uniform distribution [0–0.003] (except for patients 3 and 4 in the Roche -dataset, [0–0.01]) was chosen for β while a uniform distribution [0–0.0000001] was chosen for l. It was assumed that the measured viral loads were lognormally distributed around the true viral loads [48] with standard deviation 0.3 log10 (this was based on data provided in [49], but preliminary studies show that the assumed standard deviation of the lognormal distribution does not significantly affect the parameter estimates). For each individual dataset, 8.1 × 105 sampling iterations were performed, of which the first 104 iterations were discarded. To reduce autocorrelation, every 500th sample was recorded [50]. Convergence was assessed visually from the traces of each parameter.
2.3. Infection-related quantities
We derive either exact or approximate solutions of nine infection-related quantities that can help us to interpret the viral load scores in terms of infection severity at both the individual and the population level. The quantities considered are the following: the basic reproduction ratio R0 and the viral growth rate r0, which indicate the speed at which the virus disseminates within the human host at the early stage of infection; the generation time Tg [51], which indicates the speed at which the infection spreads in the population; the area under the viral load curve AV [9,33], which indicates the infectiousness of the host; the peak viral load PV [33], which is thought to be correlated with symptom scores; the time to peak viral load  [10,17], which can be used for assessing infection progression; the viral decay rate rd, which is the rate of viral clearance at the late stage of infection; the duration of infection td [33], which shows the time scale of the infection and can indicate the time in which the patient is infectious; and the fraction of dead cells at the end of the infection D [10,33], which indicates the damage to the tissue caused by the infection, the occurrence of some respiratory symptoms and the time to recovery. The quantities are summarized in the schematic in figure 2. For more information about these quantities and the importance of each of them, see electronic supplementary material, S2.
[10,17], which can be used for assessing infection progression; the viral decay rate rd, which is the rate of viral clearance at the late stage of infection; the duration of infection td [33], which shows the time scale of the infection and can indicate the time in which the patient is infectious; and the fraction of dead cells at the end of the infection D [10,33], which indicates the damage to the tissue caused by the infection, the occurrence of some respiratory symptoms and the time to recovery. The quantities are summarized in the schematic in figure 2. For more information about these quantities and the importance of each of them, see electronic supplementary material, S2.
Figure 2.

Summary of the infection-related quantities: the basic reproduction ratio, the initial viral growth rate, the generation time, the peak viral load, the time to peak viral load, the infection duration (time interval in which the viral load is above a threshold), the area under the viral load curve above a threshold and the fraction of dead cells. (Online version in colour.)
3. Results
The TV model fits to the placebo data and the uncertainty in the model solution is illustrated in figure 3. Despite the simplification of the complex biological process, the TV model provides a good description of the data for most of the subjects.
Figure 3.

TV model fit: the black line represents the median estimate of viral dynamics (in log10 scale) and yellow lines are viral dynamic curves based on 10 000 samples from the posterior distribution of the parameters. Red squares are viral load data points. (a) Placebo-group patients from the oseltamivir trial (Roche dataset); (b) placebo-group patients from the zanamivir trial (GSK dataset).
3.1. Infection-related quantities
In table 2, we summarize how the infection-related quantities in the TV model are affected by the administration of treatments that decrease the rate of infection (e.g. adamantane antiviral drugs [17,25]) and the viral production rate (e.g. neuraminidase inhibitors [7,8,17,23]) and hypothetical treatments that increase the virus clearance rate (e.g. monoclonal antibodies [52]). We also present the influence of the initial viral load. The results presented are for cases where the basic reproduction ratio R0 is greater than 1. A general conclusion is that the time of treatment during the infection is very important and, based on the models considered, its effect is highly dependent on the number of the remaining susceptible cells at this time. In table 3, we summarize the main determinants of some of the infection-related quantities as defined by the mathematical expressions derived. In table 4, we show the estimates of the infection-related quantities in the TV model based on the parameter estimates presented in the electronic supplementary material, table S5. We present both the numerical and analytical solution showing the good accuracy of the analytical results based on novel approximations. Figures 4 and 5 illustrate, respectively, the variability of the parameter estimates and the variability of the infection-related quantities between patients.
Table 2.
Sensitivity of infection-related quantities to changes in parameter values in the TV model.
| infection-related quantities in the TV model |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| basic reproduction ratio | initial viral growth rate | late viral decay rate | area under the viral load curve | peak viral load | time to peak viral load | duration of infection | generation time | fraction of dead cells | ||
| action | decreasing β decreasing r decreasing T0 |
decrease | decrease | almost constant | decreasea | decrease | increase | increaseb | increase | decrease |
| increasing γ | decrease | decrease | increase | decrease | decrease | increasec | decreased | increasee | decrease | |
| decreasing V0 | constant | almost no effect | almost no effect | decreasef | decreasef,g | increase | increasef | increase | decreasef | |
aAbove a threshold of β, all the target cells become infected and further increase of β does not affect AV.
bAs r decreases, a decrease is observed when R0 is relatively large and all (or almost all) cells get infected.
cA decrease is observed when R0 is large and by the time of the peak the majority of cells die due to infection.
dAn increase is observed in cases where R0 is relatively small and there is availability of target cells.
eA decrease is observed for relatively large values of R0 when the majority of the susceptible cells become infected.
fThe variation is small.
gPeak viral load increases almost as much as V0 increases.
Table 3.
Summary of the main parameters determining the infection-related quantities as derived from analytical results.
| infection-related quantities | area under the viral load curve | fraction of dead cells | time to peak viral load | peak viral load | viral decay rate |
|---|---|---|---|---|---|
| mainly influenced by | R0 | R0 | all parameters, including V0 | viral production rate (TV model) | death rate of infected cells, virus clearance rate |
Table 4.
Numerical (NS) and analytical (AS) solution of the infection-related quantities based on the posterior medians of the parameters of the TV model presented in the electronic supplementary material, table S5. The numerical solutions of the area under the viral load curve and fraction of dead cells have been compared with the simplified approximations (S3A.15) and (S3B.5) in the electronic supplementary material, S3A and S3B, respectively.
| estimates of the infection-related quantities |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| basic reproduction ratio (dimensionless) | initial viral growth rate (day−1) | late viral decay rate (day−1) | area under the viral load curve (TCID50 ml−1) | peak viral load (TCID50 ml−1) | time to peak viral load (days) | duration of infection (days) |
generation time (days) | fraction of dead cells (dimensionless) | ||||
| human influenza | A/Texas/36/91 H1N1 (Roche) | patient 1 | NS | 2.3764 | 7.7769 × 104 | 1.0676 × 105 | 2.2369 | 4.3051 | 2.3369 | 0.8750 | ||
| AS | 7.6915 | 5.5881 | 7.8805 × 104 | 1.0676 × 105 | 2.1109 | 4.2552 | 2.6827 | 0.8866 | ||||
| patient 2 | NS | 4.0559 | 6.6616 × 104 | 7.5683 × 104 | 2.0584 | 5.4530 | 2.2789 | 0.9813 | ||||
| AS | 8.3454 | 2.7309 | 6.6629 × 104 | 7.5683 × 104 | 1.9673 | 6.2280 | 2.4577 | 0.9815 | ||||
| patient 3 | NS | 5.9974 | 1.4972 × 104 | 1.4289 × 104 | 1.8299 | 5.9046 | 2.1974 | 0.9975 | ||||
| AS | 8.8971 | 1.7803 | 1.4976 × 104 | 1.4289 × 104 | 1.7592 | 7.3469 | 1.7292 | 0.9975 | ||||
| patient 4 | NS | 2.8085 | 4.2186 × 104 | 5.2681 × 104 | 2.2267 | 4.5206 | 2.3626 | 0.9257 | ||||
| AS | 7.5676 | 4.1844 | 4.2360 × 104 | 5.2681 × 104 | 2.1077 | 4.8000 | 2.6650 | 0.9295 | ||||
| patient 5 | NS | 1.6335 | 1.6271 × 105 | 2.6616 × 105 | 2.4222 | 3.7942 | 2.4662 | 0.6595 | ||||
| AS | 7.8186 | 12.3417 | 1.8319 × 105 | 2.6616 × 105 | 2.2766 | 3.3192 | 2.7704 | 0.7425 | ||||
| patient 6 | NS | 4.3905 | 6.6905 × 104 | 7.2093 × 104 | 2.0627 | 5.8110 | 2.3130 | 0.9869 | ||||
| AS | 8.2828 | 2.4429 | 6.6912 × 104 | 7.2093 × 104 | 1.9743 | 6.7194 | 2.4321 | 0.9869 | ||||
| A/Texas/91 H1N1 (GSK) | patient 1 | NS | 3.0683 | 4.0336 × 105 | 3.7139 × 105 | 3.2494 | 7.3740 | 3.4535 | 0.9449 | |||
| AS | 5.8292 | 2.8184 | 4.0419 × 105 | 3.7139 × 105 | 3.1010 | 7.7957 | 4.0286 | 0.9469 | ||||
| patient 2 | NS | 5.2974 | 6.7209 × 105 | 7.8319 × 105 | 1.9805 | 6.8742 | 2.2529 | 0.9949 | ||||
| AS | 10.0341 | 2.3349 | 6.7209 × 105 | 7.8319 × 105 | 1.9138 | 7.8943 | 2.3814 | 0.9949 | ||||
| patient 3 | NS | 3.6412 | 1.7133 × 105 | 1.9739 × 105 | 2.2544 | 5.7490 | 2.4501 | 0.9708 | ||||
| AS | 7.9745 | 3.0193 | 1.7141 × 105 | 1.9739 × 105 | 2.1541 | 6.3284 | 2.7785 | 0.9713 | ||||
| patient 4 | NS | 4.5396 | 2.2585 × 104 | 2.0012 × 104 | 2.2452 | 6.2417 | 2.5590 | 0.9888 | ||||
| AS | 6.9450 | 1.9621 | 2.2589 × 104 | 2.0012 × 104 | 2.1415 | 7.4055 | 2.5296 | 0.9888 | ||||
| patient 5 | NS | 2.0543 | 4.6104 × 109 | 7.6978 × 109 | 3.1775 | 6.9649 | 3.2438 | 0.8110 | ||||
| AS | 8.7712 | 8.3197 | 4.7642 × 109 | 7.6978 × 109 | 3.0597 | 5.8517 | 4.0535 | 0.8381 | ||||
| patient 6 | NS | 1.9183 | 2.0410 × 106 | 2.0939 × 106 | 3.6680 | 7.1460 | 3.7643 | 0.7730 | ||||
| AS | 5.2350 | 5.7006 | 2.1434 × 106 | 2.0939 × 106 | 3.4649 | 6.1130 | 4.4482 | 0.8118 | ||||
| average | NS | 3.4818 | 3.8451 × 108 | 6.4182 × 108 | 2.4510 | 5.8449 | 2.6399 | 0.9091 | ||||
| AS | 7.7827 | 4.4353 | 3.9734 × 108 | 6.4182 × 108 | 2.3359 | 6.1714 | 2.9131 | 0.9230 | ||||
Figure 4.

Estimated posterior medians of individual parameters and the corresponding 95% credible intervals for the 12 patients in the two datasets. The estimated values of the individual parameters are presented in the electronic supplementary material, table S5.
Figure 5.

Scatter plots illustrating the variability of the infection-related quantities between the 12 patients in the two datasets. Each colour represents a patient. Numerical solutions are represented by filled circles and analytical solutions by asterisks. The estimated values of the quantities are presented in table 4.
3.1.1. Basic reproduction ratio, R0
In the TV model, the basic reproduction ratio is given by  . In our study, the within-host R0 has been estimated to be approximately 3.5 (table 4). R0 is highly dependent on the model choice and the assumptions about the immune responses following infection. Moderate to high values of R0 are expected in influenza A as the virus disseminates very rapidly within the host.
. In our study, the within-host R0 has been estimated to be approximately 3.5 (table 4). R0 is highly dependent on the model choice and the assumptions about the immune responses following infection. Moderate to high values of R0 are expected in influenza A as the virus disseminates very rapidly within the host.
3.1.2. Viral growth rate, r0
Assuming that at the initial stage of the infection the number of target cells remains constant, it can be shown that the initial viral growth rate in the TV model can be described very well by rβT0 − γ. The rate at which the virus population grows in the patient over the initial period of infection is essentially independent of V0. Typically, the viral growth rate of influenza A is high, suggesting a high value of R0 [7]. In our study, the average r0 has been calculated to be approximately equal to 7.8 day−1 (table 4).
3.1.3. Generation time, Tg
An approximate solution for the average generation time (at the population level) during the course of the infection for the TIV and TV models is derived in the electronic supplementary material, S3F. Increasing the infection rate, the viral production rate, the initial number of target cells or the initial viral load yields lower Tg values. When R0 is relatively large, the increase in the viral clearance rate and the death rate of infected cells also results in lower values for Tg. Moreover, as R0 decreases and the number of cells that become infected decreases, the increase in these two rates yields higher Tg values. The TV model predicts an average generation time of influenza A infection at the population level of around 2.6 days (table 4). This agrees with previous estimations using different datasets [9,53].
3.1.4. Area under the viral load curve, AV
In the electronic supplementary material, S3A, a formula for the area under the viral load curve, AV, in the target cell-limited models TIV and TV is derived. From the approximation derived, it is clear that the basic reproduction ratio is the main determinant of the value of AV. In the TV model, as β increases AV increases exponentially and converges to (V0 + rT0)/γ, which corresponds to the maximum AV (further increase is not possible due to the depletion of susceptible cells). AV increases almost linearly with the viral production rate (especially when the parameter values are such that all susceptible cells eventually get infected) and T0, and it has an inverse relationship with the viral clearance rate and the death rate of infected cells. AV is not affected much by varying V0 as long as V0 remains low (as will normally be the case in practice).
3.1.5. Peak viral load, PV
An exact formula of the peak viral load in the TV model, PV(TV), is derived in the electronic supplementary material, S3C (an approximation of PV(TIV) is also derived in the electronic supplementary material, S3C). From the solution of PV(TV), it can be observed that, although the peak viral load is affected by all model parameters, its value is mainly influenced by the viral production rate r.
As the infection rate β increases, PV increases and converges to a constant, which corresponds to the maximum viral load when all the cells are already infected before the start of the decay phase. If PV reaches this value, then any antiviral treatment for the protection of susceptible cells that is administered after the time of the peak will have very little or no effect during the viral decay period. By contrast, PV increases linearly with the viral production rate and T0. Hence, an infection by a viral strain with a high replication rate might be more severe than an infection with a higher cell infection rate (see also [12]). Therefore, depending on the other parameter values, therapeutic interventions to reduce viral replication will be more effective than those that reduce viral cell infectivity. Similarly, therapeutic, or prophylactic, interventions that aim at the limitation of target cells, e.g. by inducing resistance to virus, may be more effective. PV has an inverse relationship with the viral clearance rate (and the death rate of infected cells in the TIV model, with the increase of the first being slightly more important than that of the second). There is almost no variation of PV with V0.
3.1.6. Time to peak viral load, 
An approximation of the time to the peak of viral load for the TIV and TV models is given in the electronic supplementary material, S3D.  decreases as the infection rate, the viral production rate and the initial number of susceptible cells increase. In cases where the virus disseminates quickly and infects the majority of susceptible cells, a decrease of
decreases as the infection rate, the viral production rate and the initial number of susceptible cells increase. In cases where the virus disseminates quickly and infects the majority of susceptible cells, a decrease of 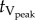 is also observed with increasing viral clearance and infected cell death rates. Otherwise, after a certain value, an increase in these rates delays the onset of the infection and therefore the time to peak. The decrease of V0 also yields higher
is also observed with increasing viral clearance and infected cell death rates. Otherwise, after a certain value, an increase in these rates delays the onset of the infection and therefore the time to peak. The decrease of V0 also yields higher  . An infection where the peak viral load occurs either much earlier or later is not necessarily a more severe infection and needs to be considered with other measures of infection severity such as the peak viral load and the area under the viral load curve. For example, in table 2 it can be observed that the late occurrence of the peak viral load does not imply a severe infection with respect to the value of the peak. In the standard influenza A virus kinetic pattern, viral load reaches its maximum level approximately 2 days after the initiation of the infection [53], which is in good agreement with our model prediction (table 4).
. An infection where the peak viral load occurs either much earlier or later is not necessarily a more severe infection and needs to be considered with other measures of infection severity such as the peak viral load and the area under the viral load curve. For example, in table 2 it can be observed that the late occurrence of the peak viral load does not imply a severe infection with respect to the value of the peak. In the standard influenza A virus kinetic pattern, viral load reaches its maximum level approximately 2 days after the initiation of the infection [53], which is in good agreement with our model prediction (table 4).
3.1.7. Viral decay rate, rd
A derivation of an approximate solution of the viral load during the decay phase for the TIV model is shown in [14] (see also [54]). In the TV model, after the infection of all target cells, the viral load decreases exponentially at rate γ (in the late decay phase the main or the only process that takes place is the clearance of the virus). This clearance rate is a composite of the natural death rates of infected cells and free virus and the action of the immune system in enhancing the clearance of both. Although all model parameters can affect the early phase of virus growth, the decay of the virus at the later stage of the infection depends almost exclusively on the death rate of infected cells and the virus clearance rate (in the TIV model, the parameter that has the lower value dominates towards the end of the infection [14,54]). This dependence suggests that a treatment acting on the infection rate and/or the viral production rate will only be effective if it is administered at the early stage of the infection. A treatment administered after the peak viral load will be most effective if it acts on the death rate of infected cells and/or the viral clearance rate. The average viral decay rate in this study was calculated to be approximately 4.4 day−1 (table 4).
3.1.8. Duration of infection, td
An approximate solution of the duration of infection in the TV model is derived in the electronic supplementary material, S3E. Increasing the infection rate and the number of target cells results in the increase of the basic reproduction ratio and the rapid dissemination of the virus. The rapid infection of the target cells results in the fast convergence to the infection-free steady state where viral load falls to zero. When the infection rate gets too large, a further increase has no significant effect on the infection duration, which then corresponds to the minimum time needed for the infection of all susceptible cells, their death and the clearance of the virus. A similar situation is observed when the initial number of target cells gets very large. An increase in the viral production rate also decreases the infection duration. However, when the rate of virus production is very large, further increase yields higher durations of infection, as then all (or almost all) cells get infected quickly. In this case, the increase in the rate at which free virus is produced by each infected cell results in increased time needed for virus clearance. For relatively large values of R0, the infection duration also decreases as the viral clearance rate and the death rate of infected cells increase. After a certain value and as R0 decreases, the virus disseminates at lower rates and, although a significant decrease in peak viral load and the number of dead cells is observed, the time needed for the virus to be cleared increases due to the high availability of susceptible cells. Therefore, based on the simple models considered, a long infection is not necessarily a severe infection. The observed increase in the infection duration with the decrease of the basic reproduction ratio is counter-intuitive, as an infection with low rates of transmissibility between cells should last for less time. This might be considered a limitation of models without an explicit description of immune responses. The TV model predicts that the infection duration is about 5.8 days (table 4), which is approximately the infectious period of a typical influenza A infection recorded in clinical studies where the date of infection is known (household transmission studies [55]).
3.1.9. Fraction of dead cells at the end of the infection, D
In the electronic supplementary material, S3B, we derive an approximate solution of D for both the TIV and TV models. The solutions of D also show that R0 is the main determinant of the fraction of dead cells at the end of the infection. In particular, decreasing the infection rate, the viral production rate, the initial number of target cells or the initial viral load decreases the total level of cell death due to infection. A decrease in the number of dead cells can also be achieved by increasing the viral clearance rate and the death rate of infected cells. It can be concluded that, although infections with lower infection and virus production rate can be less severe with respect to the peak viral load and the damage they cause to the tissue, they can last longer. The models considered in this paper predict that the majority of epithelial cells will die due to infection (table 4).
3.2. Problems in the parameter estimation due to limitations in data availability
Clinical data availability limits the development and validation of more complex models of acute viral infections that can describe the immunological mechanisms and cellular dynamics in more detail. The question therefore arises of whether the quality and quantity of available viral load data are sufficient to support even the simplest models developed to predict viral dynamics and the impact of therapeutic interventions.
3.2.1. Quantity of viral load measurements
For the simplest within-host viral dynamics mathematical model (2.5)–(2.7), at least five viral load measurements at distinct time points (ideally at least one in each day) are required to estimate the unknown model parameters [47]. In this study, 174 out of 191 individual patients were excluded from the analysis because of either the limited number of viral load measurements made in the clinical observation periods post infection or the large number of missing data points (for example, the data points during the initial phase of the infection). The maximum number of viral load measurements in one patient was eight in 7 days of infection, which is sufficient for making predictions but not entirely adequate for deriving precise parameter estimates and estimates of the infection-related quantities. As illustrated in figure 3, more measurements should be taken during the nonlinear phase, around the peak viral load, where in most cases high uncertainty occurs. Two (precise) viral load measurements at distinct time points in each of the initial viral growth and late viral decay phases might be enough to accurately approximate the rates of increase and decrease of the viral load curve. However, although the durations of these phases have been estimated [54], these vary between patients, even within the fairly homogeneous sample of patients with respect to age used in parameter estimation (table 4 and figure 5).
3.2.2. Precision of viral load measurements
In no dataset was there information about the error in the sampling and measurement methods. Consequently, this introduces a degree of uncertainty regardless of the model accuracy to describe given data (and, thus, more frequent measurements alone will not necessarily reduce the uncertainty in the model predictions, figure 3). Ideally, replicating both the viral load assays and sampling at one time point from the same patient would enable us to estimate the error in the measurement of viral load and improve the model predictions. Hence, given that we take the minimum number of measurements required to estimate the parameters (five in the TV model), it might be better to acquire replicate measurements and get more precise values of the viral load instead of taking samples more frequently at different time points. Ideally, both should be done in clinical epidemiological studies of acute infections. However, we acknowledge that, apart from the practical issues in taking multiple samples from the patients in time points close to each other, other sources of variance might affect the accuracy of the measured viral load. For example, the virus shed by patients may differ at different times of the day.
Note that, in the absence of independent information about parameter values, more precise viral load measurements alone will not necessarily reduce the uncertainty of parameter estimates due to the correlations between them (see §3.2.3).
3.2.3. Availability of data other than viral load
Although the model describes the viral load data well, the absence of information about the values of key biological parameters and the dynamics of uninfected and infected cells makes parameter estimation difficult. The high correlation between the model parameters results in equivalent predictions of the model for a wide parameter space. The individual posterior medians and the corresponding 95% credible intervals for the four unknown parameters of the TV model are summarized in the electronic supplementary material, table S5. In the electronic supplementary material, figure S4.2, we provide plots of the posterior distributions of each of these parameters.
The correlations between the parameters for each patient are illustrated by pairwise scatter plots of the parameters (electronic supplementary material, figures S4.3–S4.8). There is correlation between almost all parameters. In particular, in most of the cases the virus clearance rate γ is inversely correlated with parameter β and changes linearly with parameter l, while l is inversely proportional to β. Although the relationships of β, γ and l with the initial viral load V0 are not clear, it seems that V0 is inversely proportional to β and l and not particularly related to γ. Moreover, variation in V0 does not influence the model fit much, unless V0 varies from very small to very large values. The correlations between the model parameters are not surprising as virus infects cells and in turn infected cells produce more virus. Therefore, the virus dynamics can be controlled by just protecting the cells not yet infected, which can be achieved either by decreasing the infection rate or the rate at which virus is produced by each infected cell or by increasing either the killing rate of infected cells or the clearance rate of the virus. Independent measurements of some unknown model parameters would therefore help to produce more precise estimates of the remaining parameters.
Despite the high uncertainty in the estimates of the values of single parameters, their product may be estimated more precisely, resulting in the derivation of more accurate estimates of some of the infection-related quantities. For example, figure S4.1 in the electronic supplementary material S4 illustrates that the basic reproduction ratio R0 can be estimated precisely irrespective of the high uncertainty in individual parameter estimates (R0 = lT0/γ is eventually equal to the slope of the line showing the linear relationship between l and γ, illustrated in the electronic supplementary material, figure S4.7, multiplied by the initial number of target cells T0).
3.2.4. Unknown lower limit of quantification
The different viral load assays differ in the reported lower limits of quantitation (LLOQ) and detection. In our analysis, we had no information on the LLOQ and set it to 0.7 TCID50 ml−1. However, the choice of this value might significantly influence both the model fit and the uncertainty in the parameter estimates.
3.2.5. Unknown sampling time
During our study, the exact sampling time during a day in the data we used was unknown. Given the available information, we assumed that in the Roche dataset samples were taken every 0.5 days between days 1 and 3 and every 1 day afterwards. In the GSK dataset, we assumed that samples were taken exactly every 24 h. However, the precise sampling times can significantly affect the model predictions and therefore they should be recorded. Recording whether the sample was taken in the morning or afternoon is not enough, as variations in the sampling time by some hours might be influential.
4. Discussion
In the past few decades, mathematical model development and its use in the study of infectious diseases in general, and influenza A in particular, has largely focused on disease spread and control within populations of hosts [56]. Models of viral dynamics within an individual person serve many purposes, including creating a better understanding of what determines the temporal trajectory of viral load over time, identifying what needs to be measured experimentally and facilitating the choice of endpoints in clinical trials of possible therapies. Precise mathematical description of viral dynamics also facilitates understanding of the immune response and the assessment of the efficacy of antiviral treatments. It also plays an indirect role in understanding virus transmission between hosts, due to the relationship between viral load and infectiousness, and it can help in the evaluation of therapeutic treatments as part of the development of effective strategies for the mitigation and control of epidemics.
Mathematical models with varying complexity have been developed and analysed by analytical and numerical approaches to provide a description of the growth and decay of the influenza A virus within the patient. However, gaps in knowledge, especially concerning the role of different immune system components and the lack of detailed biological data on, for example, the life expectancies of cells and free virus at different stages of infection, limit model validation.
In this paper, we revisited and extended simple classic models of viral kinetics to study the influenza A infection within the human body. Despite their simplicity, models that exclude the explicit representation of the immune responses can adequately explain observed patterns of viral growth and decay in patients and facilitate an understanding of the processes that have the greatest impact on the course of infection. One of the advantages of using simple models to describe the infection dynamics is that a number of quantities that reflect the severity of the infection, and some of them can be considered as clinical endpoints used in the assessment of treatments, can be derived analytically. Owing to the variability of the measurements of viral load, and clinical outcome, as well as the limited number of measurements, the derivation of reliable estimates of such quantities directly from the raw data would be difficult. We derived approximate expressions and estimations of a series of morbidity and viral growth and decay related measures, such as the area under the viral load curve, the duration of infection, time to peak viral load and the slope of the viral decay curve, and focused on identifying their key determinants. New results are presented on some of these measures. This focus is a first step in developing tools to aid in the design of clinical trials of candidate therapies to treat infected patients or susceptible individuals to lessen the impact of infection. Despite the limited information that each of these quantities can provide independently, and difficulties in measuring them, the assessment of all quantities together provides insights into how different interventions will act on the observed course of infection when applied at different times post infection and post the initiation of treatment. Most importantly, they also help define what to measure in clinical trials of therapies.
Although we focused on influenza A virus infection, the models and results are applicable to other acute viral infections where measurable viral load persists for a few days to a few weeks in the patient. The fast dynamics of such infections and the short duration of viral replication create problems in the design and conducting of quantitative clinical studies of possible therapies involving sampling from infected patients. However, problems in the estimation of the model parameters and the accurate prediction of the viral load dynamics can be resolved by improving data quality. Based on our analysis, we suggest that frequent sampling at defined time points is essential to creating a deeper understanding of viral kinetics and between-patient variation. More frequent viral load measurements will be more useful if they are accompanied by measurement error data. Reduction of the measurement error of viral load could be achieved by running replicate measurements and replicating sampling of the same patient at one time point. This reduction in the measurement error will also be very important in using these models to guide the design of clinical trials of therapeutic interventions. Determining the importance of variance in viral load between patients and during therapy requires understanding the variation inherent in the sampling and measurements methods. Describing these two sources of variation (between patients and within the sampling and measurement method) is essential in the determination of the efficacy of a therapeutic intervention.
Improving the precision of viral load measurements alone will not necessarily result in more accurate parameter estimates. A major need in future clinical, animal model and in vitro studies is the determination of basic population dynamic parameters, such as cell life expectancies with and without infection and the virus clearance rate. At present their accurate estimation from the time course of infection in the patient is fraught with difficulty, due to the correlations between them, and independent measurements of at least some of them is required.
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Acknowledgements
We would like to thank F. Hoffmann-La Roche Ltd and GlaxoSmithKline Ltd for providing access to their data for this analysis.
Ethics
For the oseltamivir trial conducted by Roche, written informed consent was obtained from each participant in a form approved by the institutional review boards of the University of Virginia, Charlottesville, VA, and the University of Rochester, Rochester, NY, and subjects were compensated for participation [46]. A signed informed consent form for each subject and institutional review board approval to conduct the zanamivir trial, conducted by GlaxoSmithKline (GSK) were obtained by the principal investigator in accordance with the US Food and Drug Administration regulations set forth in Parts 50 and 56, respectively of Title 21 of the Code of Federal Regulations. The study was conducted in accordance with Good Clinical Practice Sponsor Standard Operating Procedures on the conduct and monitoring of clinical trials.
Data accessibility
Raw data are available from https://clinicalstudydatarequest.com for researchers who meet the criteria for access to confidential data.
Authors' contributions
R.M.A. and C.H. designed the study; C.H. developed and analysed the mathematical models, derived the solutions of the infection-related quantities and wrote the paper; E.C., C.V. and C.H. fitted the models to the data; E.C., E.L., C.V. and C.H. acquired and analysed the data; R.M.A. and F.d.W. approved the final manuscript.
Competing interests
R.M.A. is a non-executive board member of GlaxoSmithKline.
Funding
This research has been funded by the Janssen Prevention Center.
References
- 1.Taubenberger J, Morens D. 2008. The pathology of influenza virus infections. Annu. Rev. Pathol. 3, 499–522. ( 10.1146/annurev.pathmechdis.3.121806.154316) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simonsen L, Fukuda K, Schonberger LB, Cox NJ. 2000. The impact of influenza epidemics on hospitalizations. J. Infect. Dis. 181, 831–837. ( 10.1086/315320) [DOI] [PubMed] [Google Scholar]
- 3.Thompson WW, et al. 2003. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289, 179–186. ( 10.1001/jama.289.2.179) [DOI] [PubMed] [Google Scholar]
- 4.Ferguson NM, Cummings DAT, Cauchemez S, Fraser C, Riley S, Meeyai A, Iamsirithaworn S, Burke DS. 2005. Strategies for containing an emerging influenza pandemic in Southeast Asia. Nature 437, 209–214. ( 10.1038/nature04017) [DOI] [PubMed] [Google Scholar]
- 5.Renaud C, Boudreault AA, Kuypers J, Lofy KH, Corey L, Boeckh MJ, Englund JA. 2011. H275Y mutant pandemic (H1N1) 2009 virus in immunocompromised patients. Emerg. Infect. Dis. 17, 653–660; quiz 765 ( 10.3201/eid1704.101429) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Vries E, et al. 2013. Prolonged influenza virus shedding and emergence of antiviral resistance in immunocompromised patients and ferrets. PLoS Pathog. 9, e1003343 ( 10.1371/journal.ppat.1003343) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baccam P, Beauchemin C, Macken CA, Hayden FG, Perelson AS. 2006. Kinetics of influenza A virus infection in humans. J. Virol. 80, 7590–7599. ( 10.1128/jvi.01623-05) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Handel A, Longini IM Jr, Antia R. 2007. Neuraminidase inhibitor resistance in influenza: assessing the danger of its generation and spread. PLoS Comput. Biol. 3, e240 ( 10.1371/journal.pcbi.0030240) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Canini L, Carrat F. 2011. Population modeling of influenza A/H1N1 virus kinetics and symptom dynamics. J. Virol. 85, 2764–2770. ( 10.1128/jvi.01318-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dobrovolny HM, Baron MJ, Gieschke R, Davies BE, Jumbe NL, Beauchemin CA. 2010. Exploring cell tropism as a possible contributor to influenza infection severity. PLoS ONE 5, e13811 ( 10.1371/journal.pone.0013811) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Handel A, Longini IM Jr, Antia R. 2010. Towards a quantitative understanding of the within-host dynamics of influenza A infections. J. R. Soc. Interface 7, 35–47. ( 10.1098/rsif.2009.0067) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miao H, Hollenbaugh JA, Zand MS, Holden-Wiltse J, Mosmann TR, Perelson AS, Wu H, Topham DJ. 2010. Quantifying the early immune response and adaptive immune response kinetics in mice infected with influenza A virus. J. Virol. 84, 6687–6698. ( 10.1128/jvi.00266-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saenz RA, et al. 2010. Dynamics of influenza virus infection and pathology. J. Virol. 84, 3974–3983. ( 10.1128/JVI.02078-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pawelek KA, Huynh GT, Quinlivan M, Cullinane A, Rong L, Perelson AS. 2012. Modeling within-host dynamics of influenza virus infection including immune responses. PLoS Comput. Biol. 8, e1002588 ( 10.1371/journal.pcbi.1002588) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bocharov GA, Romanyukha AA. 1994. Mathematical model of antiviral immune response III. Influenza A virus infection. J. Theor. Biol. 167, 323–360. ( 10.1006/jtbi.1994.1074) [DOI] [PubMed] [Google Scholar]
- 16.Hancioglu B, Swigon D, Clermont G. 2007. A dynamical model of human immune response to influenza A virus infection. J. Theor. Biol. 246, 70–86. ( 10.1016/j.jtbi.2006.12.015) [DOI] [PubMed] [Google Scholar]
- 17.Lee HY, et al. 2009. Simulation and prediction of the adaptive immune response to influenza A virus infection. J. Virol. 83, 7151–7165. ( 10.1128/jvi.00098-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tridane A, Kuang Y. 2010. Modeling the interaction of cytotoxic T lymphocytes and influenza virus infected epithelial cells. Math. Biosci. Eng. 7, 171–185. ( 10.3934/mbe.2010.7.171) [DOI] [PubMed] [Google Scholar]
- 19.De Clercq E. 2006. Antiviral agents active against influenza A viruses. Nat. Rev. Drug Discov. 5, 1015–1025. ( 10.1038/nrd2175) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gubareva LV, Kaiser L, Hayden FG. 2000. Influenza virus neuraminidase inhibitors. Lancet 355, 827–835. ( 10.1016/s0140-6736(99)11433-8) [DOI] [PubMed] [Google Scholar]
- 21.Jefferson T, Demicheli V, Rivetti D, Jones M, Di Pietrantonj C, Rivetti A. 2006. Antivirals for influenza in healthy adults: systematic review. Lancet 367, 303–313. ( 10.1016/S0140-6736(06)67970-1) [DOI] [PubMed] [Google Scholar]
- 22.Hayden FG. 2006. Antivirals for influenza: historical perspectives and lessons learned. Antiviral Res. 71, 372–378. ( 10.1016/j.antiviral.2006.05.016) [DOI] [PubMed] [Google Scholar]
- 23.Canini L, Conway JM, Perelson AS, Carrat F. 2014. Impact of different oseltamivir regimens on treating influenza A virus infection and resistance emergence: insights from a modelling study. PLoS Comput. Biol. 10, e1003568 ( 10.1371/journal.pcbi.1003568) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamal MA, Gieschke R, Lemenuel-Diot A, Beauchemin CA, Smith PF, Rayner CR. 2015. A drug-disease model describing the effect of oseltamivir neuraminidase inhibition on influenza virus progression. Antimicrob. Agents Chemother. 59, 5388–5395. ( 10.1128/aac.00069-15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beauchemin CA, McSharry JJ, Drusano GL, Nguyen JT, Went GT, Ribeiro RM, Perelson AS. 2008. Modeling amantadine treatment of influenza A virus in vitro. J. Theor. Biol. 254, 439–451. ( 10.1016/j.jtbi.2008.05.031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei X, et al. 1995. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373, 117–122. ( 10.1038/373117a0) [DOI] [PubMed] [Google Scholar]
- 27.Nowak MA, Bonhoeffer S, Hill AM, Boehme R, Thomas HC, McDade H. 1996. Viral dynamics in hepatitis B virus infection. Proc. Natl Acad. Sci. USA 93, 4398–4402. ( 10.1073/pnas.93.9.4398) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 271, 1582–1586. ( 10.1126/science.271.5255.1582) [DOI] [PubMed] [Google Scholar]
- 29.Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. 1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373, 123–126. ( 10.1038/373123a0) [DOI] [PubMed] [Google Scholar]
- 30.Nowak M, May RM. 2000. Virus dynamics: mathematical principles of immunology and virology. Oxford, UK: Oxford University Press. [Google Scholar]
- 31.Perelson AS. 2002. Modelling viral and immune system dynamics. Nat. Rev. Immunol. 2, 28–36. ( 10.1038/nri700) [DOI] [PubMed] [Google Scholar]
- 32.Smith AM, Perelson AS. 2011. Influenza A virus infection kinetics: quantitative data and models. Wiley Interdiscip. Rev. Syst. Biol. Med. 3, 429–445. ( 10.1002/wsbm.129) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dobrovolny HM, Reddy MB, Kamal MA, Rayner CR, Beauchemin CA. 2013. Assessing mathematical models of influenza infections using features of the immune response. PLoS ONE 8, e57088 ( 10.1371/journal.pone.0057088) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beauchemin CAA, Handel A. 2011. A review of mathematical models of influenza A infections within a host or cell culture: lessons learned and challenges ahead. BMC Public Health 11, S7 ( 10.1186/1471-2458-11-S1-S7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nowak MA, Bonhoeffer S, Shaw GM, May RM. 1997. Anti-viral drug treatment: dynamics of resistance in free virus and infected cell populations. J. Theor. Biol. 184, 203–217. ( 10.1006/jtbi.1996.0307) [DOI] [PubMed] [Google Scholar]
- 36.Ikeda H, de Boer RJ, Sato K, Morita S, Misawa N, Koyanagi Y, Aihara K, Iwami S. 2014. Improving the estimation of the death rate of infected cells from time course data during the acute phase of virus infections: application to acute HIV-1 infection in a humanized mouse model. Theor. Biol. Med. Modell. 11, 22 ( 10.1186/1742-4682-11-22) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo S, Reed M, Mattingly JC, Koelle K. 2012. The impact of host immune status on the within-host and population dynamics of antigenic immune escape. J. R. Soc. Interface 9, 2603–2613. ( 10.1098/rsif.2012.0180) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anderson RM, May RM. 1992. Infectious diseases of humans: dynamics and control. Oxford, UK: Oxford University Press. [Google Scholar]
- 39.Diekmann O, Heesterbeek H, Britton T. 2013. Mathematical tools for understanding infectious disease dynamics. Princeton, NJ: Princeton University Press. [Google Scholar]
- 40.Diekmann O, Heesterbeek JAP. 2000. Mathematical epidemiology of infectious diseases: model building, analysis and interpretation. New York, NY: Wiley. [Google Scholar]
- 41.Rawlins EL, Hogan BL. 2008. Ciliated epithelial cell lifespan in the mouse trachea and lung. Am. J. Physiol. 295, L231–L234. ( 10.1152/ajplung.90209.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boers JE, den Brok JL, Koudstaal J, Arends JW, Thunnissen FB. 1996. Number and proliferation of neuroendocrine cells in normal human airway epithelium. Am. J. Respir. Crit. Care Med. 154, 758–763. ( 10.1164/ajrccm.154.3.8810616) [DOI] [PubMed] [Google Scholar]
- 43.Smith AM, Adler FR, McAuley JL, Gutenkunst RN, Ribeiro RM, McCullers JA, Perelson AS. 2011. Effect of 1918 PB1-F2 expression on influenza A virus infection kinetics. PLoS Comput. Biol. 7, e1001081 ( 10.1371/journal.pcbi.1001081) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stray SJ, Air GM. 2001. Apoptosis by influenza viruses correlates with efficiency of viral mRNA synthesis. Virus Res. 77, 3–17. ( 10.1016/S0168-1702(01)00260-X) [DOI] [PubMed] [Google Scholar]
- 45.Martin K, Helenius A. 1991. Transport of incoming influenza virus nucleocapsids into the nucleus. J. Virol. 65, 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hayden FG, et al. 1999. Use of the oral neuraminidase inhibitor oseltamivir in experimental human influenza: randomized controlled trials for prevention and treatment. JAMA 282, 1240–1246. ( 10.1001/jama.282.13.1240) [DOI] [PubMed] [Google Scholar]
- 47.Miao H, Xia X, Perelson AS, Wu H. 2011. On identifiability of nonlinear ODE models and application in viral dynamics. SIAM Rev. 53, 3–39. ( 10.1137/090757009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wulff NH, Tzatzaris M, Young PJ. 2012. Monte Carlo simulation of the Spearman-Kaerber TCID50. J. Clin. Bioinform. 2, 5 ( 10.1186/2043-9113-2-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ward CL, Dempsey MH, Ring CJ, Kempson RE, Zhang L, Gor D, Snowden BW, Tisdale M. 2004. Design and performance testing of quantitative real time PCR assays for influenza A and B viral load measurement. J. Clin. Virol. 29, 179–188. ( 10.1016/s1386-6532(03)00122-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carlin BP, Louis TA. 1996. Bayes and empirical Bayes methods for data analysis. New York, NY: Chapman and Hall. [Google Scholar]
- 51.Svensson Å. 2007. A note on generation times in epidemic models. Math. Biosci. 208, 300–311. ( 10.1016/j.mbs.2006.10.010) [DOI] [PubMed] [Google Scholar]
- 52.Ekiert DC, Wilson IA. 2012. Broadly neutralizing antibodies against influenza virus and prospects for universal therapies. Curr. Opin. Virol. 2, 134–141. ( 10.1016/j.coviro.2012.02.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carrat F, Vergu E, Ferguson NM, Lemaitre M, Cauchemez S, Leach S, Valleron AJ. 2008. Time lines of infection and disease in human influenza: a review of volunteer challenge studies. Am. J. Epidemiol. 167, 775–785. ( 10.1093/aje/kwm375) [DOI] [PubMed] [Google Scholar]
- 54.Smith AM, Adler FR, Perelson AS. 2010. An accurate two-phase approximate solution to an acute viral infection model. J. Math. Biol. 60, 711–726. ( 10.1007/s00285-009-0281-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ip DKM, Lau LLH, Chan K-H, Fang VJ, Leung GM, Peiris MJS, Cowling BJ. 2015. The dynamic relationship between clinical symptomatology and viral shedding in naturally acquired seasonal and pandemic influenza virus infections. Clin. Infect. Dis. 62, 431–437. ( 10.1093/cid/civ909) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murillo LN, Murillo MS, Perelson AS. 2013. Towards multiscale modeling of influenza infection. J. Theor. Biol. 332, 267–290. ( 10.1016/j.jtbi.2013.03.024) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data are available from https://clinicalstudydatarequest.com for researchers who meet the criteria for access to confidential data.