

Abstract
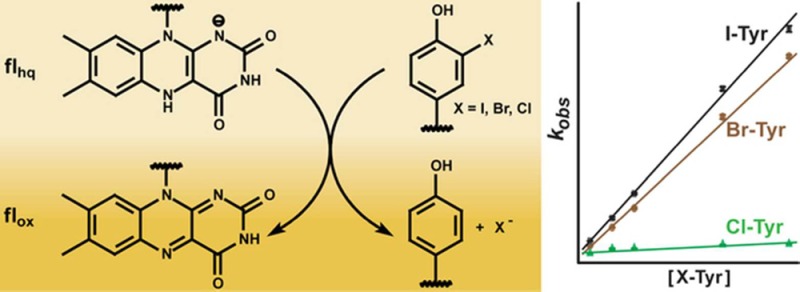
Reductive dehalogenation such as that catalyzed by iodotyrosine deiodinase (IYD) is highly unusual in aerobic organisms but necessary for iodide salvage from iodotyrosine generated during thyroxine biosynthesis. Equally unusual is the dependence of this process on flavin. Rapid kinetics have now been used to define the basic processes involved in IYD catalysis. Time-dependent quenching of flavin fluorescence was used to monitor halotyrosine association to IYD. The substrates chloro-, bromo-, and iodotyrosine bound with similar rate constants (kon) ranging from 1.3 × 106 to 1.9 × 106 M–1 s–1. Only the inert substrate analogue fluorotyrosine exhibited a significantly (5-fold) slower kon (0.3 × 106 M–1 s–1). All data fit a standard two-state model and indicated that no intermediate complex accumulated during closure of the active site lid induced by substrate. Subsequent halide elimination does not appear to limit reactions of bromo- and iodotyrosine since both fully oxidized the reduced enzyme with nearly equivalent second-order rate constants (7.3 × 103 and 8.6 × 103 M–1 s–1, respectively) despite the differing strength of their carbon–halogen bonds. In contrast to these substrates, chlorotyrosine reacted with the reduced enzyme approximately 20-fold more slowly and revealed a spectral intermediate that formed at approximately the same rate as the bromo- and iodotyrosine reactions.
Flavoproteins promote a wide-range of essential reactions in primary and secondary metabolism, and our appreciation of their chemistry continues to expand as their versatility becomes increasingly apparent.1−4 Once a new role for flavin is identified, related processes are often discovered in quick succession. For example, the relatively recent isolation of a flavoprotein promoting an oxidative halogenation in the biosynthesis of chlortetracycline was soon followed by reports of other flavoproteins promoting similar halogenations of other natural products.5 Surprisingly, the range of flavoproteins involved in reductive dehalogenation has remained quite limited since the first report over 30 years ago of flavin in iodotyrosine deiodinase (IYD).6,7 This enzyme is responsible for iodide salvage in vertebrates through the release of iodide from mono- and diiodotyrosine (I-Tyr, I2-Tyr) (Scheme 1). These iodinated derivatives are generated in vivo as side products of thyroid hormone biosynthesis,8 and their iodide must be reclaimed to avoid iodide deficiency and thyroid disease.9,10
Scheme 1. Catalytic Dehalogenation Promoted by IYD.

The use of reduction to drive dehalogenation is common in anaerobes and typically depends on cobalamin or other metal-containing cofactors rather than flavin.11 Conversely, in aerobes, hydrolytic and oxidative processes are primarily used for dehalogenation.12 IYD represents one of only two enzymes yet discovered in vertebrates that promote the reductive process. Native IYD is anchored in cell membranes by a single N-terminal sequence and utilizes the reductant NADPH indirectly through coordinated action of a presumed membrane-bound reductase.13,14 Crystal structures of IYD lacking its membrane anchor reveal no apparent site for NADPH binding.15 Characterization of IYD to date has relied instead on dithionite as the reductant.16 Three isozymes of iodothyronine deiodinase (ID) responsible for deiodinating the thyroid hormones thyroxine (T4) and triiodothyronine (T3) provide the remaining example of reductive dehalogenation in vertebrates.17,18 Despite the obvious similarities between the respective substrates for IYD and ID, dehalogenation follows two very different strategies. ID catalyzes a thiol-dependent deiodination involving an active site selenocysteine rather than flavin.19,20 Additionally, ID is a member of the thioredoxin superfamily, whereas IYD is a member of the nitro-FMN reductase superfamily.13,21,22
Numerous questions on the mechanism of IYD remain to be answered after initial characterization of its binding, redox, and steady-state properties.23 The pH dependence of I2-Tyr binding suggests that the deprotonated phenolate form of the halotyrosine preferentially coordinates to IYD. Comparable binding was also observed for I-Tyr as well as bromotyrosine (Br-Tyr) and chlorotyrosine (Cl-Tyr). These halotyrosines were further shown to undergo dehalogenation by the reduced flavin hydroquinone (flhq)-containing IYD, although no kinetic experiments were performed previously.23,24 Fluorotyrosine (F-Tyr) binds IYD an order of magnitude more weakly and is not subject to defluorination. However, association between IYD and any of the halotyrosines is essential for organizing the active site structure. In the absence of an appropriate ligand, residues that cover the active site are not evident by X-ray diffraction, as expected for a disordered or highly dynamic region.23 Significant solvent accessibility can be expected as well under these conditions since the flavin exhibits a midpoint potential (Em) of −200 mV that is very similar to the Em of −205 mM for free flavin.23,25 As I-Tyr binds to the active site, it acts as a template for assembly and closure of the lid-forming sequences by direct coordination between its zwitterion and the protein side chains.23,26 Both the ligand and flavin become sequestered from the solvent. The resulting environment surrounding the flavin favors the formation of a single-electron-reduced neutral flavin semiquinone (flsq) and disfavors formation of the two-electron-reduced flhq as is evident from their respective Em values of −156 and −310 mV.23 Thus, substrate binding actively participates in the control of the redox properties of bound FMN.
The spectral changes associated with flavin oxidation and reduction provide a convenient approach in our search for intermediates formed during catalysis. Similarly, the fluorescence of oxidized flavin (flox) within the IYD active site provides a sensitive reporter for ligand binding. Both physical and chemical processes involved in dehalogenation have the potential to limit the rate of catalysis. The rather slow turnover of IYD measured earlier by steady-state experiments may reflect the extensive conformational changes within the active site that are required for turnover. Alternatively, a slow transfer of electrons to the electron-rich phenolate form of the substrates may instead control enzyme turnover. Rapid kinetics of ligand binding and substrate reduction for IYD is now described below in an initial effort to define the rate-limiting processes that contribute to the ability of flavin to promote reductive dehalogenation.
Materials and Methods
Materials
Reagents of the purest grade available were obtained commercially and used without further purification. Human IYD (hIYD) was expressed without its N-terminal membrane binding region (residues 1–31) and fused to SUMO as described previously.23 Purification of this construct and subsequent isolation of hIYD lacking SUMO followed standard protocols.23
Methods
All kinetic measurements were recorded with a Hi-Tech Scientific stopped-flow spectrophotometer, model SF-61DX, and performed at 25 °C using enzyme solutions containing 500 mM NaCl, 10% glycerol, 1 mM DTT, and 50 mM sodium phosphate, pH 7.4. For the ligand binding experiments, equal volumes of the enzyme and substrate solutions were mixed in the stopped-flow instrument under an ambient atmosphere. For single-turnover measurements, equal volumes of enzyme and substrate solutions were mixed in the stopped-flow instrument under anaerobic conditions. Oxygen was excluded from the apparatus by flushing with an anaerobic solution of 100 μM protocatechuate and 1 μM protocatechuate dioxygenase in 50 mM sodium phosphate, pH 7.4.27 Prior to measurements, the flow unit was rinsed with anaerobic reaction buffer. All solutions were prepared in gas-tight glass tonometers. Oxygen was removed via repeated cycles of evacuation and equilibration with oxygen-free argon. hIYD·flox was reduced by titration with a stoichiometric concentration of dithionite under anaerobic conditions and monitored with a Shimadzu UV-2501PC spectrophotometer.24,28 Oxygen was excluded from substrate solutions by bubbling with oxygen-free argon for a minimum of 10 min. For reactions between hIYD·flhq and oxygen, the mixing solution of 500 mM NaCl, 10% glycerol, 1 mM DTT, and 50 mM sodium phosphate, pH 7.4, was alternatively equilibrated with air (21% O2) and certified oxygen/nitrogen gas mixtures (10, 50, and 100% O2) to produce oxygen solutions of 130, 60, 305, and 610 μM, respectively.
Data Processing and Analysis
Apparent rate constants were determined from the appropriate single- and double-exponential fits (eqs 1–3) of the kinetic traces by Kinetic Studio Software (TgK Scientific, Bradford-on-Avon, UK). Concentration dependence of the observed kinetic constants was fit by least-squares analysis using Origin 7.0 (OriginLab, Northampton, MA). Errors represent the standard deviation provided by the programs above. Global analysis to resolve spectra of intermediates used the SpecFit program from Spectrum Software Associates.
| 1 |
| 2 |
| 3 |
Results
Kinetics of Halotyrosine Binding to hIYD
Equilibrium binding of halotyrosines to IYD was previously monitored by a concomitant quenching of flox fluorescence. This quenching is consistent with the intimate complex formed between the ligand and flox that is stabilized by stacking of the two aromatic ring systems and direct association between the ligand’s zwitterion and the pyrimidine region of flavin.23 Fluorescence quenching may similarly be used to report on the rate of active site binding (kon). To date, attention has focused on the flox form of hIYD, but the order of substrate binding and flavin reduction is not yet known. Turnover studies under physiological conditions await isolation of the partner reductase to promote the relevant transfer of reducing equivalents from NADPH to IYD. Current analysis with the flox form of hIYD examines the ability of the enzyme to discriminate among the various halotyrosines and the potential for intermediate complexes to accumulate during the conformational changes necessary to establish the most stable complex among ligand, flavin, and protein. These investigations utilize hIYD lacking its N-terminal membrane anchor, as described in an earlier report on generating a soluble derivative of the enzyme.23 Although this form no longer associates with membranes in vitro or in vivo, its active site properties are not perturbed.14
The rate of halotyrosine binding to IYD was monitored from 1 ms to 1 s after rapidly mixing equal volumes of enzyme and ligand in buffer (Scheme 2 and Figure 1A). The resulting fluorescence decay conformed well to a single-exponential function. These results are consistent with a two-state system with no detectable intermediates or alternative kinetic pathways. Fluorescence measurements were repeated for halotyrosine concentrations of 5–50 μM to determine the concentration dependence of the kobs values. A linear dependence on concentration was observed for all of the halotyrosines and provided values for the second-order kon (Figure 1B and Table 1). Even the kobs values measured for ligand in a small excess (2.5-fold) over enzyme still fit well to the concentration dependence of reaction. The substrates for IYD turnover (Cl-, Br-, and I-Tyr) bind with similar high rates. The basis for their small differences in rate is not obvious and does not reflect their relative affinities for hIYD or the size of the halogen substituent. The binding rate of F-Tyr is approximately 5-fold slower than the other halotyrosines. This analogue containing the smallest halogen substituent is not a substrate for IYD. Moreover, F-Tyr expresses the weakest affinity for IYD and the highest phenolic pKa of the halotyrosine series. Rates of ligand dissociation (koff) were also estimated from the Kd and kon values (Table 1).
Scheme 2. Halotyrosine Association with hIYD.

Figure 1.
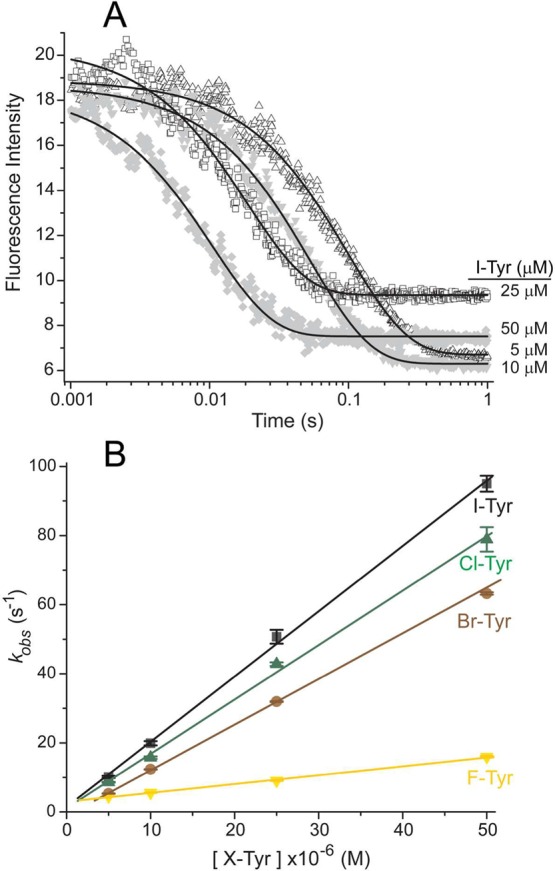
Rate of halotyrosines binding to hIYD·flox. (A) Solutions of hIYD·flox (2 μM final) in 500 mM NaCl, 10% glycerol, 1 mM DTT, and 50 mM sodium phosphate, pH 7.4, were mixed with an equal volume of I-Tyr to final concentrations of 5–50 μM in the same buffer solution. The fluorescence of the bound flox was monitored over time using λex = 450 nm and λem > 530 nm. The solid black lines represent fits to a single-exponential model (eq 1) that yield the first-order rate constants (kobs). (B) This analysis was repeated for the indicated halotyrosines as a function of concentration to determine the second-order binding rate constants (kon) summarized in Table 1. Data points represent the average of three independent measurements, and the standard deviations are illustrated by error bars. The solid lines were generated by linear best fits to the data.
Table 1. Rate Constants for Ligand Association with hIYD·flox and Oxidation of hIYD·flhq.
Kinetics of hIYD (flhq) Oxidation by the Halotyrosines
The ability of Cl-Tyr and Br-Tyr to oxidize the reduced form of IYD containing flhq provided the first indication that these analogues were also substrates, and subsequent detection of stoichiometric quantities of tyrosine confirmed their dehalogenation.24 Comparable reactions have now been monitored by rapid kinetic analysis to determine the effect of the halogen on the rate of flavin oxidation. The increasing bond energy of the C–X bond for X = I < Br < Cl has the potential to influence the rate-limiting step of catalysis. Even the relatively weak benzyl halide bond increases by more than 20 kcal/mol after substitution of Cl for I.29 A further increase of an additional 25 kcal/mol30 after substitution of F for Cl exceeds the capacity of IYD catalysis and rendered F-Tyr inert to IYD.24
To measure the kinetics of the active Cl-, Br-, and I-Tyr, hIYD was initially reduced with dithionite under anaerobic conditions and subsequently mixed rapidly with anaerobic solutions of the substrates in buffer at 25 °C (Scheme 3). Reactions were monitored from 10 ms to beyond 7 s by the increase in absorbance at the λmax of flox (446 nm). Concentrations of I-Tyr were varied between 20 and 200 μM, and the data for each fit well to a single-exponential increase of the flox form of IYD (Figure 2A). At this wavelength, the simple first-order increase in absorbance provided no evidence of chemical or kinetic intermediates during flavin oxidation. A plot of kobs versus I-Tyr concentration provided the second-order kox value (Figure 2B and Table 1). Equivalent analysis was repeated for Br- and Cl-Tyr, and, again, data from A446 fit to single-exponential increases of flox (Figures 2B and Table 1). The kobs values for all three substrates remained linear throughout the range of concentrations examined, including at the low excess (2.5-fold) of substrate over enzyme. The lack of obvious saturation of the enzyme suggests that substrate binds more weakly to the flhq form of IYD used in these kinetic experiments than to its flox form used for measuring the strong affinity of the halotyrosines (Kd < 0.2 μM).23
Scheme 3. Halotyrosine-Promoted Oxidation of hIYD Containing flhq.

Figure 2.
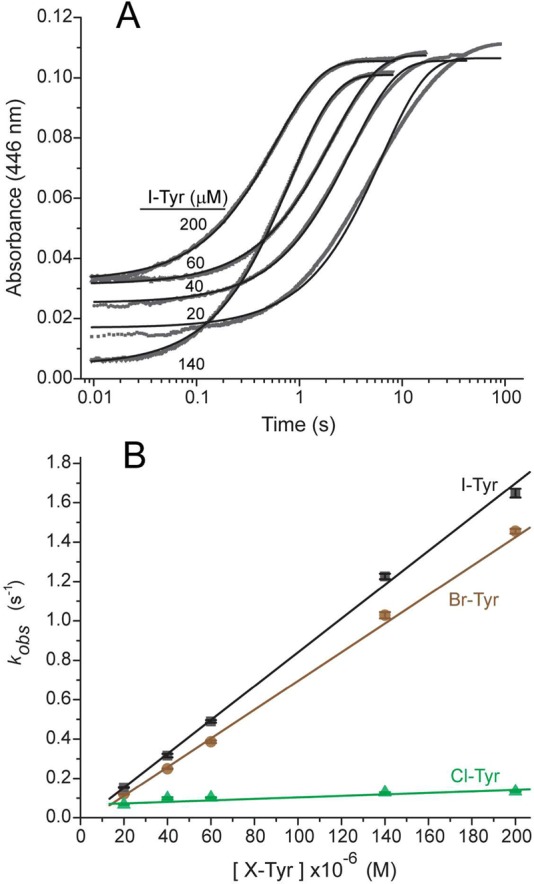
Oxidation of hIYD·flhq by halotyrosines. (A) Solutions of hIYD·flhq (8 μM final) in 500 mM NaCl, 10% glycerol, 1 mM DTT, and 50 mM sodium phosphate, pH 7.4, were mixed with an equal volume of I-Tyr in the same buffer solution under anaerobic conditions. Oxidation of hIYD·flhq was monitored by absorbance at 446 nm. The solid black lines represent the best fits to a single-exponential model (eq 2) to yield kobs. (B) This analysis was repeated for the indicated halotyrosines as a function of concentration (Figure S1 to determine the second-order rate constants for oxidation (kox) summarized in Table 1). Data points represent the average of three independent measurements, and the standard deviations are illustrated by error bars. The solid lines were generated by linear best fits to the data.
The relative differences in kox for I-, Br-, and Cl-Tyr do not reflect their respective affinities23 or association rates with hIYD (Table 1), and they do not correlate with differences in C–X bond strength or phenolic pKa. The Br- and I-Tyr substrates support very similar rates of hIYD (flhq) oxidation in contrast to the approximately 20-fold smaller rate for Cl-Tyr. As expected from earlier investigation,24 oxidation of flhq by F-Tyr was at least 160-fold slower than that with I-Tyr, as established by the detection threshold of the stopped-flow instrument. Full spectra of the reaction mixtures containing I-, Br-, or Cl-Tyr were also recorded beginning after 4 ms for substrate concentrations of 200 μM (Figure S2). No spectral intermediates were apparent between flhq and flox for Br- and I-Tyr. However, a low level of the neutral flavin semiquinone (flsq) accumulated, as is evident from the absorbance centered at approximately 600 nm. This had similarly been detected when Br- and Cl-Tyr were first discovered as substrates.24 At that time, the neutral flsq was confirmed by EPR and found to be stable for days under aerobic conditions as if it may form as a nonproductive side product. Turnover of Cl-Tyr generated the greatest fraction of flsq, as expected if dehalogenation and flsq formation were competitive. Nevertheless, the yield of dechlorination remained high (∼94%), as detected previously by HPLC analysis.24 The spectra recorded during Cl-Tyr turnover also revealed the presence of an additional species that was not evident during turnover of Br- or I-Tyr.
Detection of a Transient Intermediate Formed during Oxidation of hIYD·flhq by Cl-Tyr
The investigations above relied on just one of two absorption bands that characterize flox formation (A446). The second band, typically in the region of 370 nm, can be used alternatively to monitor formation of flox, although complications can arise from the concurrent formation of other species such as the anionic flsq and 4a-adducts of flavin that absorb light in this region as well.31,32 Measurements at A360 to monitor oxidation of flhq by either Br- or I-Tyr generated results comparable to those using A446. Progress curves recorded at 360 nm fit well to single-exponential increases in flox, with kobs values that were nearly identical to those determined at 446 nm (Figure 3A and Table 2). These results are consistent with detection of only two species, flhq and flox during IYD reaction with Br- and I-Tyr. In contrast, the absorbance changes at 360 nm (and shorter wavelengths) for the reaction with Cl-Tyr did not fit to a single exponential and instead follow a double-exponential function (Figure 3A). The slower phase (k2(obs)) is the same as that determined by A446 under equivalent conditions (Table 2). This similarity is expected for the net conversion of flhq to flox (Table 2). The faster phase observed at wavelengths shorter than 360 nm (k1(obs)) is an order of magnitude larger than the slower phase and suggests an intermediate forms prior to the rate-limiting step that controls formation of flox. Interestingly, this fast phase is characterized by a kobs that is similar to the kobs values for Br- and I-Tyr (Table 2). Accordingly, all three halogenated substrates may follow similar mechanisms for dehalogenation with only a change in a rate-determining step for dechlorination of Cl-Tyr.
Figure 3.
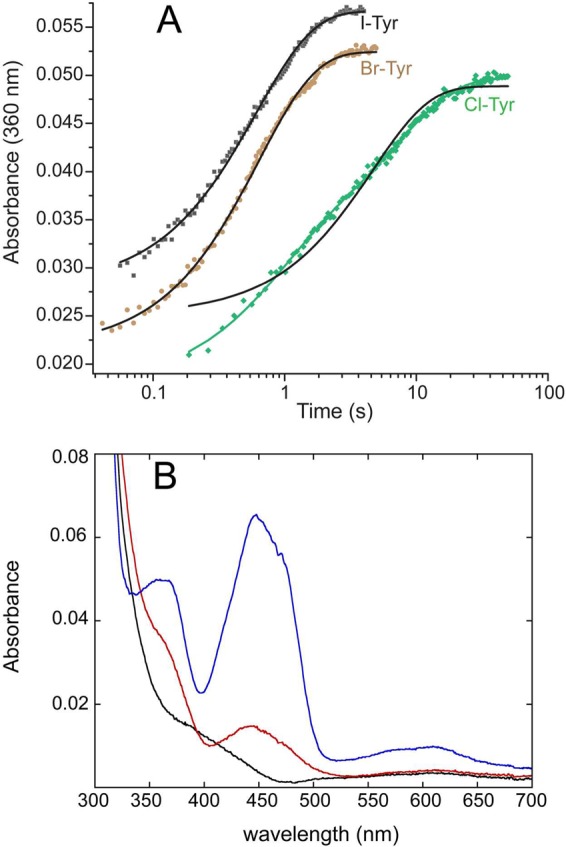
Oxidation of hIYD·flhq by Cl-Tyr generates a spectral intermediate. Solutions of hIYD·flhq (5 μM final) in 500 mM NaCl, 10% glycerol, 1 mM DTT, and 50 mM sodium phosphate, pH 7.4, were mixed with an equal volume of the indicated halotyrosine (200 μM final) in the same buffer. (A) Oxidation of hIYD·flhq was monitored at 360 nm. Solid black lines represent fits to a single-exponential model (eq 2), and the solid green line represents fit to a double exponential model (eq 3). (B) Spectral data generated by reaction between Cl-Tyr and hIYD·flhq in the stopped-flow instrument, as monitored in the diode array mode was subject to global analysis (see Figure S2). Results of fitting with a double-exponential model suggested rate constants of k1 = 1.5 s–1 and k2 = 0.134 s–1 and yielded spectra of the final flox (blue), the starting spectrum of flhq (black), and the transient intermediate (red). Residual spectra fit to within ≤0.002 absorbance units.
Table 2. Observing hIYD·flhq Oxidation by Halotyrosines at Two λmax.
The significance of the biphasic kinetics of flhq oxidation by Cl-Tyr became more obvious from a global analysis of the diode array data for the full spectra over time (Figure 3B). An intermediate with a spectrum containing a peak at ∼440 nm and broad absorbance between 350 and 380 nm is evident. This analysis also indicates that no semiquinone of the flavin has formed until this intermediate converts to the flox form. The possible identity of this transient intermediate is considered in the Discussion.
Reaction of hIYD·flhq with Oxygen
IYD belongs to a structural superfamily that includes enzymes with activities ranging from oxygen-insensitive nitroreductases33 to oxygen-dependent flavin “destructases”.34 This latter flavoprotein has been named BluB and promotes oxidative degradation of its own active site flavin to form 5,6-dimethylbenzimidazole as part of the biosynthesis of vitamin B12. Measuring the ability of IYD to suppress or activate its flavin for reaction with oxygen consequently provides an important reference for differentiating structure and function within this superfamily. Anaerobic solutions of the reduced flhq form of hIYD were rapidly mixed with buffer containing fixed concentrations of oxygen. Full spectra from 300–700 nm were recorded between 4 ms and 7 s after mixing (Figure 4). Only flhq and flox were apparent during reaction. No characteristic absorbance of anionic or neutral flsq (370 and 600 nm, respectively)31 or 4a-adducts of flavin (∼380–390 nm)32 was observed. Similarly, the full spectra do not suggest accumulation of the intermediate detected previously during reaction with Cl-Tyr. The absence of this intermediate is additionally confirmed by the lack of biphasic kinetic data from A360. A single-exponential function was sufficient to describe these data, as expected from the presence of only flhq and flox (Figure S3).
Figure 4.

Oxidation of hIYD·flhq by O2. A solution containing hIYD·flhq (5 μM final) in 500 mM NaCl, 10% glycerol, 1 mM DTT, and 50 mM sodium phosphate, pH 7.4, was mixed with an equal volume of an oxygenated solution containing 500 mM NaCl, 10% glycerol, 1 mM DTT, and 50 mM sodium phosphate, pH 7.4. (A) Spectra of hIYD·flhq oxidation by air-saturated buffer were recorded from 4 ms to 7 s with a diode array spectrophotometer. The arrow indicates the direction of spectral change as a function of time. (B) Oxidation of hIYD·flhq was monitored by absorbance at 446 nm, and the resulting kobs values (see Figure S3) were plotted against oxygen concentration to determine the second-order rate constant (kox). Data points represent the average of three independent measurements, and the standard deviations are illustrated by error bars. The solid line was generated by a linear best fit to the data.
The rapid mixing experiments were next monitored at the single wavelength of 446 nm. The resulting data fit well to a single exponential, and the corresponding kobs values varied linearly with the concentration of dissolved oxygen (Figures 4B and S3). From this analysis, the second-order rate constant kox for reaction between hIYD·flhq and oxygen was calculated to be (9.3 ± 0.3) × 103 M–1 s–1. This value is only slightly larger than that measured when I-Tyr was used as the oxidant (Table 1), but it is in the lower range of rate constants for reaction between oxygen and reduced flavin oxidases.4,35 Similar to IYD, the oxidases also do not generate intermediates such as a flsq or a 4a-adduct of flavin at detectable levels. Although the flavin ring of IYD appears to have few contacts with the protein in the absence of active site ligands, reaction of its reduced flhq with oxygen is considerably faster than the comparable reaction for free flavin and oxygen.4
Discussion
Active Site Binding
This first characterization of IYD by rapid kinetics focuses on substrate association and oxidation of its reduced flavin (flhq). Complementary processes such as initial enzyme reduction will be the subject of future investigations once the native reductase can be identified. Regardless of the order of enzyme reduction and substrate binding, the substrate–enzyme complex itself is key for inducing catalytic activity. In the absence of substrate, much of the active site is unstructured. The flavin is exposed to solvent under these conditions, and its redox properties mimic those of free flavin in solution.23 An active site ligand such as I-Tyr provides the necessary template for stabilizing a helix and loop that combine to form a lid over the active site and sequester substrate and flavin from solvent.15,23,26 Central to this reorganization is a chelation of the zwitterion of the halotyrosine between the pyrimidine ring of the flavin and side chains of the lid domain. Additional stabilization of the substrate–enzyme complex includes stacking between the aromatic systems of I-Tyr and the flavin and hydrogen bonding between the side chain of a Thr and the N5 of flavin.23 Despite this high level of restructuring induced by substrate, its binding is relatively fast, with second-order rate constants on the order of 106 M–1 s–1 for Cl-, Br-, and I-Tyr (Table 1). No intermediates were observed during this binding process since the time-resolved quenching of flavin fluorescence by substrate matched a first-order monophasic process. Even the inert substrate analogue F-Tyr binds to IYD with only a ∼5-fold slower rate constant compared to the average rate for the other halotyrosines. Affinity for the dehalogenated product Tyr is orders of magnitude weaker than that for its halogenated derivatives, and this characteristic most likely facilitates product release. Solubility limits prevented an accurate measure of Tyr binding, and its Kd could be estimated only at greater than 1.0 mM.23
Reactivity toward Oxygen
BluB represents the closest known structural relative to IYD. These proteins share a similar architecture and topology even though their sequence identity is low. Both have lids to cover their active sites, but BluB relies solely on protein contacts to control the chemistry of its flavin, in contrast to the need for both substrate and protein contacts in IYD.23,34 Recently, the active site environments of hIYD and a homologue of BluB were shown to stabilize their one-electron-reduced flsq form during redox titration.23,36 BluB also appears to stabilize the 4a-hydroperoxide adduct of its flavin, as anticipated from the proposed oxygen-dependent mechanism for generating the product dimethylbenzimidazole.36 In the absence of a halotyrosine, the reduced (flhq) form of hIYD is readily reoxidized by oxygen, and the flox form of IYD is regenerated for further catalytic turnover (Figure 4). This process may additionally involve a 4a-hydroperoxide intermediate, although sufficient quantities do not accumulate for detection in IYD and many other flavoproteins.4,35 Alternatively, reoxidation of the flhq might have included one-electron transfer to form superoxide, but, again, no accumulation of flsq was evident. In either event, IYD promotes moderately rapid reoxidation of its flavin without the degradation that is synonymous with BluB.
Catalytic Dehalogenation by IYD
The chemistry associated with kox appears to limit the consumption of the flhq form of hIYD (Table 1). On the basis of the tight binding of halotyrosine for hIYD, dissociation of substrate from hIYD·flox is slow and not competitive with turnover. The kox value for I-Tyr measured in this study by rapid kinetics (8.6 × 103 M–1 s–1) is also very similar to the kcat/Km for I2-Tyr measured by steady-state kinetics (6.7 × 103 M–1 s–1),23 implying that product release does not likely contribute to the rate-determining step(s). This observation is consistent with the inability of the relatively nonpolar binding pocket to stabilize the anionic product iodide.15 Although thermodynamics may not always correlate with kinetics, the unmeasurably weak affinity of hIYD for Tyr would be consistent with its rapid dissociation from the active site. The limits of catalysis probably derive from one or more of the chemical steps required in reductive dehalogenation. The reaction mechanism that most readily explains the current data on IYD involves an initial protonation to a nonaromatic intermediate followed by electron transfer, halide release, and a second electron transfer (Scheme 4). Any of these steps has the potential to control kox, although the final electron transfer should be relatively rapid based on the proximity of the two radical species and the stability of nonradical products.
Scheme 4. Possible Mechanism for Catalytic Dehalogenation.

An initial protonation has been proposed in the dehalogenation process to diminish the electrostatic repulsion between the electron donated from flhq and the phenolate form of halotyrosine that binds preferentially to hIYD.23 Consistent with this proposal, a series of N-alkyl pyridones that mimic the nonaromatic keto form of this intermediate bind to the active site with a Kd as low as 24 nM.37 Reduced flavin (flhq) is competent at both stepwise electron transfer and hydride transfer,4 but single-electron transfer seems to be most likely for the reaction of IYD. Association of a halotyrosine to IYD stabilizes the flsq form of IYD and hence could induce single-electron transfers from flhq.23 The resulting midpoint potentials of a halotyrosine·IYD complex are similar to those of an electron transferase such as ferredoxin reductase.23,38 Hydrogen atom transfer from flhq to the halotyrosine provides an alternative path to the ketyl radical proposed to form after stepwise proton and electron transfer (Scheme 4). Both e–aq and hydrogen atoms are highly efficient at dehalogenating iodophenol during pulse radiolysis, although e–aq is considered to be the major mechanism of dehalogenation under these conditions.39 Electron transfer rather than hydrogen atom transfer is also favored for IYD based on the lack of precedent for flhq to serve as a hydrogen atom donor.4 Interestingly, the low yields of dechlorination and defluorination from radiolysis were previously rationalized by the slow dehalogenation of the radical anion intermediate.39
For IYD, Br- and I-Tyr reduction is likely controlled by either initial proton transfer and/or the first electron transfer since their kox values are nearly equal (Table 1) despite the difference in their C–X bond energies and leaving group potentials.29 In contrast, the kox for Cl-Tyr, as measured at A446, is ∼20-fold lower than the kox for Br- and I-Tyr and indicates a possible change in the rate-determining step. The most significant chemical differences among Cl-, Br-, and I-Tyr are the stronger C–Cl bond and weaker leaving group ability of chloride. Initial protonation or electron transfer is not expected to be greatly affected by the nature of the halogen substituent, but halide elimination may be slowed considerably for the chloro derivative. The inability of IYD to defluorinate F-Tyr24 may be an extension of this trend since fluorine forms the strongest C–X bond and fluoride is the weakest leaving group of the halides. One caveat on the effects of the halogens should be noted. A competing mechanism involving direct electron transfer to the halotyrosine might follow a trend established by the second-order rate constants for consumption of e–aq by halobenzenes that decrease from 12 × 109 M–1 s–1 (iodobenzene) to 4.3 × 109 M–1 s–1 (bromobenzene) and 0.5 (chlorobenzene) × 109 M–1 s–1.40
The transient intermediate detected by the biphasic kinetics of reaction between hIYD·flhq and Cl-Tyr may ultimately be most revealing for the mechanism of reductive dehalogenation once its structure can be identified (Figure 3B). This intermediate forms at nearly the same rate as the generation of flox by Br- and I-Tyr, and its accumulation becomes evident when the second phase of reaction with Cl-Tyr is slowed by 10-fold relative to the reactions with Br- and I-Tyr (Figures 3B and S2 and Table 2). The absorbance spectrum of the intermediate is quite broad and exhibits one λmax of ∼440–450 nm. While this may appear to be consistent with flox, the second band at 350–380 nm is not equivalent to the additional absorbance of flox that centers near 370 nm (Figure 3B). The spectral characteristics of this intermediate also do not correspond to the phenoxy radical of tyrosine that has a much narrower absorbance band and a λmax of ∼410 nm.41 Similarly, this transient is not easily ascribed to either the anionic or neutral forms of flsq since they respectively produce a narrow absorbance band with a λmax of ∼370 nm and a broad absorbance band with a λmax of ∼600 nm.31 Likewise, the proposed nonaromatic keto intermediate (λmax ∼ 330 nm)42 and its one-electron-reduced ketyl radical derivative (λmax ∼360 nm)43 are not likely responsible for the transient spectrum (Scheme 4). Instead, the absorption properties may derive from an intimate association of two chromophores in the active site that may include those considered above. Alternatively, these data may suggest formation of a new intermediate that has not yet been considered in the working mechanism for dehalogenation.
Conclusions
The rapid kinetics described above indicates that substrates bind to IYD and establish order within the active site relatively quickly compared to their subsequent turnover. Dehalogenation is not likely to be rate-determining for Br- or I-Tyr since both produce the fully oxidized enzyme with similar rates despite the greater strength of the C–Br vs C–I bond. Dehalogenation may become limiting when the C–X bond strength increases further and creates a greater barrier for its cleavage as illustrated by the kinetic behavior of IYD with Cl-Tyr. Additional strengthening of the C–X bond can ultimately exceed the catalytic ability of IYD as is evident with F-Tyr. This derivative binds relatively tightly to the active site and helps to stabilize the adjacent flsq, but it remains inert to dehalogenation. The kinetics of flavin oxidation by the halotyrosines has now provided the first tangible evidence for an intermediate during reductive dehalogenation by a flavoprotein, but further investigations will be required to confirm the identity of this species.
Acknowledgments
We thank Jimin Hu for preparing the vector for hIYD expression.
Glossary
Abbreviations
- Br-Tyr
bromotyrosine
- Cl-Tyr
chlorotyrosine
- I2-Tyr
diiodotyrosine
- F-Tyr
fluorotyrosine
- hIYD
human IYD
- ID
iodothyronine deiodinase
- I-Tyr
iodotyrosine
- IYD
iodotyrosine deiodinase
- flsq
neutral flavin semiquinone
- flox
oxidized flavin
- flhq
reduced flavin hydroquinone
Supporting Information Available
Time-dependent oxidation of IYD·flhq (A446) by chloro- and bromotyrosine; spectral change (325–700 nm) during IYD·flhq oxidation by chloro-, bromo-, and iodotyrosine; and kinetics of IYD·flhq oxidation by oxygen. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b00410.
The research was supported in part by a grant from the National Institutes of Health (DK084186 to S.E.R.).
The authors declare no competing financial interest.
Supplementary Material
References
- Massey V. (2000) The chemical and biological versatility of riboflavin. Biochem. Soc. Trans. 28, 283–296. 10.1042/0300-5127:0280283. [DOI] [PubMed] [Google Scholar]
- Fraaije M. W.; Mattevi A. (2000) Flavoenzymes: diverse catalysts with recurrent features. Trends Biochem. Sci. 25, 126–132. 10.1016/S0968-0004(99)01533-9. [DOI] [PubMed] [Google Scholar]
- Mansoorabadi S. O.; Thibodeaux C. J.; Liu H.-w. (2007) The diverse roles of flavin coenyzmes--nature’s most versatile thespians. J. Org. Chem. 72, 6329–6342. 10.1021/jo0703092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan R. L., and Palfey B. A. (2010) Flavin-dependent enzymes, in Comprehensive Natural Products II (Begley T. P., Ed.) Chapter 3, pp 37–114, Elsevier, Oxford. [Google Scholar]
- Vaillancourt F. H.; Yeh E.; Vosburg D. A.; Garneau-Tsodikova S.; Walsh C. T. (2006) Nature’s inventory of halogenation catalysts: oxidative strategies predominate. Chem. Rev. 106, 3364–3378. 10.1021/cr050313i. [DOI] [PubMed] [Google Scholar]
- Rosenberg I. N.; Goswami A. (1979) Purification and characterization of a flavoprotein from bovine thyroid with iodotyrosine deiodinase activity. J. Biol. Chem. 254, 12318–12325. [PubMed] [Google Scholar]
- Rokita S. E. (2013) Flavoprotein dehalogenases, in Handbook of Flavoproteins (Hille R., Miller S. M., and Palfey B., Eds.) pp 337–350, DeGruyter, Berlin. [Google Scholar]
- Nunez J.; Pommier J. (1982) Formation of thyroid hormones. Vitam. Horm. 39, 175–229. [DOI] [PubMed] [Google Scholar]
- Moreno J. C.; Klootwijk W.; van Toor H.; Pinto G.; D’Alessandro M.; Lèger A.; Goudie D.; Polak M.; Grüters A.; Visser T. J. (2008) Mutations in the iodotryosine deiodinase gene and hypothyroidism. N. Engl. J. Med. 358, 1811–1818. 10.1056/NEJMoa0706819. [DOI] [PubMed] [Google Scholar]
- Afink G.; Kulik W.; Overmars H.; de Randamie J.; Veenboer T.; van Cruchten A.; Craen M.; Ris-Stalpers C. (2008) Molecular characterization of iodotyrosine dehalogenase deficiency in patients with hypothyroidism. J. Clin. Endocrinol. Metab. 93, 4894–4901. 10.1210/jc.2008-0865. [DOI] [PubMed] [Google Scholar]
- Wackett L. P.; Schanke C. A. (1992) Mechanisms of reductive dehalogenation by transition metal cofactors found in anaerobic bacteria. Metals in Biology 28, 329–356. [Google Scholar]
- van Pée K.-H.; Unversucht S. (2003) Biological dehalogenation and halogenation reactions. Chemosphere 52, 299–312. 10.1016/S0045-6535(03)00204-2. [DOI] [PubMed] [Google Scholar]
- Friedman J. E.; Watson J. A. Jr.; Lam D. W.-H.; Rokita S. E. (2006) Iodotyrosine deiodinase is the first mammalian member of the NADH oxidase/flavin reductase superfamily. J. Biol. Chem. 281, 2812–2819. 10.1074/jbc.M510365200. [DOI] [PubMed] [Google Scholar]
- Watson J. A. Jr.; McTamney P. M.; Adler J. M.; Rokita S. E. (2008) The flavoprotein iodotyrosine deiodinase functions without cysteine residues. ChemBioChem 9, 504–506. 10.1002/cbic.200700562. [DOI] [PubMed] [Google Scholar]
- Thomas S. R.; McTamney P. M.; Adler J. M.; LaRonde-LeBlanc N.; Rokita S. E. (2009) Crystal structure of iodotyrosine deiodinase, a novel flavoprotein responsible for iodide salvage in thyroid glands. J. Biol. Chem. 284, 19659–19667. 10.1074/jbc.M109.013458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg I. N. (1970) Purification of iodotyrosine from bovine thyroid. Metab., Clin. Exp. 19, 785–798. 10.1016/0026-0495(70)90076-4. [DOI] [PubMed] [Google Scholar]
- Bianco A. C.; Salvatore D.; Gereben B.; Berry M. J.; Larsen P. R. (2002) Biochemistry, cellular and molecular biology and physiological roles of the iodothyronine selenodeiodinases. Endocr. Rev. 23, 38–89. 10.1210/edrv.23.1.0455. [DOI] [PubMed] [Google Scholar]
- Schweizer U.; Schlicker C.; Braun D.; Köhrle J.; Steegborn C. (2014) Crystal structure of mammalian selenocysteine-dependent iodothyronine deiodinase suggests a peroxiredoxin-like catalytic mechanism. Proc. Natl. Acad. Sci. U. S. A. 111, 10526–10531. 10.1073/pnas.1323873111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry M. J.; Banu L.; Larsen P. R. (1991) Type I iodothyronine deiodinase is a selenocysteine-containing enzyme. Nature 349, 438–440. 10.1038/349438a0. [DOI] [PubMed] [Google Scholar]
- Berry M. J.; Kieffer J. D.; Harney J. W.; Larsen P. R. (1991) Selenocysteine confers the biochemical properties characteristic of the type I iodothyronine deiodinase. J. Biol. Chem. 266, 14155–14158. [PubMed] [Google Scholar]
- Gnidehou S.; Caillou B.; Talbot M.; Ohayon R.; Kaniewski J.; Noël-Hudson M.-S.; Morand S.; Agnangji D.; Sezan A.; Courtin F.; Virion A.; Dupuy C. (2004) Iodotyrosine dehalogenase 1 (DEHAL1) is a transmembrane protein involved in the recycling of iodide close to the thyroglobulin iodination site. FASEB J. 18, 1574–1576. 10.1096/fj.04-2023fje. [DOI] [PubMed] [Google Scholar]
- Callebaut I.; Curcio-Morelli C.; Mornon J.-P.; Gereben B.; Buettner C.; Huang S.; Castro B.; Fonseca T. L.; Harney J. W.; Larsen P. R.; Bianco A. C. (2003) The iodothyronine selenodeiodinases are thioredoxin-fold family proteins containing a glycoside hydrolase clan GH-A-like structure. J. Biol. Chem. 278, 36887–36896. 10.1074/jbc.M305725200. [DOI] [PubMed] [Google Scholar]
- Hu J.; Chuenchor W.; Rokita S. E. (2015) A switch between one- and two-electron chemistry of the human flavoprotein iodotyrosine deiodinase is controlled by substrate. J. Biol. Chem. 290, 590–600. 10.1074/jbc.M114.605964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTamney P. M.; Rokita S. E. (2009) A mammalian reductive deiodinase has broad power to dehalogenate chlorinated and brominated substrates. J. Am. Chem. Soc. 131, 14212–14213. 10.1021/ja906642n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper R. D.; Ingraham L. L. (1968) A potentiometic study of flavin semiquinone equilibrium. Arch. Biochem. Biophys. 125, 802–8808. 10.1016/0003-9861(68)90517-1. [DOI] [PubMed] [Google Scholar]
- Buss J. M.; McTamney P. M.; Rokita S. E. (2012) Expression of a soluble form of iodotyrosine deiodinase for active site characterization by engineering the native membrane protein from Mus musculus. Protein Sci. 21, 351–361. 10.1002/pro.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil P. V.; Ballou D. P. (2000) The use of protocatechuate dioxygenase for maintaing anaerobic conditions in biochemical experiments. Anal. Biochem. 286, 187–192. 10.1006/abio.2000.4802. [DOI] [PubMed] [Google Scholar]
- Goswami A.; Rosenberg I. N. (1979) Characterization of a flavoprotein iodotyrosine deiodinase from bovine thyroid. J. Biol. Chem. 254, 12326–12330. [PubMed] [Google Scholar]
- McMillen D. F.; Golden D. M. (1982) Hydrocarbon bond dissociation energies. Annu. Rev. Phys. Chem. 33, 493–532. 10.1146/annurev.pc.33.100182.002425. [DOI] [Google Scholar]
- Blanksby S. J.; Ellison G. B. (2003) Bond dissociation energies of organic molecules. Acc. Chem. Res. 36, 255–263. 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]
- Massey V.; Palmer G. (1966) On the existence of spectrally distinct classes of flavoprotein semiquiones. A new method for the quantitative production of flavoprotein semiquinones. Biochemistry 5, 3181–3189. 10.1021/bi00874a016. [DOI] [PubMed] [Google Scholar]
- Chakraborty S.; Ortiz-Maldonado M.; Entsch B.; Ballou D. P. (2010) Studies on the mechanism of p-hydroxylphenylacetate 3-hydroxylase from Pseudomonas aeruginosa: a system composed of a small flavin reductase and a large flavin-dependent oxygenase. Biochemistry 49, 372–385. 10.1021/bi901454u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roldán M. D.; Pérez-Reinado E.; Castillo F.; Moreno-Vivián C. (2008) Reduction of polynitroaromatic compounds: the bacterial nitroreductases. FEMS Microbiol. Rev. 32, 474–500. 10.1111/j.1574-6976.2008.00107.x. [DOI] [PubMed] [Google Scholar]
- Taga M. E.; Larsen N. A.; Howard-Jones A. R.; Walsh C. T.; Walker G. C. (2007) BluB cannibalizes flavin to form the lower ligand of vitamin B12. Nature 446, 449–453. 10.1038/nature05611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey V. (1994) Activation of molecular oxygen by flavins and flavoproteins. J. Biol. Chem. 269, 22459–22462. [PubMed] [Google Scholar]
- Collins H. F.; Biedendieck R.; Leech H. K.; Gray M.; Escalante-Semerena J. C.; McLean K. J.; Munro A. W.; Rigby S. E. J.; Warren M. J.; Lawrence A. D. (2013) Bacillus megaterium has both a functional BluB protein required for DMNB synthesis and a related flavoprotein that forms a stable radical species. PLoS One 8, e55708. 10.1371/journal.pone.0055708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunishima M.; Friedman J. E.; Rokita S. E. (1999) Transition-state stabilization by a mammalian reductive dehalogenase. J. Am. Chem. Soc. 121, 4722–4723. 10.1021/ja990693n. [DOI] [Google Scholar]
- Bowsher C. G.; Eyres L. M.; Gummadova J. O.; Hothi P.; McLean K. J.; Munro A. W.; Scrutton N. S.; Hanke G. T.; Sakakibara Y.; Hase T. (2011) Identification of N-terminal regions of wheat leaf ferredoxin NADP+ oxidoreductase important for interactions with ferredoxin. Biochemistry 50, 1778–1787. 10.1021/bi1014562. [DOI] [PubMed] [Google Scholar]
- Glowa G. A.; Mezyk S. P. (1998) The radiation chemistry of iodophenols. Radiat. Phys. Chem. 53, 127–135. 10.1016/S0969-806X(98)00011-5. [DOI] [Google Scholar]
- Anbar A.; Hart E. J. (1964) The reactivity of aromatic compounds toward hydrated electrons. J. Am. Chem. Soc. 86, 5633–5637. 10.1021/ja01078a046. [DOI] [Google Scholar]
- Seyedsayamdost M. R.; Reece S. Y.; Nocera D. G.; Stubbe J. (2006) Mono-, di- tri- and tetra-substituted fluorotyrosines: new probes for enzymes that use tyrosyl radicals in catalysis. J. Am. Chem. Soc. 128, 1569–1579. 10.1021/ja055926r. [DOI] [PubMed] [Google Scholar]
- Miller B. (1970) Acid-catalyzed [1,2] and [1,5] migrations in linearly conjugated cyclohexadienones. Further evidence for differing types of migration from n- and π-protonated cyclohexadienones. J. Am. Chem. Soc. 92, 6246–6252. 10.1021/ja00724a024. [DOI] [Google Scholar]
- Das T. N. (1998) Redox Chemistry of 3-iodotyrosine in aqueous medium. J. Phys. Chem. A 102, 426–433. 10.1021/jp9716344. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.