

Abstract
Glomerular visceral epithelial cells (podocytes) play a critical role in the maintenance of glomerular permselectivity. Podocyte injury, manifesting as proteinuria, is the cause of many glomerular diseases. We reported previously that calcium-independent phospholipase A2γ (iPLA2γ) is cytoprotective against complement-mediated glomerular epithelial cell injury. Studies in iPLA2γ KO mice have demonstrated an important role for iPLA2γ in mitochondrial lipid turnover, membrane structure, and metabolism. The aim of the present study was to employ iPLA2γ KO mice to better understand the role of iPLA2γ in normal glomerular and podocyte function as well as in glomerular injury. We show that deletion of iPLA2γ did not cause detectable albuminuria; however, it resulted in mitochondrial structural abnormalities and enhanced autophagy in podocytes as well as loss of podocytes in aging KO mice. Moreover, after induction of anti-glomerular basement membrane nephritis in young mice, iPLA2γ KO mice exhibited significantly increased levels of albuminuria, podocyte injury, and loss of podocytes compared with wild type. Thus, iPLA2γ has a protective functional role in the normal glomerulus and in glomerulonephritis. Understanding the role of iPLA2γ in glomerular pathophysiology provides opportunities for the development of novel therapeutic approaches to glomerular injury and proteinuria.
Keywords: autophagy, mitochondria, Phospholipase A, podocyte, proteinuria, cell injury, glomerulonephritis
Introduction
Phospholipase A2 (PLA2)4 enzymes catalyze the cleavage of the sn-2-ester bond of phospholipids to yield free fatty acids, such as arachidonic acid, and lysophospholipids (1–3). Both products represent precursors for signaling molecules that can exert multiple biological functions. Calcium-independent PLA2s (iPLA2s) are members of the group VI family of PLA2 enzymes (3, 4). Group VIB (iPLA2γ) is homologous to iPLA2β in the C-terminal catalytic domain but shows no similarities in the N-terminal region; accordingly, iPLA2γ has regulatory properties distinct from those of iPLA2β (3, 5, 6).
Genetic ablation of iPLA2γ in mice resulted in the generation of viable progeny that demonstrated reduced growth rate as well as cold intolerance due to impaired fat burning in brown adipose tissue and mitochondrial dysfunction (3, 7). Several studies showed that iPLA2γ KO mice display multiple bioenergetic dysfunctional phenotypes. For example, iPLA2γ deletion induced profound alterations in hippocampal phospholipid metabolism and mitochondrial phospholipid homeostasis, resulting in enlarged and degenerating mitochondria, leading to enhanced autophagy and cognitive dysfunction (8). Another recent study showed that genetic ablation of iPLA2γ prevented obesity and insulin resistance during high fat feeding by mitochondrial uncoupling and increased adipocyte fatty acid oxidation (9). Taken together, these reports confirm an obligatory role for iPLA2γ in mitochondrial lipid metabolism and membrane structure, perturbation of which may profoundly influence fatty acid β-oxidation, oxygen consumption, energy expenditure, and, thus, tissue homeostasis. Despite the recent advances in studying the pathophysiological role of iPLA2γ in the brain, liver, heart, and skeletal muscles (7, 8, 10, 11), the pathophysiological roles of iPLA2γ in the kidney are poorly understood.
Visceral glomerular epithelial cells (GECs), also known as podocytes, are highly differentiated cells of the kidney glomerulus, which play a critical role in the maintenance of glomerular permselectivity (12–14). Podocytes have a complex morphology that is characterized by cell bodies with projecting foot processes, which adhere to the glomerular basement membrane (GBM). The unique shape of podocytes is supported by the actin cytoskeleton, which allows podocytes to alter their shape dynamically (12, 15). In many renal diseases, changes in the size and surface charge of podocytes contribute to the development of proteinuria. Specifically, proteinuria is associated with the disruption of the foot processes with the concomitant transformation of the actin cytoskeleton (12–15). Thus, the intricate structure of foot processes is central to their function of maintaining glomerular permselectivity (i.e. filtration of plasma but restriction of proteins). Various forms of glomerulonephritis, including those induced by antibody and complement, are associated with podocyte injury, which may lead to foot process effacement, detachment from the GBM, or apoptosis, with consequent impairment of glomerular function and permselectivity (proteinuria), and glomerulosclerosis (13, 16–18).
Previously, we characterized the expression and activity of iPLA2γ in cultured GECs and characterized the mechanisms of iPLA2γ activation by complement C5b-9 (19, 20). In GECs, we demonstrated that iPLA2γ is localized at the endoplasmic reticulum (ER) and mitochondria, and this localization was dependent on the N-terminal region of iPLA2γ (20). Complement stimulated the activation of iPLA2γ, which was mediated via the extracellular signal-regulated kinase and p38 pathways. Stimulation of iPLA2γ was dependent on phosphorylation of Ser-511 and/or Ser-515 via MNK1 (MAPK-interacting kinase 1) (20). In addition to characterizing these signaling pathways, we showed that overexpression of iPLA2γ in cultured GECs reduced complement-mediated GEC injury (19) as well as injury by tunicamycin, which was associated with augmentation of the ATF6 (activating transcription factor 6) pathway of the unfolded protein response in the ER (21). In other cells, such as primary cultures of rabbit renal proximal tubules cells, knockdown of iPLA2γ expression increased lipid peroxidation and impaired mitochondrial function (22). Likewise, knockdown of iPLA2γ in C2C12 myoblasts also caused an elevation of lipid peroxidation and reduction of ATP synthesis (11). Thus, the function of iPLA2γ can be cytoprotective, and depending on the cellular context, it may involve ER stress or removal of peroxidized phospholipid from the mitochondrial membrane, thereby preserving membrane integrity.
So far, we and others have provided considerable insight into the functions of iPLA2γ in cultured cell lines, but information on the functional role of iPLA2γ in the kidney is lacking. The aim of the present study was to employ iPLA2γ KO mice to better understand the role of iPLA2γ in normal glomerular/podocyte function and in glomerular injury. We show that deletion of iPLA2γ did not cause albuminuria; however, it resulted in mitochondrial structural abnormalities and enhanced autophagy in podocytes of aging mice. Moreover, after induction of anti-GBM nephritis in young mice, iPLA2γ KO mice exhibited significantly increased levels of albuminuria, podocyte injury, and loss of podocytes compared with WT mice. Our results indicate that iPLA2γ has a protective functional role in the normal glomerulus and in glomerulonephritis.
Experimental Procedures
Materials
A mouse albumin ELISA quantification kit was purchased from Bethyl Laboratories (Montgomery, TX). The creatinine assay kit was from Cayman Chemical Co. (Ann Arbor, MI). Tissue culture media and Lipofectamine 2000 were from Invitrogen (Burlington, Canada) and Wisent (Saint-Jean-Baptiste, Canada). Electrophoresis reagents were from Bio-Rad (Mississauga, Canada), and GE Healthcare (Baie d'Urfé, Canada). Rabbit anti-Wilms tumor-1 (WT1; catalog no. 192), goat anti-synaptopodin (catalog no. 21537), rabbit anti-GRP94 (catalog no. 11402), and rat anti-GRP78 (BiP; catalog no. 13539) antibodies were purchased from Santa Cruz Biotechnology. Goat anti-podocalyxin antibody (catalog no. AF1556) was from R&D Systems (Minneapolis, MN). Rabbit anti-microtubule-associated protein light chain 3B (LC3; catalog no. 2775), rabbit anti-phospho-AMP-activated protein kinase (AMPK)-α (Thr-172; catalog no. 2531), and rabbit anti-AMPKα (catalog no. 2532) antibodies were from Cell Signaling Technology (Danvers, MA). FITC-conjugated rabbit anti-sheep IgG (catalog no. 31509) was from Zymed Laboratories Inc. (South San Francisco, CA). FITC-conjugated goat anti-mouse complement C3 (catalog no. 55500) was purchased from MP Biomedical (Santa Ana, CA). MitoTracker Red CMXRos was from Thermo Fisher Scientific (Burlington, ON). Plasmid pBABE-puro mCherry-EGFP-LC3B was from Addgene (catalog no. 22418) (23). Plasmid “mito-YFP” (cytochrome c oxidase-subunit IV fused with yellow fluorescent protein; catalog no. 10089272) was from the American Type Culture Collection (Manassas, VA). Sheep anti-rat GBM, and rabbit anti-nephrin antisera were described previously (24, 25).
Mice
iPLA2γ KO mice in a C57BL/6 background were produced, bred, and genotyped, as described previously (7). Animal protocols were reviewed and approved by the McGill University Animal Care Committee. Conscious animals fed ad libitum were weighed, and urine samples were collected at various time points after weaning up to 12–16 months of age.
Assays for Albuminuria and Serum Creatinine
Mouse urine albumin concentration was quantified using an enzyme-linked immunosorbent assay kit according to the manufacturer's instructions. The assay utilizes urine samples diluted 1:5000 or 1:150,000, analyzed in a plate reader set at 450 nm. Urine creatinine concentration was measured using a colorimetric assay kit according to the manufacturer's instructions. The assay measures creatinine in urine samples diluted 1:15, using a picric acid-based method, and analyzed on a plate reader set at 490–500 nm. Excretion of albumin is expressed as the albumin/creatinine ratio. Serum creatinine was measured using an enzymatic assay on the Olympus AU5822 chemistry system (Beckman Coulter) in the Biochemistry Laboratory of the McGill University Health Centre.
Induction of Anti-GBM Nephritis in Mice
Anti-GBM nephritis was induced in 3–4-month-old iPLA2γ KO or WT mice by a single tail intravenous injection of 5–10 μl of sheep anti-GBM antiserum diluted in 0.1 ml of sterile saline (24, 26). After 24 h, mice were sacrificed by CO2 asphyxiation, and kidneys were dissected. Kidney sections were collected for immunofluorescence, light, and electron microscopy (see below). Glomeruli were isolated utilizing a differential sieving technique (24).
Cell Culture and Complement Cytotoxicity Assay
Mouse GECs were derived from iPLA2γ KO mice and WT control. The detailed method and characterization of the cells was published previously (21). Briefly, by quantitative reverse polymerase chain reaction, iPLA2γ mRNA was present in cells derived from WT mice but was completely absent in cells from KO mice. WT and KO cell lines expressed the podocyte proteins, synaptopodin and nephrin (12, 14, 21). The cells were cultured on plastic substratum in K1 medium and were used at passages 2–20. Cells were transfected with pBABE-puro mCherry-EGFP-LC3B, using Lipofectamine 2000 reagent according to the manufacturer's instructions.
Rat GECs were cultured as described above and were characterized previously (19–21, 26). These cells were employed in the complement cytotoxicity assay (19, 20, 26). Briefly, rat GECs were incubated with rabbit anti-GEC antiserum (5%, v/v) in modified Krebs-Henseleit buffer containing 145 mm NaCl, 5 mm KCl, 0.5 mm MgSO4, 1 mm Na2HPO4, 0.5 mm CaCl2, 5 mm glucose, and 20 mm Hepes, pH 7.4, for 30 min at 22 °C. The cells were then incubated for 40 min at 37 °C with normal human serum (with full complement activity) or heat-inactivated (decomplemented) human serum (incubated at 56 °C for 60 min) in controls. Complement-mediated cytolysis was determined by measuring lactate dehydrogenase (LDH) release (19, 26). Specific LDH release was calculated as (NS − HIS)/(100 − HIS), where NS represents the percentage of total LDH released into cell supernatants in incubations with normal serum, and HIS represents the percentage of total LDH released in incubations with heat-inactivated serum.
Immunofluorescence Microscopy
Kidney poles were snap-frozen using HistoPrep (Fisher Scientific, Ottawa, Canada) in isopentane (−80 °C). Cryostat sections (4 μm) were cut and stored at −80 °C. Frozen sections were then fixed in 4% paraformaldehyde and blocked with 5% normal rabbit serum (for sheep IgG) or goat serum (for C3) in 5% BSA or in 5% BSA alone (for synaptopodin, podocalyxin, and nephrin antibodies). The blocking solution was replaced with a fluorophore-conjugated primary antibody or, alternatively, with an unconjugated primary antibody, followed by a fluorophore-conjugated secondary antibody. Incubations with primary antibodies ranged from 2 h to overnight (4 or 22 °C). Incubations with secondary antibodies were 1 h at 22 °C. Sections were examined with a Zeiss AxioObserver fluorescence microscope with visual output connected to an AxioCam digital camera. Images of multiple glomeruli were collected from series of images derived from different focal planes (z-stack). To allow comparisons of fluorescence intensity, all images were taken at the same exposure. Fluorescence intensity was quantified using the histogram function of Adobe Photoshop or ImageJ software (National Institutes of Health, Bethesda, MD). Results are expressed in arbitrary units.
For WT1 immunofluorescence, frozen sections were fixed with 4% paraformaldehyde and then immersed in 10 mmol/liter sodium citrate, pH 6.0, for 8 min at 100 °C. After cooling to 22 °C, sections were blocked with 10% normal goat serum and were incubated with rabbit anti-WT1 antibody (4 °C, overnight), followed by rhodamine-goat anti-rabbit IgG (22 °C, 1 h). WT1-positive cells were quantified by visual counting.
To visualize mitochondria, cells on coverslips were incubated for 15–30 min at 37 °C with MitoTracker Red CMXRos (25 nm). Hoechst H33342 (1 μg/ml) was added for the final 5 min to label nuclei. Cells were then fixed with paraformaldehyde. After washing, coverslips were mounted onto glass slides and visualized with a fluorescence microscope (20).
GFP and mCherry-LC3-II puncta in cells can be readily distinguished from background fluorescence and quantified using ImageJ software. Green and red images were separately converted to grayscale. Cell contours on grayscale images were selected with the freehand tool, and the area of the cross-section was measured. Then image histogram plots were obtained, representing the range of pixel brightness values in each image. The threshold intensity was set at the end of the histogram declining slope (thereby only including the brightest fluorescent puncta and omitting cellular features and background). The area of LC3-II puncta was set between 0.2 and 25 μm2. The number of particles/puncta and total cross-sectional area of the particles/puncta whose areas fell within the set range of 0.2–25 μm2 were measured. Results were normalized per 1000 μm2 of cell area. The image calculator function (subtract operation) was used to calculate the mCherry-LC3-II particles/puncta that did not colocalize with GFP-LC3-II.
Light and Electron Microscopy
For light microscopy, kidney tissue was fixed in formalin, and was processed and embedded in paraffin according to conventional techniques. Sections were stained with periodic acid Schiff at the Goodman Cancer Research Center Histology Facility, McGill University. Electron microscopy was carried out at the McGill University Facility for Electron Microscopy Research. Briefly, kidney tissue was fixed in 2.5% glutaraldehyde in 0.1 mol/liter sodium cacodylate buffer containing 0.1% CaCl2, pH 7.4. Samples were washed and postfixed with 1% aqueous OsO4 and 1.5% aqueous potassium ferrocyanide. Then samples were dehydrated with serially increasing concentrations of acetone (from 30 to 100%) and were infiltrated with epon/acetone. Sections of 90–100 nm were placed onto grids and stained with uranyl acetate and Reynold's lead. Samples were viewed with an FEI Tecnai 12 transmission electron microscope operating at an accelerating voltage of 120 kV and equipped with an AMT XR80C CCD camera.
Quantification of Foot Process Effacement and Glomerular Surface Area
Randomly selected electron micrographs were used for morphometric analysis of foot process effacement, as described previously (27, 28), using ImageJ software. The mean width of the foot process (FPW) was calculated according to the formula (27), FPW = (π × glomerular basement membrane length)/(4 × foot process number).
A greater foot process width value represents increased foot process effacement. Three capillary loops from 2–3 glomeruli/mouse were quantified, and the results from 3–5 mice/group were pooled and analyzed. Glomerular cross-sectional surface area was measured in light micrographs of KO and WT mice, using ImageJ software.
Immunoblot Analysis
Lysates of isolated glomeruli (24) were prepared in buffer containing 1% Triton X-100, 125 mm NaCl, 10 mm Tris (pH 7.4), 1 mm EGTA, 2 mm Na3VO4, 10 mm sodium pyrophosphate, 25 mm NaF, and protease inhibitor mixture (Bioshop, Burlington, Canada). Equal amounts of lysate proteins were dissolved in Laemmli buffer and were subjected to SDS-PAGE under reducing conditions. Proteins were then electrophoretically transferred onto nitrocellulose or polyvinyl difluoride membranes and blocked at 22 °C for 1 h with 5% BSA in buffer containing 10 mm Tris, pH 7.5, 50 mm NaCl, 2.5 mm EDTA, and 0.05% Tween 20. The membrane was then incubated with primary and secondary antibodies. Bands were detected by an enhanced chemiluminescence system (GE Healthcare, Mississauga, Canada). Quantitative densitometry was performed using ImageJ (25).
Statistics
Data are presented as mean ± S.E. One-way analysis of variance was used to determine significant differences among groups. Where significant differences were found, individual comparisons were made between groups using Student's t test and adjusting the critical value according to Bonferroni's method. Student's t test was used to determine significant differences between two groups.
Results
iPLA2γ Deletion Does Not Affect Baseline Urinary Albumin Excretion
General phenotypic features of mice null for iPLA2γ were described previously (7, 11). In keeping with the earlier report, modest growth retardation was visible in KO mice after 2 months of age. At 3–4 months, iPLA2γ KO animals (males and females) had a mean weight of 22.09 ± 0.75 g versus 26.02 ± 0.84 g for WT (p < 0.005, n = 9–10 mice/group). There was no significant difference in weight between males and females within the same groups. To determine whether deletion of iPLA2γ can affect glomerular permeability and induce proteinuria, we quantified the urine albumin/creatinine ratio. Mice at 4–7 months of age showed a low albumin/creatinine ratio, and there was no significant difference between KO and WT mice (Fig. 1A). Urine albumin tended to be slightly higher in male mice compared with females. The albumin/creatinine ratio between WT and age/sex-matched KO mice did not differ significantly up to 16 months of age (Fig. 1). Serum creatinine values were 9.3 ± 0.7 μm in 10–12-month-old iPLA2γ KO mice and 12.1 ± 0.9 μm in WT (p = 0.02; 7–8 mice/group). The lower mean value in the KO mice is consistent with lower body weight. Light microscopy (periodic acid Schiff staining) showed normal glomerular and tubular morphology in both WT and KO mice 10–12 months of age (results not shown). To confirm the visual impression, glomerular surface area was measured in light micrographs of KO and WT mice. There were no significant differences between groups (Fig. 1B).
FIGURE 1.
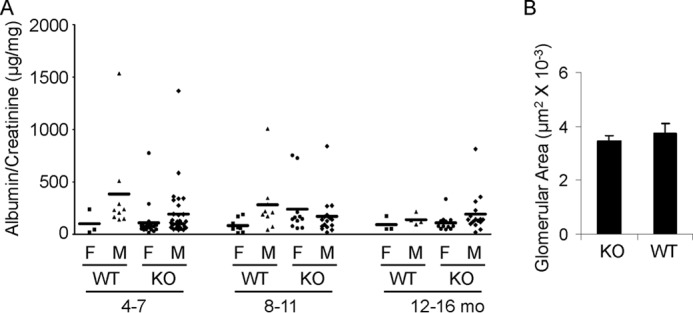
iPLA2γ deletion does not alter urinary albumin excretion in mice. A, urinary albumin/creatinine ratio. Each point represents the urine collection of a single mouse taken at monthly intervals between 4 and 16 months (mo) of age. WT and iPLA2γ KO mice were divided into groups according to sex (male (M) and female (F)). There are no significant differences among the groups. B, glomerular surface area was measured in light micrographs of KO and WT mice (age 10–11 months). There were no significant differences between groups (3 mice/group; 14–24 measurements/group). Error bars, S.E.
iPLA2γ Deletion Induces Mitochondrial Damage and Autophagy in Podocytes
Given that iPLA2γ is localized in GECs at the ER and mitochondria (20), we hypothesized that deletion of iPLA2γ may induce abnormalities in these organelles. Kidneys of iPLA2γ KO and WT control mice at age 10–11 months were examined by electron microscopy. Podocyte foot processes appeared structurally intact in both iPLA2γ KO mice (Fig. 2A) and WT controls (not shown). WT mice showed no apparent glomerular abnormalities, and in particular, podocytes showed intact cell organelles, including the ER and mitochondria (Fig. 2B). In contrast, some podocytes in iPLA2γ KO mice showed microvillous transformation and membrane vesiculation, compatible with podocyte injury (Fig. 2C). The striking observation in iPLA2γ KO mice was that podocytes showed abnormal mitochondria and the presence of autophagic vacuoles (Fig. 2, D–I). The mitochondrial abnormalities were widespread and included aggregation, disruption of membranes, and loss/rearrangement of cristae (Fig. 2, F and H), whereas some mitochondria appeared to be undergoing fission (Fig. 2F).
FIGURE 2.

Kidney cell ultrastructure in 10–11-month-old mice (electron microscopy). A, podocyte foot processes in iPLA2γ-KO mice appeared structurally intact. B, podocyte from a WT, control mouse shows intact cell organelles, including mitochondria. C, podocytes from an iPLA2γ KO mouse show microvillous transformation and membrane vesiculation (arrows). D–I, podocytes from iPLA2γ-KO mice show abnormal mitochondria and autophagic vacuoles. F and H, magnified views of the areas in E and G outlined by boxes, respectively. Mitochondria in podocytes show aggregation, disruption of membranes, and loss of cristae (arrowheads in F and H), whereas some mitochondria appear to be undergoing fission (F). In D and I, the closed arrows point to autophagosomes in podocytes. Mitochondria in a parietal epithelial cell (open arrow, D) and a glomerular endothelial cell (open arrow, H) appear normal. The ER (F and H) and nuclei (E–I) in podocytes appear intact. J, proximal tubular epithelial cell from an iPLA2γ-KO mouse shows normal mitochondria. The brush border is seen on the top right of the photomicrograph. Bar, 500 nm. Glomeruli were examined in five KO and three WT mice, 2–3 glomeruli/mouse. K, quantification of foot process width in iPLA2γ-KO and WT mice. *, p < 0.0005 KO versus WT (21–22 measurements/group). Error bars, S.E.
We analyzed all high power electron micrographs to quantify damaged mitochondria and autophagosome structures in podocytes. In 5 iPLA2γ KO mice, damaged mitochondria were observed in 54% of the micrographs, and damaged autophagosomes were seen in 17%. Forty percent of the micrographs showed both normal mitochondria and an absence of autophagosomes (35 high power electron micrographs were examined in total). In three WT mice, there were no damaged mitochondria nor any autophagosomes (17 electron micrographs were examined).
It should also be noted that, unlike podocytes, mitochondria in parietal epithelial cells, glomerular endothelial cells, and proximal tubular epithelial cells in iPLA2γ KO mice appeared normal (Fig. 2, D, H, and J). In contrast to the mitochondria in iPLA2γ KO podocytes, the ER and nuclei appeared intact (Fig. 2, E–I). Quantification of foot process width in iPLA2γ KO and WT mice demonstrated modest but significant widening of foot processes in the KO (Fig. 2K). Thus, deletion of iPLA2γ in mice induces prominent mitochondrial abnormalities, autophagy, and modest widening of the foot processes and plasma membrane changes in podocytes.
iPLA2γ Deletion Induces Podocyte Loss but Not ER Dysfunction in Podocytes
The above results showed that iPLA2γ deletion affected podocyte ultrastructure; consequently, podocyte-associated proteins that are believed to play key roles in maintaining ultrastructure and slit diaphragms (nephrin, synaptopodin, and podocalyxin) (12, 14, 26, 29) were examined by immunofluorescence microscopy and immunoblot analysis. The podocyte nuclear marker, WT1, was also studied as an index of podocyte number (12, 14, 26, 29). Kidney sections from iPLA2γ KO and WT mice (age 10–11 months) were stained with specific antibodies. Synaptopodin, nephrin, and podocalyxin showed an immunofluorescence staining pattern consistent with expression in podocytes in glomeruli of iPLA2γ KO and WT mice, although the intensity of synaptopodin was reduced in the KO (Fig. 3A). Quantification of glomerular fluorescence intensity showed that synaptopodin staining was significantly lower in iPLA2γ KO glomeruli compared with WT (Fig. 3B). Fluorescence intensities of nephrin and podocalyxin staining did not differ significantly between WT and iPLA2γ KO mouse glomeruli, although both tended to decrease in the KO (Fig. 3, C and D).
FIGURE 3.

Expression of synaptopodin, nephrin, podocalyxin, and WT1 in 10–11-month-old WT and iPLA2γ-KO mice. A, kidney sections were stained with specific antibodies and were examined by immunofluorescence microscopy. Bar, 20 μm. B–D, quantification of fluorescence intensity in arbitrary units (3–7 glomeruli/mouse in three KO and two WT mice, or 11–18 glomeruli/group, were analyzed). B, synaptopodin: p < 0.0002, KO versus WT. E, WT1-positive (pos) nuclei: p < 0.0001, KO versus WT (6–12 glomeruli/mouse in three KO and two WT mice, or 34–55 glomeruli/group, were analyzed). Error bars, S.E.
To determine whether deletion of iPLA2γ induces podocyte loss, we quantified the number of podocyte nuclei in glomeruli of iPLA2γ KO and WT mice using WT1 immunostaining. There was a ∼25% decrease in the number of WT1-positive cells in iPLA2γ KO glomeruli compared with control (Fig. 3, A and E), indicating that deletion of iPLA2γ induced a loss of podocytes. This result does not, however, exclude the possibility that a reduction in WT1-positive cells might have been related to injury instead of loss of cells.
Immunoblotting of glomerular lysates showed that expression of nephrin and podocalyxin proteins was not significantly different between iPLA2γ KO and WT mice. Synaptopodin was reduced in the KO (Fig. 4, A–D), consistent with the immunofluorescence staining (Fig. 3, B–D). Nephrin undergoes important post-translational modifications in the ER before export to the plasma membrane (25). By SDS-PAGE, nephrin migrates as a doublet of ∼180 kDa. The upper band represents the fully glycosylated, mature form of nephrin, whereas the lower band is an immature form found in the ER (25, 26). To address the role of iPLA2γ in ER function in vivo, we examined the pattern of nephrin glycosylation in isolated glomeruli by immunoblotting. There were no apparent differences in nephrin glycosylation between iPLA2γ KO and WT mice (Fig. 4B, bottom), suggesting that ER function remained intact despite iPLA2γ deletion.
FIGURE 4.

Effect of iPLA2γ deletion on expression of nephrin (fully glycosylated and ER forms), podocalyxin, GRP94, synaptopodin (synpo), phospho-AMPK (pAMPK), AMPK, and LC3-I and -II. Glomeruli were isolated from 10–11-month-old WT and iPLA2γ-KO mice. Lysates were immunoblotted, as indicated. A, representative immunoblots. The white lines in the top panel indicate reassembly of noncontiguous gel lanes. There were no adjustments made to the digital images among the lanes that would alter the information in the panels. B–G, densitometric quantification. B, C, and E, there were no significant differences in the expression of fully glycosylated nephrin (top nephrin band), nephrin maturation (ratio of top to bottom band), podocalyxin, and GRP94 between groups (3–5 mice/group). D, synaptopodin was significantly lower in KO glomeruli. *, p < 0.05, 3 mice/group. F, the ratio of phospho-AMPK/AMPK was significantly greater in KO glomeruli. *, p < 0.04, 4 mice/group. G, the ratio of LC3-II/LC3-I was significantly greater in KO mice. *, p = 0.01, 7–9 mice/group. Because the intensity of the LC3-I band in A was relatively weak, the immunoblot was repeated using a longer exposure time. This resulted in enhanced signals in LC3-I bands, although the signals of the LC3-II bands became oversaturated. Nevertheless, the LC3-II/LC3-I ratio remained significantly elevated in the KO mice compared with WT (results not shown). Error bars, S.E.
In a previous study (21), we demonstrated that in cultured GECs, iPLA2γ, via its localization at the ER, amplified the activation of the ATF6 pathway of the unfolded protein response. Amplification of ATF6 resulted in the up-regulation of the ER chaperones, GRP78 (BiP) and GRP94 (21). Conversely, deletion of iPLA2γ blunted these responses, as shown in cultured GECs derived from WT and iPLA2γ KO mice (21). Therefore, we undertook to examine the expression of ER chaperones in podocytes in vivo by immunoblotting. Unexpectedly, there was no significant difference in the glomerular GRP94 expression between WT and iPLA2γ KO mice (Fig. 4, A and E). A similar result was obtained with GRP78 (BiP) expression (result not shown). Therefore, either differences in ER chaperone expression between WT and iPLA2γ KO mice were too small to be detected in vivo, or additional factors regulate the expression of these proteins in vivo compared with in cultured cells.
iPLA2γ Deletion Reduces Mitochondrial Function
The electron micrographs of iPLA2γ KO mice showed striking mitochondrial abnormalities (Fig. 2). To determine whether these mitochondrial abnormalities were associated with impaired mitochondrial function in vivo, we monitored phosphorylation of AMPK, a ubiquitous serine/threonine kinase (30, 31). During periods of cellular stress, particularly when there is a decrease in ATP, and increases in ADP and AMP, binding of ADP or AMP activates AMPK by facilitating phosphorylation and inhibiting dephosphorylation of Thr-172 in the α-subunit. Thus, AMPK acts as a sensor of cell energy stores, and phosphorylation of AMPK is inversely related to the cell's ATP level. The mean glomerular phospho-AMPK level was clearly increased in iPLA2γ KO mice compared with control, although there was some variability among animals (Fig. 4, A and F). This result is in keeping with reduced ATP generation by mitochondria in podocytes.
To provide additional support for the conclusion that deletion of iPLA2γ impaired mitochondrial function, we examined Mitotracker Red CMXRos staining in cultured GECs produced from WT and iPLA2γ KO mice (21). MitoTracker Red CMXRos is a fluorescent dye that stains mitochondria in live cells, and its accumulation is dependent on the mitochondrial membrane potential. In WT cells, the mitochondria stained brightly and localized diffusely throughout the cytoplasm (Fig. 5, A and C). In these cells, the mitochondria appeared as short tubular structures, and they did not form long filaments. In most of the iPLA2γ KO cells, cytoplasmic staining was faint or absent, and the mitochondria appeared aggregated in a perinuclear distribution (Fig. 5, A and C). The latter resembled the staining of Mitotracker Red CMXRos in WT GECs that had been treated with antimycin A (Fig. 5A). This drug disrupts mitochondrial function by blocking the electron transport chain and results in loss of fluorescent probe from mitochondria (32). It should be noted that, in contrast to the Mitotracker Red CMXRos staining, there were no differences in the expression of transfected mito-YFP (YFP targeted to mitochondria) between iPLA2γ KO and WT GECs (Fig. 5, B and C). This reflects similar mitochondrial mass in the two cell lines. Thus, the reduced Mitotracker Red CMXRos staining in iPLA2γ KO GECs indicates impaired mitochondrial function.
FIGURE 5.
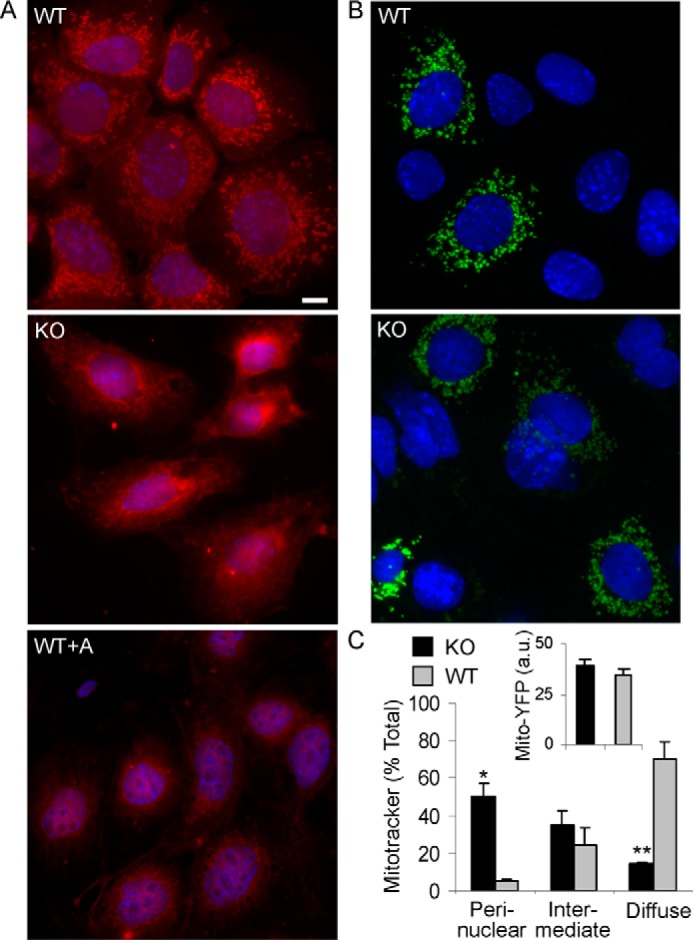
Mitochondrial function in cultured GECs. A, WT or iPLA2γ KO GECs were incubated with Mitotracker Red CMX-Ros (25 nm for 30 min). In most WT cells, the mitochondria stained brightly and were found diffusely throughout the cytoplasm. In many iPLA2γ KO cells, cytoplasmic staining was faint or absent, and the mitochondria were collapsed in a perinuclear distribution. The latter resembled the staining of Mitotracker Red in WT podocytes that had been treated with antimycin A (WT+A; 10 μm for 30 min) to induce mitochondrial dysfunction. Bar, 15 μm. Cells showing diffuse, perinuclear, and intermediate Mitotracker Red CMX-Ros staining patterns are quantified in the graph (C; intermediate pattern indicates perinuclear localization but with some diffuse staining). *, p < 0.0005; **, p < 0.0001, KO versus WT (170 KO cells and 228 WT cells). B, GECs were transiently transfected with mito-YFP. Images were obtained after 24 h. In transfected cells, expression of mito-YFP was comparable between KO and WT cells. The inset (C) shows quantification of YFP fluorescence intensity in arbitrary units (a.u.) (22–30 cells/group). Error bars, S.E.
iPLA2γ Deletion Induces Autophagy in GECs
Ultrastructural examination of iPLA2γ KO mice showed autophagic vacuoles in some podocytes (Fig. 2, C–E). The extent of lipidation of LC3 (i.e. conversion of LC3-I to LC3-II) is widely used to monitor autophagy, and typically, an increase in the ratio of LC3-II to LC3-I correlates with the number of active autophagosomes (33). We employed immunoblotting to monitor the levels of LC3-I and LC3-II in glomeruli of WT and iPLA2γ KO mice (Fig. 4A). Densitometric quantification showed that the LC3-II/LC3-I ratio was markedly greater in iPLA2γ KO glomeruli compared with WT control (Fig. 4G). These results are in keeping with the presence of autophagosomes in podocytes in the electron micrographs of iPLA2γ KO mice.
There are some limitations to quantifying enhanced autophagic flux in vivo. Thus, to confirm enhanced autophagy and the changes in LC3 in glomeruli, we measured the conversion of LC3-I to LC3-II in cultured GECs from WT and iPLA2γ KO mice (21). GECs were incubated with or without chloroquine for 2 or 6 h. Chloroquine blocks the fusion of autophagosomes with lysosomes, which forms autolysosomes, and therefore prevents autolysosomal protein degradation, allowing us to compare the rate of autophagosome formation in WT and iPLA2γ KO GECs (33). GEC lysates were immunoblotted with anti-LC3 antibody. Deletion of iPLA2γ in cultured GECs resulted in a significantly greater increase in the ratio of LC3-II/LC3-I in KO cells, compared with WT, in the presence of chloroquine at 6 h (Fig. 6A). This result is consistent with enhanced autophagy and is in keeping with the findings in vivo (Fig. 4G).
FIGURE 6.
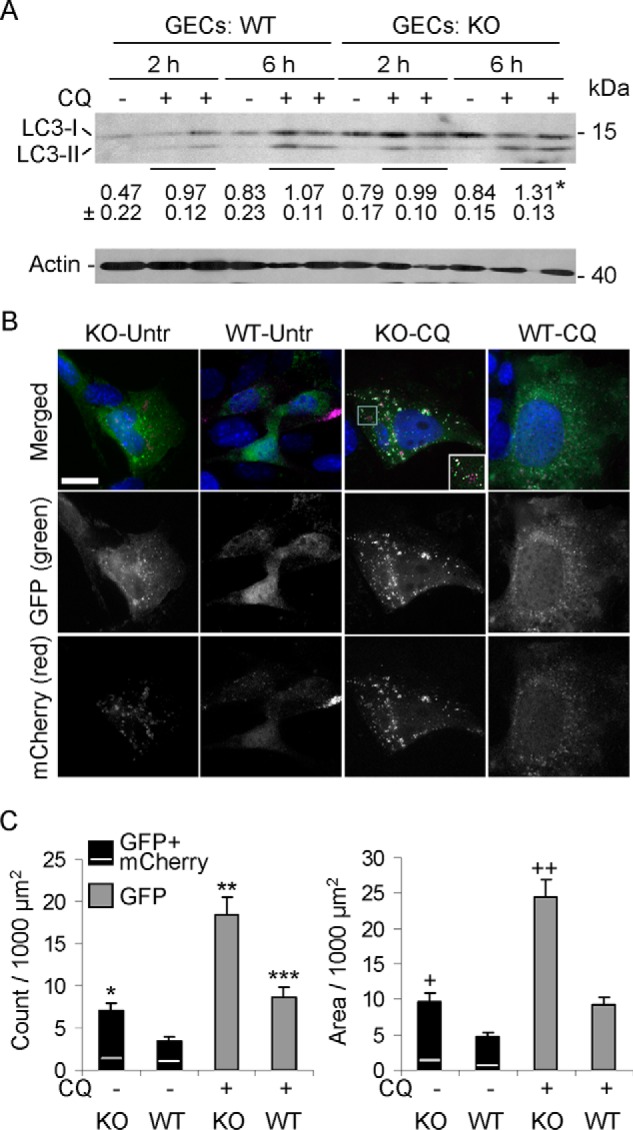
Autophagy is enhanced in iPLA2γ KO GECs in culture. A, deletion of iPLA2γ in GECs increases the ratio of LC3-II/LC3-I. WT or iPLA2γ KO GECs were incubated with or without chloroquine (CQ; 15 μm) for 2 or 6 h. Shown is a representative immunoblot of GEC lysates. Values below the LC3 immunoblot indicate mean ± S.E. ratios of LC3-II/LC3-I in the eight groups (four experiments). *, p < 0.03 KO versus WT at 6 h (+CQ). B, iPLA2γ KO GECs in culture show greater numbers of mCherry- and GFP-LC3-II puncta compared with WT cells (representative fluorescence micrographs). GECs were transiently transfected with mCherry-GFP-LC3B (0.1 μg of plasmid DNA/well). After 24 h, cells were untreated (Untr; −) or treated with 15 μm chloroquine (CQ; +). Images were obtained after 6 h. Bar, 25 μm. The inset (KO-CQ, merged) is an enlargement of the area within the square. C, quantification of GFP-LC3-II puncta and GFP-LC3-II puncta area. Results were normalized per 1000 μm2 of cell area. In the bars showing the GFP + mCherry LC3-II puncta/area, the GFP and mCherry components are above and below the white line, respectively. The mCherry component is that which did not colocalize with GFP. *, p < 0.005, KO-untreated versus WT-untreated. **, p < 0.0001, KO-CQ versus WT-CQ; p < 0.0001, KO-CQ versus KO-Untr; and ***, p < 0.01 WT-CQ versus WT-untreated. +, p < 0.002 KO-untreated versus WT-untreated; ++, p < 0.0001 KO-CQ versus WT-CQ and p < 0.0001 KO-CQ versus KO-untreated (11–12 cells/group). Error bars, S.E.
Conversion of LC3-I to LC3-II in cultured GECs was also addressed by monitoring the formation of LC3-II puncta by fluorescence microscopy (23, 33). In these experiments, GECs were transfected with mCherry-GFP-LC3B, encoding LC3 tagged with two fluorescent proteins, mCherry and GFP. mCherry- or GFP-LC3-I is cytoplasmic. GFP is quenched in an acidic environment (i.e. in lysosomes), but mCherry is acid-stable; thus, formation of GFP-LC3-II puncta reflects formation of autophagosomes, whereas mCherry-LC3-II puncta reflect formation of autophagosomes and their fusion with lysosomes (23, 33). In resting WT GECs, the distribution of mCherry- and GFP-LC3-I was in a cytoplasmic pattern (Fig. 6B). Quantification confirmed the visual impression that resting cells contained relatively few puncta and that the puncta constituted only a minimal proportion of cell area (Fig. 6C, second columns). Increased formation of GFP-LC3-II puncta and, to a lesser extent, mCherry-LC3-II puncta that did not colocalize with GFP was evident in iPLA2γ KO GECs even in the absence of chloroquine (Fig. 6, B and C, first columns). The addition of chloroquine markedly enhanced GFP-LC3-II puncta in KO cells (Fig. 6, B and C, third column), consistent with the formation of autophagosomes in the KO cells. Chloroquine slightly increased GFP-LC3-II puncta in WT cells, but the change was not significant (Fig. 6, B and C, fourth column). In the presence of chloroquine, prominent mCherry-LC3-II puncta were also increased in KO cells, and most mCherry puncta colocalized with GFP (Fig. 6B). These results reflect LC3-II at autophagosomes and to a minor extent in lysosomes and thus confirm enhanced autophagy in iPLA2γ KO GECs.
iPLA2γ Deletion Exacerbates Albuminuria in Anti-GBM Nephritis
Our previous studies in cultured GECs demonstrated a protective role for iPLA2γ in injury induced by complement (19) and tunicamycin (21). In addition, the above experiments showed that in vivo, iPLA2γ is essential to the maintenance of podocyte integrity under basal conditions. Specifically, deletion of iPLA2γ induced mitochondrial abnormalities with autophagy in older mice. In the next set of experiments, we examined whether induction of podocyte injury with anti-GBM antibody would reveal a protective phenotype of iPLA2γ in young mice. C5b-9-induced GEC/podocyte injury and proteinuria are hallmarks of heterologous mouse anti-GBM nephritis. Anti-GBM antibody binds in part to podocyte antigens in vivo, and heterologous phase proteinuria is abolished by C6 deficiency, indicating a key role of C5b-9 (24, 34, 35). In previous studies, we determined that administration of 5–10 μl of sheep anti-GBM antiserum induces mild to moderate albuminuria (26). Therefore, iPLA2γ KO and WT mice (age 3–4 months) were injected with anti-GBM antiserum, and additional mice in each group were injected with saline (control). Urine samples were collected just before administration of antibody and after 24 h. Both WT and iPLA2γ KO mice developed albuminuria 24 h after induction of anti-GBM nephritis. However, iPLA2γ KO mice exhibited significantly higher levels of albuminuria compared with WT (Fig. 7A).
FIGURE 7.
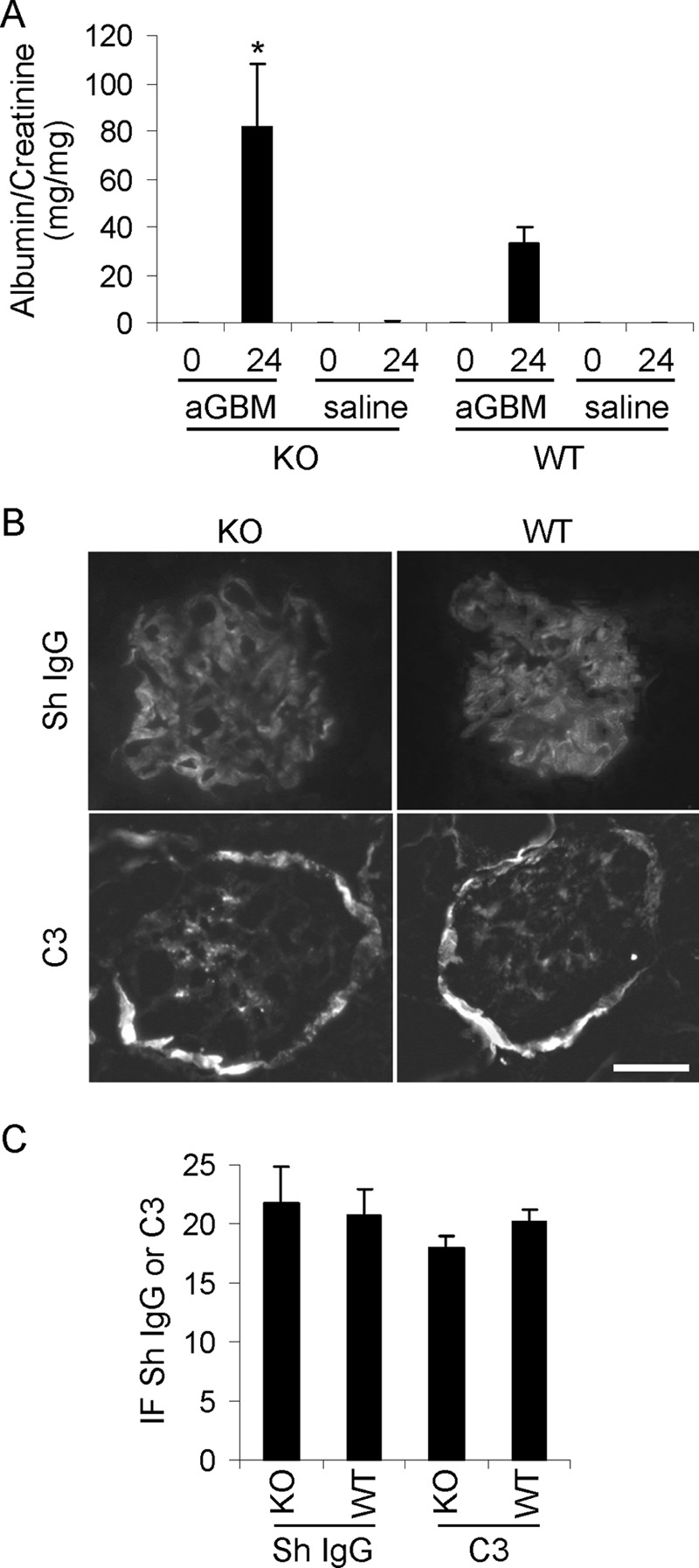
iPLA2γ deletion exacerbates albuminuria in anti-GBM nephritis. Nephritis was induced in WT and iPLA2γ KO mice (3–4 months of age) via single intravenous injection of sheep anti-rat GBM antiserum (aGBM). Control mice received saline. The urinary albumin/creatinine ratio was measured before and 24 h after the injection. A, after the induction of anti-GBM nephritis, iPLA2γ KO mice had greater albuminuria compared with WT. *, p < 0.05, iPLA2γ KO anti-GBM versus WT anti-GBM. KO anti-GBM, n = 7 mice (4 females and 3 males); WT anti-GBM, n = 7 mice (2 females and 5 males); KO control, n = 4 mice; WT control, n = 4 mice. B, immunofluorescence staining for sheep (Sh) anti-GBM IgG and mouse complement C3 in mice with anti-GBM nephritis (at 24 h). There is bright glomerular fluorescence staining for sheep IgG in both WT and iPLA2γ KO mice. Fainter glomerular C3 staining was present in both groups of mice. The C3 staining of Bowman's capsule (surrounding the glomerulus) is observed in normal mouse kidneys and is not due to the administration of anti-GBM antibody. Bar, 20 μm. C, quantification of anti-GBM antibody and complement C3 deposition (fluorescence intensity) in arbitrary units shows no significance differences between groups. Error bars, S.E.
Deposition of anti-GBM antibody and activation of complement were assessed by immunofluorescence microscopy. There was prominent linear deposition of sheep anti-GBM IgG along the glomerular capillary walls in both WT and iPLA2γ KO mice (Fig. 7B). Glomerular C3 staining was slightly fainter, compared with IgG, but was clearly present in both groups of mice (Fig. 7B). Quantification of IgG and C3 fluorescence intensity did not demonstrate significant differences between the two groups (Fig. 7C). Therefore, the increased albuminuria in iPLA2γ KO mice occurred despite equal glomerular anti-GBM antibody and C3 deposition in WT and iPLA2γ KO mice.
iPLA2γ Deletion Enhances Podocyte Ultrastructural Abnormalities in Anti-GBM Nephritis
After injection of anti-GBM antibody, glomerular and tubular morphology of WT and iPLA2γ KO mice (n = 6–7 mice) appeared normal by light microscopy; in particular, there was no inflammatory cell infiltration in the glomeruli (results not shown). Kidneys of albuminuric (anti-GBM antibody-treated) and control mice (saline-treated groups) were also examined by electron microscopy. The 3-month-old saline-treated KO mice showed no apparent glomerular abnormalities, and podocyte ultrastructure appeared normal (Fig. 8A). Similarly, the saline-treated WT mice showed no ultrastructural abnormalities (result not shown). Anti-GBM-injected WT mice showed focal abnormalities in podocyte ultrastructure (Fig. 8B). Some podocyte cell bodies appeared swollen, and there was some villous transformation and microvesiculation of the podocyte plasma membranes. In most areas of the glomerulus, foot processes appeared normal, but in other areas, there were abnormalities, ranging from short and widened foot processes to patchy effacement (Fig. 8B). Anti-GBM-injected iPLA2γ KO mice showed substantially more severe abnormalities in podocyte ultrastructure. Podocyte cell bodies were swollen with fewer organelles, and there was extensive villous transformation and microvesiculation of the plasma membranes. Foot processes were severely malformed and were effaced extensively (Fig. 8C). However, there were no apparent abnormalities in the ultrastructure of organelles, including mitochondria, and no apparent autophagosomes. There was an absence of leukocytes in the glomeruli. To quantify podocyte foot process effacement objectively, we measured foot process width in iPLA2γ KO and WT mice in both saline- and anti-GBM-injected groups. The anti-GBM-injected iPLA2γ KO mice showed the greatest foot process width, indicating severe foot process effacement (Fig. 8D). Taken together, deletion of iPLA2γ in anti-GBM nephritis induced severe abnormalities in podocyte ultrastructure, which are in keeping with the increased level of albuminuria in these mice.
FIGURE 8.
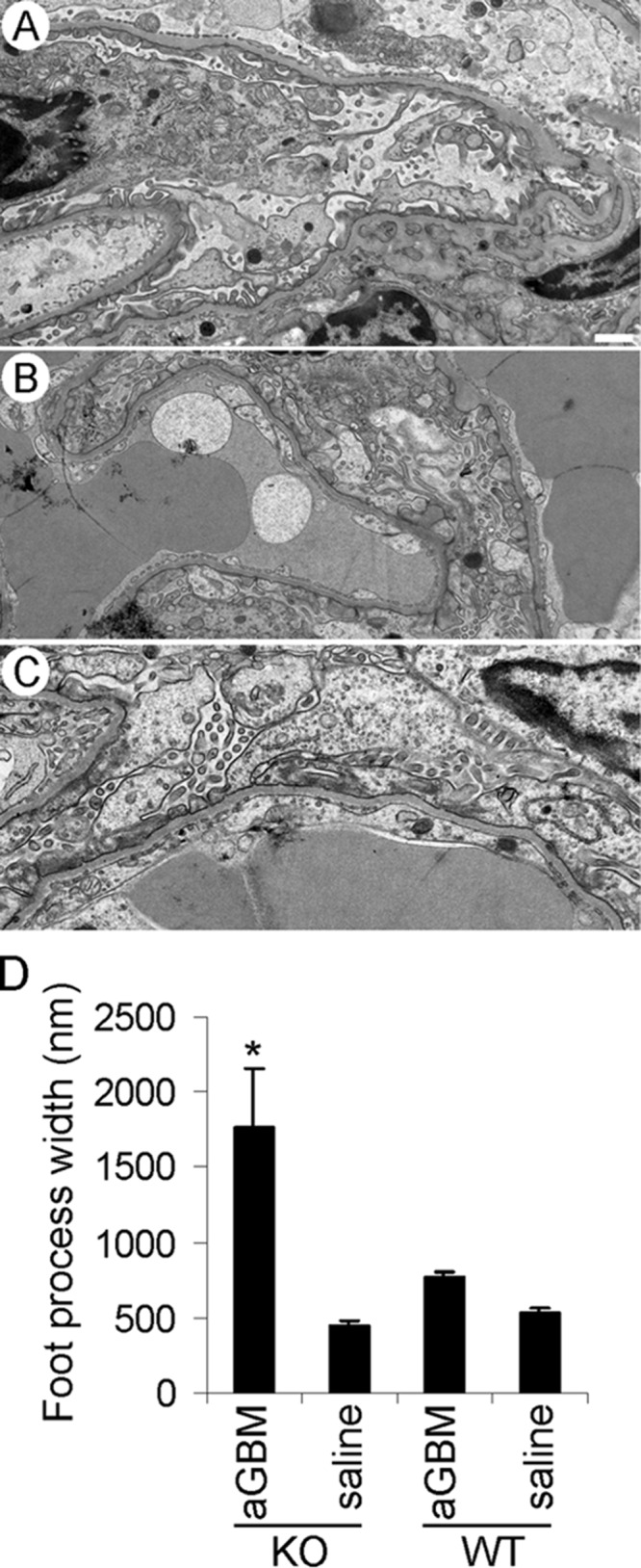
iPLA2γ deletion exacerbates abnormalities in podocyte ultrastructure in anti-GBM nephritis. Kidneys of four albuminuric mice (two iPLA2γ KO and two WT; 3–5 glomeruli/mouse) and three control mice (saline-treated groups) were examined by electron microscopy. A, an iPLA2γ KO mouse (age 3 months) injected with saline shows normal podocyte ultrastructure. Foot processes, cell bodies, and intracellular organelles appear intact (bar, 500 nm). B, WT mouse (age 3 months) injected with anti-GBM antibody. In this electron micrograph, podocyte foot processes appear widened with segmental effacement. There is some villous transformation of the podocyte plasma membranes. C, iPLA2γ KO mouse (age 3 months) injected with anti-GBM antibody. Podocyte cell bodies appear swollen, and the cell membranes show marked villous transformation and microvesiculation. Podocyte foot processes are almost completely effaced. D, quantification of foot process width in iPLA2γ-KO and WT mice. *, p < 0.0001 KO/anti-GBM versus other groups, 9–36 measurements/group. Error bars, S.E.
iPLA2γ Deletion Induces Marked Podocyte Loss in Anti-GBM Nephritis
In addition to albuminuria, glomerular injury may be associated with loss of podocytes. To provide further evidence for a cytoprotective effect of iPLA2γ in vivo, we quantified the number of podocyte nuclei in glomeruli of KO and WT mice by WT1 immunostaining. In saline-treated mice (age 3 months), deletion of iPLA2γ did not alter the number of WT1-positive cells compared with WT. Furthermore, in WT mice, injection of anti-GBM antiserum did not alter the number of WT1-positive cells. In contrast, there was a ∼60% decrease in the number of WT1-positive cells in iPLA2γ KO glomeruli after anti-GBM antiserum injection (Fig. 9, A and B). Therefore, genetic ablation of iPLA2γ induced a marked loss of podocytes after these cells were subjected to injury. Taken together, the results indicate that iPLA2γ exerts a cytoprotective effect on the podocyte and glomerular injury in vivo.
FIGURE 9.

Immunofluorescence staining for WT1 and nephrin in anti-GBM nephritis. A and B, iPLA2γ deletion reduced podocyte number in anti-GBM nephritis. WT1 immunofluorescence was assessed in two WT mice without and with anti-GBM antibody and four iPLA2γ KO mice (age 3–4 months), without and with anti-GBM antibody. Bar, 20 μm. B, quantification of WT1-positive nuclei per glomerulus (25–36 glomeruli/group). *, p < 0.001 KO/anti-GBM versus WT/anti-GBM. A and C, nephrin immunofluorescence intensity was assessed in iPLA2γ KO mice without (n = 2) and with anti-GBM antibody (n = 6) and in WT mice without (n = 2) and with anti-GBM antibody (n = 5). *, p < 0.0001 anti-GBM versus saline (14–31 measurements/group). Error bars, S.E.
Effects of iPLA2γ Deletion on Expression of Nephrin and ER Stress Proteins in Anti-GBM Nephritis
The expression of nephrin was examined by immunofluorescence microscopy and was then quantified. In parallel with albuminuria, anti-GBM antibody induced a decrease in nephrin expression in both WT and iPLA2γ KO mice; nephrin expression tended to be lower in iPLA2γ KO mice, compared with WT, but the difference did not reach statistical significance (Fig. 9, A and C). By immunoblotting, expression of the mature form of nephrin was reduced markedly in anti-GBM nephritis but comparably in WT and iPLA2γ KO mice (Fig. 10, A (top nephrin band) and B). Furthermore, the amount of mature nephrin (top band) was reduced significantly relative to the ER form (bottom band) in anti-GBM nephritic mice (Fig. 10, A and C). These results imply that in anti-GBM nephritis, there is defective glycosylation of nephrin due to ER dysfunction, although this appears to be largely independent of iPLA2γ.
FIGURE 10.
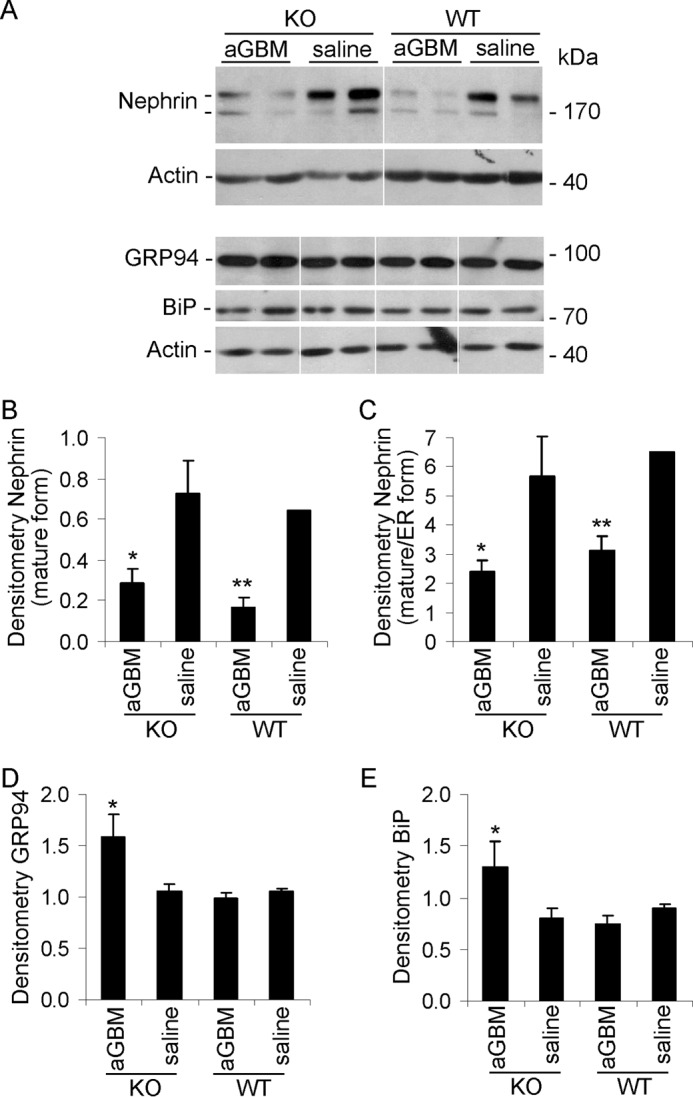
Effect of iPLA2γ on the expression of nephrin and ER stress proteins in anti-GBM nephritis. Glomeruli were isolated from 3-month-old WT and iPLA2γ KO mice that were treated with anti-GBM antibody or saline (at 24 h). Lysates were immunoblotted as indicated. A, representative immunoblots. The white lines indicate reassembly of noncontiguous gel lanes. There were no adjustments made to the digital images among the lanes that would alter the information in the panels. B–E, densitometric quantification. B and C, anti-GBM antibody reduced expression of mature nephrin (top nephrin band) as well as maturation of nephrin (ratio of top to bottom band) in both WT and iPLA2γ KO mice. B, *, p < 0.02; **, p < 0.01, anti-GBM versus saline (3–4 mice/group). C, *, p = 0.05; **, p < 0.02 anti-GBM versus saline (3–4 mice/group). D and E, anti-GBM antibody increased expression of GRP94 and BiP (GRP78) in iPLA2γ KO mice. D, *, p < 0.005 KO/anti-GBM versus KO/saline (3–4 mice/group). E, *, p < 0.02, KO/anti-GBM versus KO/saline (3–4 mice/group). Error bars, S.E.
To further address the role of iPLA2γ in ER function in vivo, we monitored the unfolded protein response (i.e. changes in the expression of ER chaperones after induction of anti-GBM nephritis). Glomerular GRP94 and GRP78 (BiP) were up-regulated significantly in the KO anti-GBM mice but not in WT (Fig. 10, A, D, and E). There were no consistent changes in CHOP (CCAAT/enhancer-binding protein homologous protein) among groups (results not shown). These results imply that deletion of iPLA2γ in the context of glomerulonephritis leads to amplification of the adaptive unfolded protein response. The dose of anti-GBM antibody used in this study was low, allowing us to ascertain differences in albuminuria between iPLA2γ and WT mice. At substantially higher doses of antibody, WT mice with anti-GBM nephritis also show comparable increases in glomerular expression of GRP94 and BiP (results not shown). However, at these higher doses, albuminuria is massive and does not allow for the study of iPLA2γ effects.
Mitochondrial Injury Enhances Complement-mediated Cytotoxicity
The primary pathological finding in aging iPLA2γ KO mice is mitochondrial damage in podocytes (Fig. 2). Compared with WT, iPLA2γ KO mice (age 3 months) showed greater albuminuria and podocyte injury after induction of anti-GBM nephritis (Figs. 7A and 8), but mitochondrial ultrastructural damage in the podocytes was not observed. Nevertheless, to determine whether enhanced complement-mediated podocyte injury in anti-GBM nephritis in the iPLA2γ KO mice may have been associated with underlying mitochondrial dysfunction, we examined the role of the mitochondria in complement-dependent cytolysis in cultured GECs. We pretreated GECs with antimycin A to impair mitochondrial function. Then both untreated and antimycin A-treated GECs were incubated with antibody and complement, and cytotoxicity was monitored by release of LDH. As expected, complement-mediated cytolysis increased with serially increasing doses of complement, and pretreatment of cells with antimycin A enhanced complement-mediated cytolysis at every dose (Fig. 11). For comparison, in analogous experiments, GECs were pretreated with tunicamycin, which induces cytotoxic ER stress (21). Tunicamycin did not, however, enhance complement cytotolysis (data not shown). These results indicate that mitochondrial dysfunction in cultured GECs enhances complement-mediated cytotoxicity and support the view that exacerbation of GEC injury in anti-GBM nephritis in the iPLA2γ KO mice was associated with underlying mitochondrial dysfunction.
FIGURE 11.
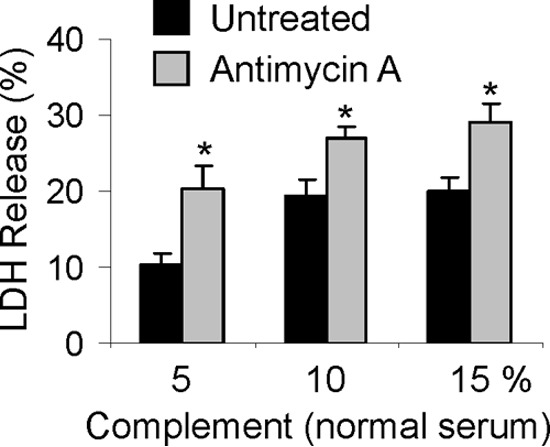
Mitochondrial injury enhances complement-mediated cytotoxicity. Cultured GECs were preincubated with or without antimycin A (10 μm, 30 min). Then untreated and antimycin A-treated GECs were incubated with anti-GEC antibody (40 min) and complement (5, 10, and 15% normal serum; heat-inactivated serum in controls) for 40 min. Cytolysis was monitored by release of LDH. *, p < 0.001 antimycin A versus untreated (four experiments). Antimycin A had no significant independent effect on cytolysis. Error bars, S.E.
Discussion
In this study, we demonstrated that deletion of iPLA2γ resulted in striking mitochondrial abnormalities in GECs/podocytes of 10–11-month-old mice (Fig. 2). These ultrastructural abnormalities were widespread and included disruption of membranes, loss of cristae, fission, and aggregation (Fig. 2). Aging iPLA2γ KO mice also demonstrated podocyte plasma membrane damage and slight but significant widening of foot processes compared with WT (Fig. 2). Expression of synaptopodin was lower in iPLA2γ KO glomeruli (Figs. 3 and 4), and there was a ∼25% decrease in the number of WT1-positive cells in iPLA2γ KO glomeruli compared with control (Fig. 3), implying that deletion of iPLA2γ induced a loss of podocytes. In association with mitochondrial damage, phosphorylation of AMPK was increased in iPLA2γ KO glomeruli (Fig. 4), consistent with reduced ATP generation by mitochondria in KO podocytes leading to podocyte injury. In the aging iPLA2γ KO mice, some podocytes contained prominent autophagic vacuoles (Fig. 2). There was also an increase in the glomerular ratio of LC3-II/LC3-I protein expression in KO mice, in keeping with accumulation of autophagic vacuoles (Fig. 4). Surprisingly, the ultrastructural abnormalities in aging podocytes were not associated with development of albuminuria (Fig. 1) or renal failure, although these could potentially appear at a more advanced age. Studies in cultured GECs derived from WT and iPLA2γ KO mice showed that deletion of iPLA2γ impaired mitochondrial function and enhanced autophagy (Figs. 5 and 6). Indeed, the clustering of mitochondria in the perinuclear regions of cells (as observed in iPLA2γ KO GECs) is reported to be associated with autophagy (36). Thus, the GEC culture studies recapitulate the phenotype in vivo and support the functional importance of the ultrastructural findings and low cell energy stores in iPLA2γ KO glomeruli.
In cells, iPLA2γ is localized at the mitochondria, ER, and peroxisomes (3). There are several variants of iPLA2γ, and subcellular localization may, at least in part, depend on the variant expressed in the specific cell type (3, 20, 37). Previously, we demonstrated that iPLA2γ mRNA and protein of 88 kDa are expressed in the glomerulus in vivo (19). GFP-tagged iPLA2γ was localized at the ER and mitochondria in cultured GECs, and this pattern of localization was dependent on the N-terminal domain of iPLA2γ (20). Our results in podocytes are in keeping with previous studies, where deletion of iPLA2γ in mice resulted in mitochondrial disruption. For example, deletion of iPLA2γ induced mitochondrial dysfunction in the heart, skeletal muscle, liver, and brain (7, 8, 10, 11). iPLA2γ KO mice displayed cold intolerance, a defect in mitochondrial cytochrome oxidase, and multiple bioenergetic dysfunctional phenotypes at 4–6 months of age (7). In skeletal muscle of 4-month-old KO mice, there was mitochondrial dysfunction, oxidative stress, and lipid peroxidation (11). These mice exhibited growth retardation. iPLA2γ deletion induced significant changes in mitochondrial phospholipid homeostasis (see below). In aging mice (10 months), this was associated with ultrastructural evidence for enlarged and degenerating mitochondria in the brain, autophagy, and cognitive dysfunction (8). These studies identified an obligatory role for iPLA2γ in mitochondrial structure.
Increased autophagy in podocytes of 10–11-month-old iPLA2γ KO mice (Fig. 2) may have been triggered by reduced ATP levels and activation of AMPK (Fig. 4). Actually, AMPK has been implicated as an important positive regulator of autophagy (30, 31). Autophagy was less likely to have been due to ER stress, since there was no evidence for significant ER dysfunction in iPLA2γ KO mice (Fig. 4). It has been suggested that in podocytes, reduced ubiquitin-proteasome function might result in increased autophagy (30, 38). We did not, however, observe any differences in polyubiquitination of glomerular proteins between iPLA2γ KO and WT mice (result not shown). Determination of whether cross-talk between these two systems plays a mechanistic role in injury will require further study. In iPLA2γ KO podocytes, autophagy may promote survival by improving cell energy levels. Enhanced autophagy in podocytes as well as in the brains of iPLA2γ KO mice may also be a cytoprotective mechanism to clear damaged mitochondria from the cytoplasm (30, 31, 38). Based on a study in Atg5 KO mice, autophagy appears to be a homeostatic mechanism to maintain podocyte integrity and a protective mechanism against aging and glomerular injury (30).
It should be noted that in contrast to podocytes, mitochondria in other kidney cell types, including parietal epithelial cells and proximal tubular epithelial cells in iPLA2γ KO mice, showed normal ultrastructure (Fig. 2). This finding in proximal tubular epithelial cells was unexpected because the function of these cells is highly dependent on mitochondrial energy production, and depletion of iPLA2γ in cultured proximal tubular epithelial cells in previous studies resulted in metabolic abnormalities (see below) (22). In contrast to podocytes, which appear to contain relatively few, small mitochondria, tubular cells in vivo contain numerous, large mitochondria (Fig. 2). Perhaps a more robust mitochondrial structure allowed the tubular cells to withstand potential damage from the deletion of iPLA2γ in vivo. Alternatively, there may be differences in iPLA2γ variants, expression, or activation among the renal cell types. Finally, certain cells may activate alternate mechanisms to compensate for iPLA2γ deficiency, or the podocyte, a cell with limited proliferative capacity (12, 14), may not be able to withstand mitochondrial damage, unlike proximal tubular cells, which have high potential for proliferation and repair from injury (39).
In regard to its role in lipid homeostasis, deletion of iPLA2γ resulted in a decrease in cardiolipin content and altered cardiolipin molecular species distribution in heart and skeletal muscle mitochondria, but there were no significant changes in phospholipids and triglycerides (7, 9, 11). In brain hippocampal lipid extracts, iPLA2γ KO mice showed an increase in various cardiolipin molecular species, increases in arachidonoyl-phosphatidylcholine and oxidized phosphatidylethanolamine, an increase in ceramide, and a decrease in plasmalogen-phosphatidylethanolamine (8). Decreased iPLA2γ expression in cultured renal proximal tubular cells mildly enhanced phosphatidylcholine and phosphatidylethanolamine levels (22). Cardiolipin is an important mitochondrial lipid, and proper remodeling of cardiolipin is essential for efficient electron transport chain function. Defects in cardiolipin have also been associated with aging. Thus, abnormal cardiolipin remodeling in iPLA2γ-deficient cells would be expected to result in defective electron coupling and bioenergetic inefficiency (8). It should be noted that although distinct directional changes in cardiolipin levels were observed in the heart and brain, mitochondrial function was impaired in both instances. Furthermore, disruption of iPLA2γ-mediated release of fatty acids may impair important signaling that is required for mitochondrial function (9).
The podocyte plays a key role in maintaining glomerular permselectivity. In normal podocytes, actin filaments are the core structural components of foot processes. Studies predominantly in cell culture models suggest that podocytes require a high energy supply to maintain various cellular functions, including the organization of cytoskeletal and GBM proteins (40). Condensation of the actin cytoskeleton at the base of effaced podocyte foot processes, together with alterations in filtration slits and displacement and disruption of slit diaphragms, are features of established proteinuric glomerular diseases (12–14, 17). As noted above, podocyte injury was observed in aging iPLA2γ KO mice (Fig. 2), but this was not associated with albuminuria (Fig. 1). Possibly, there was sufficient mitochondrial reserve, and the podocytes were able to maintain their permselective function. Despite the ∼25% loss of podocytes, the remaining podocytes may have been able to stretch and/or hypertrophy to compensate for the decreased number. Actually, the loss of podocytes may have been below the threshold required to produce extensive GBM denudation, proteinuria, and sclerotic lesions (16, 41, 42). In another mouse model of mitochondrial dysfunction, due to deletion of the Mpv17 gene, which is expressed in podocytes, albuminuria and mild glomerular sclerotic lesions developed only at 18 months of age (43).
Induction of podocyte injury with anti-GBM antibody revealed a protective phenotype of iPLA2γ in young mice. In heterologous anti-GBM nephritis, activation of complement and assembly of C5b-9 induces podocyte “stress” (24, 34, 35). iPLA2γ KO mice with anti-GBM nephritis had increased albuminuria and a remarkable reduction in podocytes compared with WT mice (Figs. 7 and 9; see below). Moreover, iPLA2γ KO mice showed substantially more severe abnormalities in podocyte ultrastructure, including swelling of cell bodies, marked villous transformation, and microvesiculation of plasma membranes, and almost complete foot process effacement (Fig. 8). Although structural abnormalities in podocyte organelles were not evident, studies in cultured GECs demonstrated that mitochondrial dysfunction can enhance complement-mediated cytotoxicity (Fig. 11), implying that podocyte injury in anti-GBM nephritis in iPLA2γ KO mice was most likely exacerbated due to underlying mitochondrial dysfunction. The association of podocyte mitochondrial dysfunction with proteinuria has been reported (40). By analogy to our study, after induction of anti-GBM nephritis in mice with mitochondrial dysfunction (Mpv17 gene deletion), the KO mice had greater levels of albuminuria compared with control, although the anti-GBM model employed in this study was more chronic (4 days) (43). We focused on anti-GBM nephritis at 24 h (the phase when mediation of the disease is more restricted to complement). In acute anti-GBM nephritis, ultrastructural changes in mitochondria may take a longer time to develop, or alternatively, the podocytes with injured mitochondria may have been those lost into the urine due to detachment from the GBM. Based on these results, it is reasonable to conclude that iPLA2γ is cytoprotective. This in vivo result is consistent with earlier studies, which showed that increased expression of iPLA2γ in cultured GECs reduced complement-mediated GEC injury (19).
As noted above, deletion of iPLA2γ resulted in a decrease in podocyte number in aging mice (Fig. 2) and a more marked decrease in podocytes in younger mice with anti-GBM nephritis (Fig. 9). There is both experimental and clinical evidence to support the view that podocyte apoptosis leads to glomerular disease and proteinuria (14, 16, 41, 42). The presence of iPLA2γ at the mitochondria may therefore provide an anti-apoptotic effect in the aging kidney and in anti-GBM nephritis. A second possibility is that loss of podocytes is due to podocyte detachment from the GBM and the subsequent appearance of viable cells in the urine (44, 45). iPLA2γ deletion potentially impairs the energy required for the production or function of proteins involved in mediating podocyte foot process adhesion to the GBM. Additional proof of these mechanisms will require further study.
Although we previously showed that the presence of iPLA2γ at the ER is functionally important in terms of ER signaling in cultured GECs (21), deletion of iPLA2γ in aging mice did not lead to any apparent abnormalities in ER morphology or function (Figs. 2–4). However, glomerular expression of the ER chaperones, BiP and GRP94, was increased in iPLA2γ KO mice with anti-GBM nephritis (Fig. 10), implying that deletion of iPLA2γ in the context of nephritis enhanced the adaptive unfolded protein response, particularly activation of the ATF6 pathway (38, 46, 47). This result is apparently discrepant from an earlier study, where it was demonstrated that overexpression of iPLA2γ enhanced ATF6 activation, whereas deletion reduced activation in cultured GECs (21). The reason for this discrepancy will require further elucidation; however, it is likely that multiple signaling pathways are activated by complement in the in vivo anti-GBM nephritis model (18), and a number of these could potentially impact the unfolded protein response. Activation of the adaptive unfolded response in anti-GBM nephritis is similar to experimental membranous nephropathy, a glomerular disease mediated by complement C5b-9 (17, 38, 46).
In conclusion, the results of this study demonstrate an important role for iPLA2γ in maintaining podocyte integrity both in the normal kidney and in glomerular disease. In the future, modulation of iPLA2γ enzymatic activity may potentially represent a novel approach to limiting GEC injury and proteinuria.
Author Contributions
H. E., A. V. C., and T. T. designed the study and wrote the paper. H. E., J. P., J. G., and L. A. conducted experiments. D. R. K. designed the experiments in Figs. 4 and 5. R. W. G. produced the knock-out mice. All authors analyzed the results and approved the manuscript.
This work was supported by Canadian Institutes of Health Research Grants MOP-53264, MOP-133492, MOP-125988, and MOP-53335; National Institutes of Health Grant RO1 HL118639; and the Catherine McLaughlin Hakim Chair (to A. V. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PLA2
- phospholipase A2
- iPLA2
- calcium-independent phospholipase A2
- AMPK
- AMP-activated protein kinase
- ER
- endoplasmic reticulum
- GBM
- glomerular basement membrane
- GEC
- glomerular epithelial cell
- LC3
- microtubule-associated protein light chain 3
- LDH
- lactate dehydrogenase
- WT1
- Wilms tumor-1.
References
- 1.Murakami M., and Kudo I. (2002) Phospholipase A2. J. Biochem. 131, 285–292 [DOI] [PubMed] [Google Scholar]
- 2.Six D. A., and Dennis E. A. (2000) The expanding superfamily of phospholipase A2 enzymes: classification and characterization. Biochim. Biophys. Acta 1488, 1–19 [DOI] [PubMed] [Google Scholar]
- 3.Ramanadham S., Ali T., Ashley J. W., Bone R. N., Hancock W. D., and Lei X. (2015) Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J. Lipid Res. 56, 1643–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schaloske R. H., and Dennis E. A. (2006) The phospholipase A2 superfamily and its group numbering system. Biochim. Biophys. Acta 1761, 1246–1259 [DOI] [PubMed] [Google Scholar]
- 5.Mancuso D. J., Jenkins C. M., and Gross R. W. (2000) The genomic organization, complete mRNA sequence, cloning, and expression of a novel human intracellular membrane-associated calcium-independent phospholipase A2. J. Biol. Chem. 275, 9937–9945 [DOI] [PubMed] [Google Scholar]
- 6.Tanaka H., Takeya R., and Sumimoto H. (2000) A novel intracellular membrane-bound calcium-independent phospholipase A2. Biochem. Biophys. Res. Commun. 272, 320–326 [DOI] [PubMed] [Google Scholar]
- 7.Mancuso D. J., Sims H. F., Han X., Jenkins C. M., Guan S. P., Yang K., Moon S. H., Pietka T., Abumrad N. A., Schlesinger P. H., and Gross R. W. (2007) Genetic ablation of calcium-independent phospholipase A2γ leads to alterations in mitochondrial lipid metabolism and function resulting in a deficient mitochondrial bioenergetic phenotype. J. Biol. Chem. 282, 34611–34622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mancuso D. J., Kotzbauer P., Wozniak D. F., Sims H. F., Jenkins C. M., Guan S., Han X., Yang K., Sun G., Malik I., Conyers S., Green K. G., Schmidt R. E., and Gross R. W. (2009) Genetic ablation of calcium-independent phospholipase A2γ leads to alterations in hippocampal cardiolipin content and molecular species distribution, mitochondrial degeneration, autophagy, and cognitive dysfunction. J. Biol. Chem. 284, 35632–35644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mancuso D. J., Sims H. F., Yang K., Kiebish M. A., Su X., Jenkins C. M., Guan S., Moon S. H., Pietka T., Nassir F., Schappe T., Moore K., Han X., Abumrad N. A., and Gross R. W. (2010) Genetic ablation of calcium-independent phospholipase A2γ prevents obesity and insulin resistance during high fat feeding by mitochondrial uncoupling and increased adipocyte fatty acid oxidation. J. Biol. Chem. 285, 36495–36510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moon S. H., Jenkins C. M., Kiebish M. A., Sims H. F., Mancuso D. J., and Gross R. W. (2012) Genetic ablation of calcium-independent phospholipase A2γ (iPLA2γ) attenuates calcium-induced opening of the mitochondrial permeability transition pore and resultant cytochrome c release. J. Biol. Chem. 287, 29837–29850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoda E., Hachisu K., Taketomi Y., Yoshida K., Nakamura M., Ikeda K., Taguchi R., Nakatani Y., Kuwata H., Murakami M., Kudo I., and Hara S. (2010) Mitochondrial dysfunction and reduced prostaglandin synthesis in skeletal muscle of Group VIB Ca2+-independent phospholipase A2γ-deficient mice. J. Lipid Res. 51, 3003–3015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pavenstädt H., Kriz W., and Kretzler M. (2003) Cell biology of the glomerular podocyte. Physiol. Rev. 83, 253–307 [DOI] [PubMed] [Google Scholar]
- 13.Chiang C. K., and Inagi R. (2010) Glomerular diseases: genetic causes and future therapeutics. Nat. Rev. Nephrol. 6, 539–554 [DOI] [PubMed] [Google Scholar]
- 14.Greka A., and Mundel P. (2012) Cell biology and pathology of podocytes. Annu Rev. Physiol. 74, 299–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Welsh G. I., and Saleem M. A. (2012) The podocyte cytoskeleton: key to a functioning glomerulus in health and disease. Nat. Rev. Nephrol. 8, 14–21 [DOI] [PubMed] [Google Scholar]
- 16.Pippin J. W., Brinkkoetter P. T., Cormack-Aboud F. C., Durvasula R. V., Hauser P. V., Kowalewska J., Krofft R. D., Logar C. M., Marshall C. B., Ohse T., and Shankland S. J. (2009) Inducible rodent models of acquired podocyte diseases. Am. J. Physiol. Renal Physiol. 296, F213–F229 [DOI] [PubMed] [Google Scholar]
- 17.Cybulsky A. V. (2011) Membranous nephropathy. Contrib. Nephrol. 169, 107–125 [DOI] [PubMed] [Google Scholar]
- 18.Takano T., Elimam H., and Cybulsky A. V. (2013) Complement-mediated cellular injury. Semin. Nephrol. 33, 586–601 [DOI] [PubMed] [Google Scholar]
- 19.Cohen D., Papillon J., Aoudjit L., Li H., Cybulsky A. V., and Takano T. (2008) Role of calcium-independent phospholipase A2 in complement-mediated glomerular epithelial cell injury. Am. J. Physiol. Renal Physiol. 294, F469–F479 [DOI] [PubMed] [Google Scholar]
- 20.Elimam H., Papillon J., Takano T., and Cybulsky A. V. (2013) Complement-mediated activation of calcium-independent phospholipase A2γ: role of protein kinases and phosphorylation. J. Biol. Chem. 288, 3871–3885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elimam H., Papillon J., Takano T., and Cybulsky A. V. (2015) Calcium-independent phospholipase A2γ enhances activation of the ATF6 transcription factor during endoplasmic reticulum stress. J. Biol. Chem. 290, 3009–3020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kinsey G. R., Blum J. L., Covington M. D., Cummings B. S., McHowat J., and Schnellmann R. G. (2008) Decreased iPLA2γ expression induces lipid peroxidation and cell death and sensitizes cells to oxidant-induced apoptosis. J. Lipid Res. 49, 1477–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.N'Diaye E. N., Kajihara K. K., Hsieh I., Morisaki H., Debnath J., and Brown E. J. (2009) PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep. 10, 173–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salant D. J., and Cybulsky A. V. (1988) Experimental glomerulonephritis. Methods Enzymol. 162, 421–461 [DOI] [PubMed] [Google Scholar]
- 25.Drozdova T., Papillon J., and Cybulsky A. V. (2013) Nephrin missense mutations: induction of endoplasmic reticulum stress and cell surface rescue by reduction in chaperone interactions. Physiol. Rep. 1, e00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nezvitsky L., Tremblay M. L., Takano T., Papillon J., and Cybulsky A. V. (2014) Complement-mediated glomerular injury is reduced by inhibition of protein-tyrosine phosphatase 1B. Am. J. Physiol. Renal Physiol. 307, F634–F647 [DOI] [PubMed] [Google Scholar]
- 27.van den Berg J. G., van den Bergh Weerman M. A., Assmann K. J., Weening J. J., and Florquin S. (2004) Podocyte foot process effacement is not correlated with the level of proteinuria in human glomerulopathies. Kidney Int. 66, 1901–1906 [DOI] [PubMed] [Google Scholar]
- 28.Bechtel W., Helmstädter M., Balica J., Hartleben B., Kiefer B., Hrnjic F., Schell C., Kretz O., Liu S., Geist F., Kerjaschki D., Walz G., and Huber T. B. (2013) Vps34 deficiency reveals the importance of endocytosis for podocyte homeostasis. J. Am. Soc. Nephrol. 24, 727–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kreidberg J. A. (2003) Podocyte differentiation and glomerulogenesis. J. Am. Soc. Nephrol. 14, 806–814 [DOI] [PubMed] [Google Scholar]
- 30.Huber T. B., Edelstein C. L., Hartleben B., Inoki K., Jiang M., Koya D., Kume S., Lieberthal W., Pallet N., Quiroga A., Ravichandran K., Susztak K., Yoshida S., and Dong Z. (2012) Emerging role of autophagy in kidney function, diseases and aging. Autophagy 8, 1009–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Periyasamy-Thandavan S., Jiang M., Schoenlein P., and Dong Z. (2009) Autophagy: molecular machinery, regulation, and implications for renal pathophysiology. Am. J. Physiol. Renal Physiol. 297, F244–F256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson L. V., Walsh M. L., Bockus B. J., and Chen L. B. (1981) Monitoring of relative mitochondrial membrane potential in living cells by fluorescence microscopy. J. Cell Biol. 88, 526–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mizushima N., Yoshimori T., and Levine B. (2010) Methods in mammalian autophagy research. Cell 140, 313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Couser W. G., Baker P. J., and Adler S. (1985) Complement and the direct mediation of immune glomerular injury: a new perspective. Kidney Int. 28, 879–890 [DOI] [PubMed] [Google Scholar]
- 35.Lin F., Emancipator S. N., Salant D. J., and Medof M. E. (2002) Decay-accelerating factor confers protection against complement-mediated podocyte injury in acute nephrotoxic nephritis. Lab. Invest. 82, 563–569 [DOI] [PubMed] [Google Scholar]
- 36.Gomes L. C., and Scorrano L. (2013) Mitochondrial morphology in mitophagy and macroautophagy. Biochim. Biophys. Acta 1833, 205–212 [DOI] [PubMed] [Google Scholar]
- 37.Mancuso D. J., Jenkins C. M., Sims H. F., Cohen J. M., Yang J., and Gross R. W. (2004) Complex transcriptional and translational regulation of iPLAγ resulting in multiple gene products containing dual competing sites for mitochondrial or peroxisomal localization. Eur. J. Biochem. 271, 4709–4724 [DOI] [PubMed] [Google Scholar]
- 38.Cybulsky A. V. (2013) The intersecting roles of endoplasmic reticulum stress, ubiquitin- proteasome system, and autophagy in the pathogenesis of proteinuric kidney disease. Kidney Int. 84, 25–33 [DOI] [PubMed] [Google Scholar]
- 39.Bonventre J. V., and Yang L. (2011) Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Invest. 121, 4210–4221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muller-Deile J., and Schiffer M. (2014) The podocyte power-plant disaster and its contribution to glomerulopathy. Front. Endocrinol. 5, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.D'Agati V. D. (2008) Podocyte injury in focal segmental glomerulosclerosis: Lessons from animal models (a play in five acts). Kidney Int. 73, 399–406 [DOI] [PubMed] [Google Scholar]
- 42.Wiggins R. C. (2007) The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int. 71, 1205–1214 [DOI] [PubMed] [Google Scholar]
- 43.Casalena G., Krick S., Daehn I., Yu L., Ju W., Shi S., Tsai S. Y., D'Agati V., Lindenmeyer M., Cohen C. D., Schlondorff D., and Bottinger E. P. (2014) Mpv17 in mitochondria protects podocytes against mitochondrial dysfunction and apoptosis in vivo and in vitro. Am. J. Physiol. Renal Physiol. 306, F1372–F1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petermann A. T., Krofft R., Blonski M., Hiromura K., Vaughn M., Pichler R., Griffin S., Wada T., Pippin J., Durvasula R., and Shankland S. J. (2003) Podocytes that detach in experimental membranous nephropathy are viable. Kidney Int. 64, 1222–1231 [DOI] [PubMed] [Google Scholar]
- 45.Petermann A. T., Pippin J., Krofft R., Blonski M., Griffin S., Durvasula R., and Shankland S. J. (2004) Viable podocytes detach in experimental diabetic nephropathy: potential mechanism underlying glomerulosclerosis. Nephron Exp. Nephrol. 98, e114–e123 [DOI] [PubMed] [Google Scholar]
- 46.Cybulsky A. V. (2010) Endoplasmic reticulum stress in proteinuric kidney disease. Kidney Int. 77, 187–193 [DOI] [PubMed] [Google Scholar]
- 47.Inagi R., Ishimoto Y., and Nangaku M. (2014) Proteostasis in endoplasmic reticulum: new mechanisms in kidney disease. Nat. Rev. Nephrol. 10, 369–378 [DOI] [PubMed] [Google Scholar]