

Abstract
Genomic DNA replication requires helicases to processively unwind duplexes. Although helicases encoded by positive-strand RNA viruses are necessary for RNA genome replication, their functions are not well understood. We determined structures of the hepatitis C virus helicase (NS3h) in complex with the transition state ATP mimic ADP·AlF4− and compared them with the previous nucleic acid-associated ternary complexes. The results suggested that nucleic acid binding promotes a structural change of the spring helix at the transition state, optimizing the interaction network centered on the nucleophilic water. Analysis of ATP hydrolysis with and without conformational restraints on the spring helix further supported the importance of its action for both nucleic acid-stimulated and basal catalysis. We further found that an F238P substitution, predicted to destabilize the helix, diminished viral RNA replication without significantly affecting ATP-dependent duplex unwinding. The stability of the secondary structure, thus, seems critical for additional functions of NS3h. Taken together, the results suggest that the spring helix may be central to the coordination of multiple modes of NS3h action. Further characterization centered on this element may help understand the molecular details of how the viral helicase facilitates RNA replication. This new structural information may also aid efforts to develop specific inhibitors targeting this essential viral enzyme.
Keywords: ATPase, hepatitis C virus (HCV), RNA helicase, RNA virus, structure-function, viral replication
Introduction
Helicases are vital enzymes that perform highly diverse functions in important biological processes (1, 2). They are ubiquitous in cellular organisms and are also found in many viral proteomes (1). Those encoded by positive-strand RNA viruses, many of which are medically important pathogens, are believed to function at different stages of virus life cycles (3). Studies of several viral helicases have indeed implicated a convergent theme regarding their potential roles in the action of replicase complexes for genomic RNA replication, such as initial recruitment of replication templates, and synthesis of negative-strand RNA intermediates and positive-strand RNA genomes (4–8). Although the enzymatic activities of the viral helicases are somewhat similar to those of replicative DNA helicases, which are well known to processively unwind DNA duplexes for genome replication by utilizing ATP, the details of how the viral enzymes participate in the assembly and function of RNA replicase complexes remain mysterious. This has hampered our understanding of the mechanisms of positive-strand genomic RNA replication in general as well as the development of specific inhibitors targeting this class of important viral enzymes.
Positive-strand RNA viruses in the Flaviviridae family, such as HCV3 and dengue virus, are responsible for widespread global infection with significant public health and economic impact (9). HCV chronically infects ∼3% of the world population, causing liver disease including cirrhosis and liver cancer (10). The virus encodes a superfamily (SF) 2 viral helicase in the C-terminal portion of the NS3 protein (11). Its enzymatic activity is important for the virus life cycle, as mutations engineered in motif II, the central element for metal ion binding and (d)NTP hydrolysis, prevented detectable virus replication in vivo (12). In a cell culture model, mutagenesis targeting the helicase nucleic acid binding site reduced HCV RNA replication (13). In addition, the importance of NS3h in the HCV lifecycle has also been demonstrated by mutagenesis of residues with no known roles in enzymatic activity. For example, alteration of residues involved in forming contacts in crystals reduced viral RNA replication (14), and adaptive mutations in NS3h greatly enhance replication of different HCV isolates in cultured cells (15–18).
In vitro, HCV NS3h can unwind both DNA and RNA duplexes, and the mechanism of its motion has been broadly characterized. Nucleotide triphosphates, (d)NTPs, bind between the two NS3h RecA-like domains to trigger closure of the two domains (19, 20). Hydrolysis of one (d)NTP by NS3h drives one-base translocation in the 3′-5′ direction, allowing NS3h to separate nucleic acid duplexes along its translocation path (19–22). Similar stepwise dynamics and analogous conformational changes have been described for cellular helicases and likely occur for most SF1 and SF2 helicases (2). Nucleic acid binding stimulates HCV NS3h (d)NTP hydrolysis, a general feature of most helicases (2, 23). Different mechanisms for this observation have been proposed by comparing nucleic acid-bound and unbound helicases (24–26). However, no study until the present one has compared transition-state mimic-captured enzymes in the presence and absence of nucleic acids. Such a comparison could provide new insight into a critical step along the reaction pathway and help clarify the mechanistic details of nucleic acid-stimulated catalysis.
HCV NS3h contains a spring α-helix in RecA-like domain 1. This structural element is predicted in helicases encoded by HCV-like non-primate hepaciviruses as well as the more distantly related pegiviruses and pestiviruses, suggesting conservation of an important replicative function (20, 27, 28). We have found that the HCV NS3h spring helix has to be structurally flexible for catalysis, and meanwhile the α-helical form of this element is important for virus replication. We think the spring helix probably plays a central role in coordinating multiple modes of NS3 action. Our results justify further investigation on NS3h to understand the molecular details of viral RNA replication and aid discovery of potent and selective viral helicase inhibitors that has thus far proven challenging.
Results
Nucleic Acid Binding Promotes Elongation of the Spring Helix and an Associated Overall Conformational Change
DNA- or RNA-stimulated nucleotide hydrolysis is a general feature of most active helicases (2). In order to interrogate the molecular basis of HCV NS3h-catalyzed nucleic acid-dependent (d)NTP hydrolysis, we performed the first structural comparison of transition-state mimic-captured NS3h complexes in the presence and absence of nucleic acids. We crystallized binary complexes containing NS3h and transition-state ATP mimics (ADP·AlF4−), allowing comparison to the previously determined ternary complex structures that included NS3h and ATP mimics as well as nucleic acids (20). Two complex structures were determined at 2.1 Å resolution from a single crystallographic asymmetric unit (Table 1). The two binary NS3h-ADP·AlF4− complexes are similar to each other despite small differences in conformation (root mean square deviation ∼0.84 Å for all the NS3h atoms). Complex A is better ordered than complex B in the crystal, as its electron density (simulated annealing composite omit map) exhibits more pronounced structural features; different parts of the structure, especially those in domain 1, are better ordered with lower temperature factors (Fig. 1). We analyzed both binary complexes to reveal the structural impact of the lack of nucleic acids.
TABLE 1.
Crystallographic data and refinement statistics
Numbers in parentheses indicate the range of the highest resolution bin and statistics for data in this resolution bin. NSLS, Brookhaven National Synchrotron Light Source.
| Complex | NS3h-ADP·AlF4− |
|---|---|
| PDB ID | 5E4F |
| Data collection | |
| Source | NSLS X26C |
| Wavelength (Å) | 1.0809 |
| Resolution (Å) | 50-2.1 (2.18-2.1) |
| Space group | P21 |
| Unit cell | |
| a, b, c (Å) | 95.5, 46.1, 109.1 |
| α, β, γ (°) | 90, 104, 90 |
| Observations | 262,634 (16582) |
| Unique reflections | 53,280 (5056) |
| Redundancy | 4.9 (3.1) |
| Completeness (%) | 98.1 (94.9) |
| Mean I/σI | 17.3 (5.6) |
| Rmerge on Ia | 0.05 (0.23) |
| Cut-off criteria I/σI | −0.5 |
| Refinement statistics | |
| Resolution limits (Å) | 50-2.1 (2.23-2.1) |
| Number of reflections | 51,003 (7,087) |
| Completeness (%) | 93.7 (83.5) |
| Cut-off criteria I/σI | 0 |
| Number of atoms | |
| Protein | 6,505 |
| Ligand | 66 |
| Solvent | 498 |
| Rcrystb | 0.214 (0.254) |
| Rfree (5% of data) | 0.252 (0.306) |
| Bonds (Å)/angles (°)c | 0.006/1.2 |
| Mean B value (Å2) | 25.6 |
| Complex A | 19.5 |
| Complex B | 31.0 |
| Coordinate error (Å)d | 0.26 |
a Rmerge = ΣhklΣi|I(hkl)i − 〈I(hkl)〉|/ΣhklΣi〈I(hkl)i〉.
b Rcryst = Σhkl|Fo(hkl) − Fc(hkl)|/Σhkl|Fo(hkl)|, where Fo and Fc are the observed and calculated structure factors, respectively.
c Values indicate root mean square deviations in bond lengths and bond angles.
d Estimated coordinate error from cross-validated Luzzati Plot.
FIGURE 1.
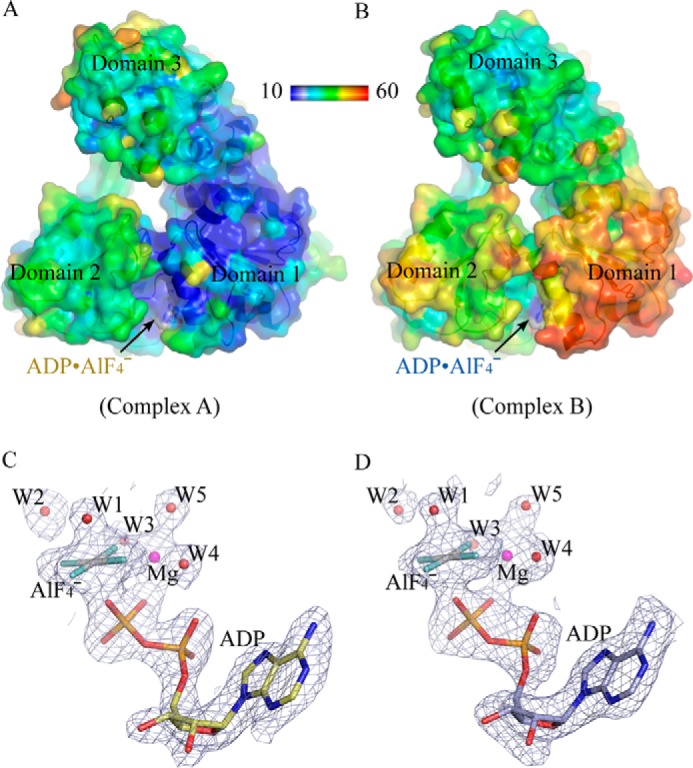
Structures of the NS3h-ADP·AlF4− binary complexes. A and B, schematic and surface presentation of the two NS3h-ADP·AlF4− binary complexes. The residues are color-coded by their B-factor values. Lower B factors indicate lower vibration. The minimum and maximum values were set to be 10 (blue) and 60 Å2 (red), respectively, in the reference bar. C and D, the simulated-annealing composite omit electron density contoured at 1.0 σ around the ATP mimics. The ATP mimics are modeled by sticks. Water molecules (W) and magnesium (Mg) ions are shown as spheres. The carbon atoms of complex A are colored in yellow, whereas those of complex B are blue. The non-carbon atoms are color-coded according to the elements defined in PyMOL. Complexes A and B are noted in parentheses under each panel. The structure presentations were prepared in PyMOL.
Structural alignment of complex A and the NS3h-ADP·BeF3-ssDNA ground-state complex revealed that the NS3h structures are largely superimposable, except that the Glu-291 residue in motif II adopts different rotamers in the two structures (Figs. 2, A and C, and 3, A–C). Comparison of complex A and the NS3h-ADP·AlF4−-ssDNA transition-state structure revealed that domains 1 and 3 are arranged in different conformations (Fig. 2B). The structural change of the spring helix and shift of the motif II loop can only be crystallographically captured in the ternary complex with ssDNA (Fig. 2, C and D). This suggests that ssDNA binding facilitates adoption of an appropriate structural arrangement of NS3h as ATP hydrolysis proceeds to the transition state. Further structural analysis of the motif II loop revealed nucleic acid-dependent rotamer changes of the essential residues Asp-290, Glu-291, and His-293 (Figs. 2C and 3, A–C). This is presumably important for remodeling the (d)NTPase active site for catalysis.
FIGURE 2.
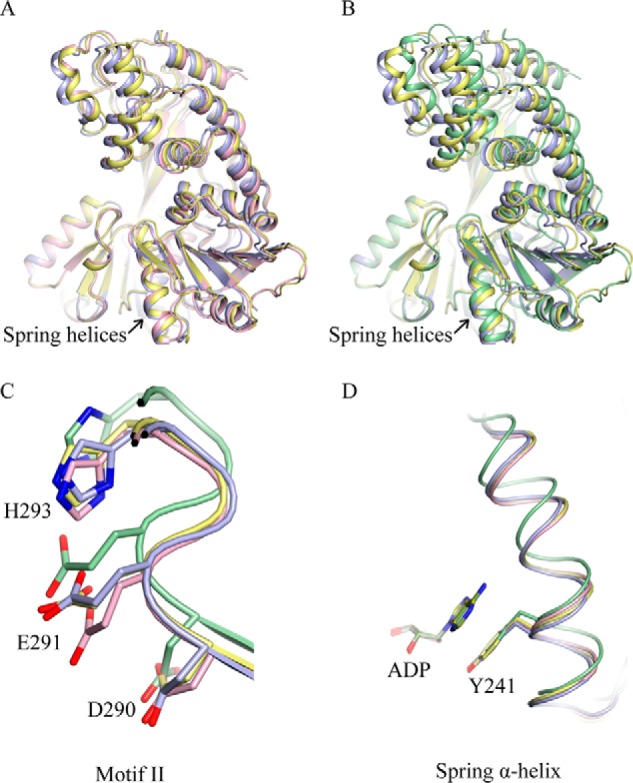
Comparison of the binary and ternary complexes. A, NS3h-ADP·AlF4− complex A (yellow) and B (light blue) aligned with NS3h-ADP·BeF3-ssDNA (pink, PDB ID 3KQU) through domain 2 (residues 329–432 and 450–481). The NS3h proteins are presented by thin ribbons (α-helices) and arrowed strands (β-strands). The spring helices are noted by arrows. B, complex A and B aligned with NS3h-ADP·AlF4−-ssDNA (green, PDB ID 3KQL) through domain 2. The structures are presented as those in A. C, close-up view of the motif II elements. The residues in motif II (Asp-290, Glu-291, and His-293) are presented as sticks. Protein main chains are shown as tubes. D, close-up view of the spring α-helices (tubes). The Tyr-241 residues and superimposed ADP molecules are presented by sticks. The change of the spring helix in this presentation does not represent the elongation distance. The two binary structures and two ternary structures were aligned through ADP in C and D. The amino acid numbering is according to the NS3 sequence.
FIGURE 3.

Active-site arrangement with and without ssDNA. A, active-site arrangement of the NS3h-ADP·AlF4− complexes and NS3h-ADP·AlF4−-ssDNA ternary complex. The active-site residues and ATP mimics are modeled by sticks. The water molecules (W) and magnesium ions (Mg) are modeled by spheres. The non-carbon atoms are color-coded according to elements defined in PyMOL. The carbon atoms are colored yellow (complex A), light blue (complex B), and green (ternary complex). The yellow dashed lines represent probable atomic interactions. The three angles centered on the carbonyl oxygen are schematically highlighted by black lines, with sizes of the angles noted. The Glu-291 carbonyl groups involved in water coordination are highlighted by spheres. B, comparison of the carboxylate groups of Glu-291 in the presence and absence of ssDNA. The simulated annealing omit composite maps were contoured at 1.5 σ. C, coordination of the nucleophilic waters (W1). The structures in B and C are presented as those in A. D, binding of fluorophore-labeled GDP to NS3h. NS3h was used to titrate BODIPY-labeled GDP in the presence of NaF, AlCl3, and MnCl2. To analyze the impact of nucleic acid enhanced transition-state mimic binding, ssDNA (dT12) was added. Fluorescence polarization values derived from single measurements were plotted against NS3h concentrations.
Nucleic Acid Binding Facilitates Optimization of a Hydrogen-bond Network
We analyzed the arrangement of the (d)NTPase active site with and without ssDNA (Fig. 3, A–C). The coordination of the ADP·AlF4− moiety, magnesium ion, and most water molecules (W3, W4, and W5) does not significantly change upon ssDNA binding. The major differences are centered on the Glu-291 residue of motif II and the nucleophilic water (W1). In the presence of ssDNA, the Glu-291 residue is oriented to interact with His-293 (Nϵ) (Fig. 3, A and C). In this orientation, the two water molecules W1 and W5 approximately lie in the plane of the carboxylate group of Glu-291 (Fig. 3B). They are positioned in symmetry with respect to the Glu-291 carbonyl bond and, therefore, are able to form linear hydrogen bonds simultaneously with the carbonyl oxygen (Glu-291 Oϵ) (Fig. 3, A and B). In contrast, in the absence of ssDNA, Glu-291 is not properly positioned to establish the hydrogen bonding network.
We then analyzed the atomic coordination on the W1 water molecule, which presumably acts as a nucleophilic water involved in pentacoordination of the transition-state planar γ-phosphate. In binary complex A without ssDNA, the distances (2.63 and 2.92 Å) between W1 and the two coordinating oxygen atoms (Oϵ of Glu-291 and Gln-460) are longer and ∼0.3 Å different, whereas those in the ternary transition-state complex are shorter and approximately equal (2.57 and 2.59 Å) (Fig. 3C). This suggests that in the nucleic acid-containing ternary complex, the nucleophilic water is coordinated by symmetric and stronger hydrogen bonds, which can appropriately orient the activated water toward the γ-phosphorus. The structural analysis predicted that transition-state mimics should bind to NS3h better in the presence of ssDNA. We, therefore, probed the binding of GDP (BODIPY FL-labeled) and AlF4− to NS3h by measuring fluorescence polarization, which increases when small fluorophores bind to macromolecules. In the presence of ssDNA, fluorescence polarization increased, and the estimated dissociation constant for GDP decreased >2-fold (∼94.6 μm to ∼39.1 μm), indicating enhancement of GDP·AlF4− formation in the (d)NTPase active site (Fig. 3D).
Probable Structural Transition in the Absence of Nucleic Acids
Like complex A described above, complex B does not exhibit elongation in the spring helix, and its overall conformation is different from the transition state ternary complex (Fig. 2B). However, complex B is also slightly different from the ground state ternary complex (Fig. 2A). Although the central Glu-291 residue of complex B is not properly oriented to interact with His-293 and does not simultaneously form linear hydrogen bonds with W1 and W5, it is arranged in a better orientation than that of complex A (Fig. 3, A–C). In addition, the distances between W1 and two coordinating oxygen atoms approximate those in the transition state ternary complex and only exhibit a 0.13 Å difference, smaller than the difference seen in complex A (Fig. 3C). It is worth noting that complex B shows more thermal mobility, especially in domain 1 (Fig. 1, A and B). We speculate that complex B represents a snapshot of NS3h approximating to the transition state with optimal structural arrangements. In the absence of nucleic acids, this probably occurs at low frequency.
The Action of the Spring α-Helix Is Essential for Nucleic Acid-stimulated Nucleotide Hydrolysis
To examine the importance of the structural transition of the spring helix, we conformationally restrained this structural element and analyzed the impact on ssDNA-dependent ATP hydrolysis. We introduced cysteine-pair mutations within the spring helix (CC1 and CC2 mutants) as well as an irrelevant helix (CCi) and cross-linked the paired cysteines using 1,4 bismaleimidyl-2,3-dihydroxybutane (BMDB), a compound with two maleimides that can react with sulfhydryl groups of cysteine residues (Fig. 4, A and B). No native cysteine pairs seem to allow efficient cross-linking by BMDB. Although adding BMDB to the wild-type or CCi mutant protein did not change ssDNA-dependent ATP hydrolysis, cross-linking the cysteine pairs in the CC1 and CC2 mutants significantly suppressed the activity (Fig. 4C, Table 2). The suppression was not observed when the individual cysteines in the CC1 and CC2 mutants were targeted by N-ethylmaleimide (NEM), which contains a single maleimide group and thus did not cross-link cysteine pairs (Fig. 4, B and D, Table 2). The results suggest that covalent attachment of small molecules on the spring helix or other structural elements containing exposed cysteines does not significantly affect nucleic acid-coupled ATP hydrolysis, whereas the cross-linking-mediated conformational restraint on the spring helix strongly suppresses enzymatic activity. It is worth noting that the CC1 cross-linking covered the two helical turns responsible for elongation, whereas CC2 seemed to cover only one (Figs. 2D and 4A). This may explain the stronger suppression associated with the CC1 cross-linking (Fig. 4C, Table 2), although different cross-linking efficiencies cannot be ruled out.
FIGURE 4.

The spring α-helix is important for enzymatic activity. A, design of cysteine pairs. NS3h in complex with ssDNA (dA6) (PDB ID 3KQH) is presented in a schematic. The cysteine mutations are highlighted in yellow. The cysteine pairs designed for cross-linking are connected by black lines. CC1 and CC2 represent cysteine pairs designed within the spring α-helix. CCi represents a pair of cysteine mutations in an irrelevant α-helix. B, chemical structure of BMDB and NEM. The two BMDB maleimide groups that can react with sulfhydryls are separated by ∼10.2 Å. The structures were prepared in Medchem Designer. C, ssDNA dT12-stimulated ATP hydrolysis in the presence (red) and absence (blue) of BMDB. Averaged initial velocities (V, pmol of phosphate released/min/pmol of enzyme) from three independent assays were plotted against increasing concentrations of ssDNA dT12. Error bars are S.D. D, ssDNA dT12-stimulated ATP hydrolysis in the presence (red) and absence (blue) of NEM. The data are plotted as those in B. E, ATP hydrolysis in the absence of ssDNA. The velocities (V, pmol of phosphate released/min/pmol of enzyme) at increasing concentrations of ATP were averaged from three independent reactions and plotted against the ATP concentrations. The error bars represent S.D.
TABLE 2.
Kinetics of ATP hydrolysis
| Enzymes | Nucleic acid-stimulated ATPase |
Basal ATPase |
||
|---|---|---|---|---|
| kcata | KNAb | kcata | KATPc | |
| WT | 822.4 ± 51.1 | 47.4 ± 6.8 | 32.29 ± 4.60 | 0.14 ± 0.06 |
| WT+BMDB | 882.4 ± 99.3 | 65.2 ± 14.7 | 33.01 ± 5.35 | 0.11 ± 0.05 |
| CCi | 776.2 ± 51.9 | 55.1 ± 8.0 | 31.82 ± 4.71 | 0.13 ± 0.06 |
| CCi+BMDB | 794.1 ± 42.9 | 43.2 ± 5.6 | 32.99 ± 6.69 | 0.10 ± 0.05 |
| CC1 | 683.7 ± 41.2 | 40.0 ± 6.0 | 32.11 ± 4.06 | 0.12 ± 0.05 |
| CC1+BMDB | 125.5 ± 17.1 | 73.9 ± 19.2 | 8.79 ± 0.41 | 0.80 ± 0.15 |
| CC2 | 697.7 ± 69.6 | 42.9 ± 10.4 | 32.57 ± 6.47 | 0.11 ± 0.06 |
| CC2+BMDB | 268.5 ± 23.6 | 35.9 ± 8.3 | 13.73 ± 1.99 | 0.13 ± 0.06 |
| CC1 | 615.0 ± 49.3 | 33.7 ± 7.3 | ||
| CC1+NEM | 556.7 ± 29.6 | 29.6 ± 4.5 | ||
| CC2 | 627.3 ± 26.8 | 28.7 ± 3.5 | ||
| CC2+NEM | 678.3 ± 41.6 | 44.8 ± 6.5 | ||
a Values are maximum turnover rates (pmol ATP hydrolyzed/min/pmol of protein).
b Values are concentrations of dT12 (μm) at 50% of the maximum velocities.
c Values are concentrations of ATP (mm) at 50% of the maximum velocities.
The Action of the Spring Helix Is Also Required for Basal (d)NTPase Activity
As the transition state ATP mimic can bind in the (d)NTPase active site even in the absence of ssDNA, we wondered whether the suboptimal active site arrangement is sufficient for (d)NTP hydrolysis to proceed across the transition state barrier and also how the nucleic acid-independent “basal” (d)NTP hydrolysis occurs. We, therefore, analyzed ATP hydrolysis in the absence of ssDNA. When the cross-linking reagent BMDB was used to conformationally restrain the spring helix in the CC1 and CC2 mutants, the velocities of ATP hydrolysis dramatically decreased. In contrast, the addition of BMDB to the wild-type protein and CCi mutant did not change ATP hydrolysis (Fig. 4E). The Km value for the CC1 cross-linking was a little higher, allowing us to speculate a possible change of ATP association in the active site leading to the stronger suppression of the basal activity (Table 2). However, we think that data-fitting might deviate because of the weak data points derived from the extremely slow reactions at low concentrations of ATP. Overall, the results indicate that the basal ATPase activity is also dependent on the flexibility of the spring α-helix and the associated overall conformational change; it is likely, from inefficient autonomous structural transitions of NS3h in the absence of ssDNA. We think the complex B structure probably highlights an initial step of the probable basal transition. The results also suggest that the same structural change of the spring helix should occur when NS3h translocates along ssRNA, although a transition state structure with ssRNA has not yet been determined (details under “Discussion”).
The α-Helical Structure of the Spring Helix Is Critical for Virus Replication
As the spring helix is a critical structural element for enzymatic activity, we tried to further characterize its role in NS3h function. We selected Phe-238 in the spring helix for mutagenesis analysis and assessment of virus replication capacity. This residue is located away from the (d)NTPase and nucleic acid binding sites. It is also partially embedded within the protein and should not be directly involved in potential surface contacts between NS3h and other factors (Fig. 5A).
FIGURE 5.

The α-helical structure of the spring helix is important for virus replication. A, schematic presentation of the spring helices in the ground-state (pink) and transition-state ternary complexes (green). The structures were aligned through amino acids 240–245. Phe-238 targeted by mutagenesis and Tyr-241 are modeled by sticks. B, replication of reporter HCV viruses in Huh-7.5 cells. The wild-type (wt) virus is Jc1(p7-nsGluc2A). The GNN strain is replication incompetent due to asparagine substitution of two catalytic aspartate residues in motif VI of NS5B (the RNA dependent RNA polymerase). F238A, F238L, and F238P were introduced in the NS3 helicase domain. Virus replication was measured by accumulation of secreted luciferase activities. Every data point was averaged from three independent assays. The error bars represent S.D. C, replication of the same set of reporter HCV viruses in Huh-7.5 CD81 knockdown cells. The data are plotted as those in B. D, duplex unwinding activities of the wild-type and mutant NS3 helicase proteins. The unwinding reactions were monitored by fluorescence of a molecular beacon annealed in a duplex. Separation of the duplexes led to self-annealing of the molecular beacons and, therefore, a decrease of fluorescence. Fluorescence intensities were measured every 2 min at 37 °C. The presented data sets, which were averaged form triplicate assays, cover 15 min without ATP and 60 min with ATP. The error bars are S.D. The negative control used bovine serum albumin (BSA).
We mutated Phe-238 to an alanine, leucine, and proline in Jc1(p7-nsGluc2A), a replication competent reporter virus (29) that expresses a secreted form of luciferase. After transfection of RNA transcripts into Huh-7.5 cells, virus replication was monitored by measuring luciferase activity in the culture medium. The Phe-238 side chain is not essential, as mutation of Phe-238 to alanine (F238A) did not affect virus replication, and the F238L mutation only slightly diminished replication and can be explained by possible steric effects between the leucine side chain and the adjacent structural elements (Fig. 5B). However, when Phe-238 was substituted by a proline, which should disrupt the main chain atomic interactions in the middle of the helix and destabilize the secondary structure, virus replication was largely diminished as early as 24 h, and weak signals were detectable only 2–3 days post transfection (Fig. 5B). A similar pattern was observed after the viral RNAs were introduced into Huh-7.5 CD81low cells, which do not support virus spread and allow quantification of RNA replication without complications from effects on virion production and subsequent entry into new cells (Fig. 5C). Thus, the F238P mutation strongly affected the function of the helicase in the early stages of viral RNA replication.
We wondered whether the block of virus replication was associated with loss of the duplex unwinding activity of NS3h. We, therefore, generated recombinant NS3h proteins harboring the Phe-238 single mutations and tested their activities with a molecular beacon fluorescence assay, in which separation of duplexes would lead to a decrease of fluorescence signals as previously described in similar assays (30). Both the F238A and F238L mutants unwound duplexes slightly faster than the wild-type protein. The F238P protein also seemed to efficiently unwind duplexes in the beginning, although the reaction resulted in a slightly higher basal signal (Fig. 5D). One possibility is that the aberrantly flexible spring helix might allow the F238P protein to convert some reaction products (blunt end hairpins) back to fluorescent duplexes, slightly shifting the equilibrium of the reaction. It is conceivable that mutations at Phe-238 did not restrain the spring helix or compromise any other structural elements involved in catalysis. We speculate that the mutations might even render more flexibility in the spring helix, making it more amenable to structural transitions required for activity. Mutations that potentially enhanced the helicase unwinding activity, such as F238A and F238L, did not impact virus replication. The significant loss of virus replication seen for the F238P mutant, therefore, seemed unlikely to be due to the minor changes in the unwinding activity but was probably caused by the disruption or destabilization of the α-helical structure of the spring helix, which may be critical for multiple functions of NS3h in the early stages of replicase complex assembly and function.
Discussion
We have shown that the elongation action of the spring helix optimizes the coordination of the nucleophilic water at the transition state. Nucleic acid binding can facilitate the conformational change and, therefore, stimulate (d)NTP hydrolysis. We have also found that destabilization of the helix strongly diminished virus replication without significant impact on ATP-dependent duplex unwinding, suggesting the importance of the α-helical structure.
We speculate that the spring helix may remain conformationally stable and temporarily suppress the unwinding activity of NS3h, allowing it to participate in the formation of putative complexes required for viral RNA replication. The subsequent (d)NTP hydrolysis triggers conformational changes, allowing NS3h to modulate its dynamic associations with other complex components and perform enzymatic activity-dependent functions, such as remodeling ribonucleoprotein complexes by translocating along single-stranded nucleic acids. Thus, the spring helix may be a central element for switching between different modes of action of NS3h. Previous studies suggest that several viral and cellular factors may interact with the HCV helicase and regulate its unwinding activity in vivo (31–33). It is possible that the action of the spring helix may impact these putative regulatory networks, altering the functions of NS3h. Further efforts to characterize the NS3h spring helix in multiprotein complexes will help illuminate its role in viral RNA genome replication.
To our knowledge this is the first comparison of helicase complexes captured by transition state mimics in the presence and absence of nucleic acids, which demonstrates the importance of the spring helix for nucleic acid-dependent catalysis of (d)NTP hydrolysis. The helicases encoded by pegiviruses and pestiviruses as well as the newly discovered HCV-like non-primate hepaciviruses likely contain spring helices (20, 27, 28). These viruses and HCV may share many common principles in helicase function and viral RNA replication. Recently, crystal structures of NS3h encoded by classical swine fever virus from the Pestivirus genus revealed a putative spring α-helix flanked by motifs Ia and Ib (28). The flavivirus (genus Flavivirus) helicases do not have spring helices, although they are similar to HCV NS3h in overall structure and are also covalently linked to viral serine proteases responsible for polyprotein processing and membrane association (9, 20). Structural analysis of the dengue virus helicase has shown that ATP binding does not significantly change the conformation of this enzyme. RNA binding seems to promote partial closure of two motor domains and rearrangement of active site elements, especially the P-loop, and thus facilitate catalysis (25). Compared with HCV NS3h, the flavivirus enzymes exhibit at least one obvious functional difference as they can act as triphosphatases involved in viral RNA 5′-cap formation (9). Several structurally well characterized cellular helicases also lack spring helices. The bacterial SF1 PcrA helicase, which is essential for plasmid rolling-circle replication and repair of DNA damage, has been shown to make a conformational change to link DNA association to catalytic ion binding and repositioning of the γ-phosphate group (26). The archaeal SF2 SWI2/SNF2 ATPase core, which is the catalytic center of large complexes involved in chromatin remodeling, can switch the DEXX motif into the active conformation upon DNA binding, so that this motif can participate in polarizing the nucleophilic water during ATP hydrolysis (24). Although it is hard to predict whether other helicases with phylogenetically distant sequences have spring helices or not, further structural and functional characterizations of those enzymes may allow us to tell whether analogous structural elements exist in any other helicase clan.
The HCV helicase exhibits more potent unwinding activity on DNA duplexes than RNA (33). Our previous study showed that it is the flexible sugar backbone that allows ssDNA to associate well with the helicase so that translocation can be coupled to the catalysis of (d)NTP hydrolysis (20). Comparison of DNA- and RNA-bound complexes has shown similar overall conformational changes from the nucleotide-free state to the ADP·BeF3-bound ground state (19, 20). There is, however, no transition state structure with associated RNA for comparison, and thus it is unclear whether a similar conformational change would occur when the reaction proceeds into the transition state. Nevertheless, our analysis of the basal (d)NTPase activity of HCV NS3h supports an indispensable role for the action of the spring helix in completion of catalysis, and therefore, the transition state conformational change required for DNA translocation likely also occurs when the helicase translocates along ssRNA. In fact, other catalytically active helicases encoded by positive-strand RNA viruses also seem to be non-discriminating between DNA and RNA substrates (3). We predict that there is no significant difference between the translocation events along DNA and RNA for all those helicases, although further studies may be required to fully address this.
Like proteases and polymerases, viral helicases are attractive drug targets. Specific helicase inhibitors would potentially be potent antiviral agents and help probe the details of helicase functions. The success in identifying compounds targeting a helicase-primase protein encoded by herpes simplex virus has stimulated strong interest in searching for inhibitors targeting helicases encoded by other pathogenic viruses (34–36). Drug screens indeed have found several promising HCV NS3h inhibitors, although the potency and specificity remain relatively low, and more evidence is required to define NS3h as the true target in cells (37). Our study suggests that small molecules that can interfere with the actions of critical structural elements or block surfaces required for complex assembly may be effective inhibitors even if they do not significantly inhibit the enzymatic activity. To this end, it would be useful to design high-throughput screens to probe direct ligand-protein interactions. One such approach would be iterative computational small-molecule docking and design. To increase the chance of success, molecular dynamics simulation is required to approximate the behavior of NS3h in solution when it contacts small molecules for binding (38). The NS3h complex structures captured in different conformations, including the binary complexes here, should allow better simulations of local structural fluctuations at each conformational state and, therefore, help identify more promising lead compounds.
Experimental Procedures
Recombinant Protein Production and Purification
The recombinant HCV NS3h proteins (NS3 helicase domain, amino acids 188–625, wild type and mutants) were produced in Escherichia coli as described previously (20). Briefly, the coding sequences of NS3h were PCR-amplified from the cDNA templates of Con1 virus genome and were engineered to fuse to the C terminus of His6-tagged Smt3 (the yeast small ubiquitin-like modifier protein) in the pET28a vector (Novagen). NS3h mutations were introduced by PCR. The proteins were expressed in E. coli and purified sequentially through nickel-affinity, ion-exchange, and gel-filtration columns (Ni-Sepharose 6 Fast Flow resin, HiTrap Q HP 5 ml, and Superdex 200 10/300 GL, respectively, from GE Healthcare). His6-SUMO was cleaved by the Ulp1 protease (39). Pure NS3h fractions were pooled and concentrated to 20 mg/ml in 100 mm NaCl, 20 mm HEPES, pH 7.5, and 5 mm dithiothreitol (DTT). Aliquots were flash-frozen in liquid nitrogen and stored at −80 °C.
Crystallization, Data Collection, and Structure Determination
NS3h-ADP·AlF4− binary complexes were reconstituted by mixing 0.2 mm NS3h with 1 mm ADP, 2.5 mm MgCl2, 10 mm NaF, and 1.5 mm AlCl3 (Sigma). The mixture was incubated at room temperature for ∼2 h. Crystallization conditions were identified through sparse matrix screens (Hampton Research) followed by successive condition optimization attempts. The complexes were crystallized at 4 °C in 20% (w/v) polyethylene glycol monomethyl ether 550. Crystals appeared as clustered thin plates. Single crystals were carefully isolated from the clusters and briefly soaked in cryo-protection solution (crystallization solutions supplemented with 5–10% (v/v) glycerol, 1 mm ADP, 2.5 mm MgCl2, 10 mm NaF, and 1.5 mm AlCl3). The crystals were flash-frozen in liquid nitrogen for diffraction experiments.
Crystals were screened with a CuKα x-ray source (Rigaku/MSC MicroMax-007HF). Datasets were collected with a synchrotron x-ray source at the National Synchrotron Light Source beamline X26C (Brookhaven National Laboratory, Upton, NY). The datasets were processed with DENZO, SCALEPACK, and CCP4 (40, 41).
Molecular replacement experiments were carried out with both MOLREP and PHASER (CCP4 program suite, Ref. 40). The domain 1 and 3 of NS3h from the NS3h-ssDNA complex structure (PDB ID 3KQH) was used as a searching model in the initial molecular replacement experiment. Domain 2 was docked into the 2Fo − Fc electron density calculated in CNS (42). Rigid body refinement and simulated annealing (42) were carried out sequentially. Model building was done with the O program (43).
ATPase Assay
To design the cysteine pair mutations, we modeled the cysteine mutations in PyMOL and measured the distances between the thiol groups. We made sure that all the distances were close to the length of the cross-linking compound BMDB and local protein dynamics would allow it to easily fit between the cysteine pairs for efficient cross-linking.
Enzymes were prepared in the following way before assays. Each version of NS3h (NS3h, CCi, CC1, and CC2) was diluted in 1400 μl of 20 mm NaCl to 0.5 μm. Tris pH 7.0 (1 m, 50 μl) was added into the 1400-μl protein solutions. Each protein solution was equally divided into 2 tubes (725 μl/tube). The BMDB compound (7.25 μl of 0.2 mm) freshly prepared in DMSO (dimethyl sulfoxide) was added into 1 tube of 725 μl of NS3h/CCi/CC1/CC2, and 7.25 μl of DMSO was added into the other tube of 725 μl of NS3h/CCi/CC1/CC2. The proteins (∼0.5 μm) with BMDB (∼2 μm) were incubated at room temperature for 45–50 min. Enzymes treated and untreated with NEM were prepared in a similar way. A 10-fold molar excess of NEM was added to the enzymes, and the mixed samples were incubated at room temperature for 1 h. The reactions were quenched by ∼20 mm DTT. The final protein concentration was ∼0.48 μm for all samples. The proteins were flash-frozen in liquid nitrogen as ∼60-μl aliquots.
A previously described malachite green assay (44) was used to measure the amount of phosphate released from HCV helicase-catalyzed ATP hydrolysis. MilliQ water was used to prepare a 0.045 (w/v) % MG solution (malachite green hydrochloride (Sigma) in water), a 4.2 (w/v) % AM solution (ammonium molybdate tetrahydrate (Sigma) in 4 n HCl), and a 2.3 (w/v) % PVA solution (polyvinyl alcohol, Sigma, in water). Then, a MG-AM-PVA solution was prepared by mixing MG, AM, and PVA in a 24:8:1 volume ratio right before the assay. Finally, a 34% (w/v) sodium citrate stabilization solution (trisodium citrate dihydrate (Sigma) in water) was prepared. All solutions were filtered before use.
Helicase-catalyzed nucleic acid-dependent ATP hydrolysis assays were carried out in 10-μl reactions, which contained ∼48 nm enzyme, ssDNA (0, 1.25, 2.5, 5, 10, 20, 40, 80 μm dT12) (Integrated DNA Technology), 2.0 mm ATP (GE Healthcare), 1.35 mm MgCl2, 20 mm NaCl, 0.1 m HEPES, pH 7.0, and 2 mm DTT. The reactions were incubated at 37 °C and stopped by 80 μl of MG-AM-PVA. Then, 10 μl of 34% sodium citrate was added to stabilize color. The mixtures were incubated at room temperature for ∼15 min before measurement of absorbance values at 660 nm using an Omega FLUOstar plate reader (BMG Labtech). Initial velocities of ATP hydrolysis with increasing amounts of dT12 were measured from three independent reactions, and the averaged values were plotted against the concentrations of dT12. The Michaelis-Menten equation was fitted to the data in Qtiplot.
Helicase-catalyzed basal ATP hydrolysis assays were carried out in 10-μl reactions containing 120 nm enzyme and increasing concentrations of ATP (0.05, 0.1, 0.25, 0.5, 1, 2, 4 mm). Released phosphate amounts were also measured in triplicate assays, and the averaged data were plotted against the concentrations of ATP for curve fitting. The kcat, KNA, and KATP values with standard errors were calculated from curve fitting and are listed in Table 2.
Nucleotide Mimic Binding Assay
Because NS3h does not have strong selection upon nucleotide bases and only BODIPY- labeled GDP (Invitrogen) was commercially available, we probed binding of BODIPY-GDP by following the change of fluorescence polarization. Serial dilutions of NS3h were prepared in a dilution buffer consisting of 100 mm NaCl, 20 mm HEPES, 7.75, and 5 mm DTT. Components required for transition-state mimic formation were mixed and diluted in the same dilution buffer to reach 40 nm BODIPY-GDP, 40 mm NaF, 4 mm AlCl3, and 8 mm MnCl2. To measure binding in the presence of nucleic acids, 0.2 mm ssDNA was added into the transition-state mimic solution. Each diluted NS3h protein was mixed with the same volume of the transition-state mimic solution. The mixtures were incubated at room temperature for 30 min before fluorescence polarization was measured in a BioTek Synergy NEO plate reader. A quadratic equation (20) describing ligand binding was used to fit the data in Qtiplot.
HCV Plasmid Construction and in Vitro RNA Transcription
A plasmid encoding a full-length HCV reporter genome Jc1(p7-nsGluc2A) (29) was used to engineer mutations in NS3. The mutations and insertions were introduced by PCR and verified by sequencing a region between the SpeI and BsrGI digestion sites. The verified fragments were then released by SpeI and BsrGI and inserted into the wild-type construct to generate the mutant constructs. The final constructs were verified by sequencing.
Viral RNA genomes were generated by in vitro transcription as previously described (45). Transcribed RNA was purified by using an RNeasy kit (Qiagen) and eluted in a 1 mm sodium citrate solution pH 6.4 (Ambion). The yield was quantified by absorbance at 260 nm, and the integrity was analyzed by agarose gel electrophoresis and ethidium bromide staining. RNA aliquots were then stored at −80 °C.
RNA Transfection and Luciferase Assay
Huh-7.5 and CD81 knockdown cells (Huh-7.5 CD81low) (46) were propagated in Dulbecco's modified minimal essential medium (Gibco) supplemented with 0.1 mm nonessential amino acids and 10% heat-inactivated fetal bovine serum, referred to below as “complete medium.” Cells were grown at 37 °C in humidified 5% CO2.
Huh-7.5 or Huh-7.5 CD81low cells were transfected with viral RNA as previously described (47). Transfection efficiency was enhanced by centrifugation at 2000 rpm, 37 °C for 30 min. The cells were then incubated at 37 °C in 5% CO2 for 5 h before 50 μl of culture supernatant aliquots were harvested, and the medium was replaced by complete medium. Culture supernatant aliquots were further harvested at 24, 48, and 72 h post transfection. The luciferase activities in the supernatant aliquots were determined by using a Renilla luciferase assay system (Promega).
Duplex Unwinding Assay
Duplex unwinding activities of the NS3h enzymes were measured using a modified molecular beacon-based assay (30). A DNA duplex substrate with 3′ single-stranded tail was prepared by annealing 0.2 mm molecular beacon strands (5′-GCCTGTTTGAGGCTTATTGCCTCTTACAGGC-3′) with complementary strands (5′-GCCTGTAAGAGGCAATAAGCCTCAAACAGGCTATTTGCCTCAGGC-3′). The enzymes and duplex DNA were diluted in a reaction buffer (0.1 m HEPES, pH 7.0, 10 mm DTT, 2 mm MgCl2) to reach 0.6 μm enzyme and 0.4 μm DNA, and 40 μl of the mixture was added into a well of a 384-well black plate. The fluorescence of protein-DNA mix without ATP was measured in a BioTek Synergy NEO plate reader at 37 °C for 30 min (1 read every 2 min). The plate was taken out for the addition of 25 μl/well ATP (∼3 mm final concentration) to initiate the reactions, and it was immediately put back to resume fluorescence measurement at the same frequency.
Author Contributions
M. G. and C. M. R. designed the experiments and wrote the manuscript. M. G. performed the experiments.
Acknowledgments
We thank the staff at the resource centers in The Rockefeller University (the Structural Biology Resource Center, the High-Throughput Screening Resource Center, and the Translational Technology Core Laboratory) for helping with the instruments. We also thank the staff at the National Synchrotron Light Source, Brookhaven National Laboratory for crystal diffraction data collection. James Letts helped refine crystallization conditions during his rotation in the laboratory. We are grateful to Margaret R. MacDonald and Mayla Hsu for critical input and editing.
This work was supported, in part, by National Institutes of Health (NIH) Grant CA057973 and Center for Synchrotron Biosciences Grant P30-EB-009998 (National Institute of Biomedical Imaging and Bioengineering (NIBIB), NIH). This work was also supported by generous gifts from the Greenberg Medical Research Institute, the Starr Foundation, the Richard Salomon Family Foundation, the Ronald A. Shellow, M.D. Memorial Fund, Paul Nash and the MGM Mirage Voice Foundation, and Gregory F. Lloyd Memorial contributions. The use of the Rigaku/MSC microMax 007HF in the Rockefeller University Structural Biology Resource Center was made possible by Grant 1S10RR022321-01 from the National Center for Research Resources of the National Institutes of Health. The authors declare that they have no conflict of interest. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (code 5E4F) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- HCV
- hepatitis C virus
- SF
- superfamily
- ssDNA
- single-stranded DNA
- BMDB
- 1,4 bismaleimidyl-2,3-dihydroxybutane
- NEM
- N-ethylmaleimide
- MG
- malachite green hydrochloride
- AM
- ammonium molybdate tetrahydrate
- PVA
- polyvinyl alcohol.
References
- 1.Gorbalenya A. E., and Koonin E. V. (1993) Helicases: amino acid sequence comparisons and structure function relationships. Curr. Opin. Struct. Biol. 3, 419–429 [Google Scholar]
- 2.Singleton M. R., Dillingham M. S., and Wigley D. B. (2007) Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 76, 23–50 [DOI] [PubMed] [Google Scholar]
- 3.Kadaré G., and Haenni A. L. (1997) Virus-encoded RNA helicases. J. Virol. 71, 2583–2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahola T., den Boon J. A., and Ahlquist P. (2000) Helicase and capping enzyme active site mutations in brome mosaic virus protein 1a cause defects in template recruitment, negative-strand RNA synthesis, and viral RNA capping. J. Virol. 74, 8803–8811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barton D. J., and Flanegan J. B. (1997) Synchronous replication of poliovirus RNA: initiation of negative-strand RNA synthesis requires the guanidine-inhibited activity of protein 2C. J. Virol. 71, 8482–8489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dé I., Sawicki S. G., and Sawicki D. L. (1996) Sindbis virus RNA-negative mutants that fail to convert from minus-strand to plus-strand synthesis: role of the nsP2 protein. J. Virol. 70, 2706–2719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grassmann C. W., Isken O., and Behrens S. E. (1999) Assignment of the multifunctional NS3 protein of bovine viral diarrhea virus during RNA replication: an in vivo and in vitro study. J. Virol. 73, 9196–9205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu B., Liu C., Lin-Goerke J., Maley D. R., Gutshall L. L., Feltenberger C. A., and Del Vecchio A. M. (2000) The RNA helicase and nucleotide triphosphatase activities of the bovine viral diarrhea virus NS3 protein are essential for viral replication. J. Virol. 74, 1794–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindenbach B. D., and Rice C. M. (2003) Molecular biology of flaviviruses. Adv. Virus Res 59, 23–61 [DOI] [PubMed] [Google Scholar]
- 10.Gower E., Estes C., Blach S., Razavi-Shearer K., and Razavi H. (2014) Global epidemiology and genotype distribution of the hepatitis C virus infection. J. Hepatol. 61, S45–S57 [DOI] [PubMed] [Google Scholar]
- 11.Morikawa K., Lange C. M., Gouttenoire J., Meylan E., Brass V., Penin F., and Moradpour D. (2011) Nonstructural protein 3–4A: the Swiss army knife of hepatitis C virus. J. Viral. Hepat. 18, 305–315 [DOI] [PubMed] [Google Scholar]
- 12.Kolykhalov A. A., Mihalik K., Feinstone S. M., and Rice C. M. (2000) Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3′ nontranslated region are essential for virus replication in vivo. J. Virol. 74, 2046–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lam A. M., and Frick D. N. (2006) Hepatitis C virus subgenomic replicon requires an active NS3 RNA helicase. J. Virol. 80, 404–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mackintosh S. G., Lu J. Z., Jordan J. B., Harrison M. K., Sikora B., Sharma S. D., Cameron C. E., Raney K. D., and Sakon J. (2006) Structural and biological identification of residues on the surface of NS3 helicase required for optimal replication of the hepatitis C virus. J. Biol. Chem. 281, 3528–3535 [DOI] [PubMed] [Google Scholar]
- 15.Blight K. J., McKeating J. A., Marcotrigiano J., and Rice C. M. (2003) Efficient replication of hepatitis C virus genotype 1a RNAs in cell culture. J. Virol. 77, 3181–3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krieger N., Lohmann V., and Bartenschlager R. (2001) Enhancement of hepatitis C virus RNA replication by cell culture-adaptive mutations. J. Virol. 75, 4614–4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lohmann V., Hoffmann S., Herian U., Penin F., and Bartenschlager R. (2003) Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J. Virol. 77, 3007–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saeed M., Scheel T. K., Gottwein J. M., Marukian S., Dustin L. B., Bukh J., and Rice C. M. (2012) Efficient replication of genotype 3a and 4a hepatitis c virus replicons in human hepatoma cells. Antimicrob. Agents Chemother. 56, 5365–5373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Appleby T. C., Anderson R., Fedorova O., Pyle A. M., Wang R., Liu X., Brendza K. M., and Somoza J. R. (2011) Visualizing ATP-dependent RNA translocation by the NS3 helicase from HCV. J. Mol. Biol. 405, 1139–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gu M., and Rice C. M. (2010) Three conformational snapshots of the hepatitis C virus NS3 helicase reveal a ratchet translocation mechanism. Proc. Natl. Acad. Sci. U.S.A. 107, 521–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng W., Arunajadai S. G., Moffitt J. R., Tinoco I. Jr., and Bustamante C. (2011) Single-base pair unwinding and asynchronous RNA release by the hepatitis C virus NS3 helicase. Science 333, 1746–1749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Myong S., Bruno M. M., Pyle A. M., and Ha T. (2007) Spring-loaded mechanism of DNA unwinding by hepatitis C virus NS3 helicase. Science 317, 513–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suzich J. A., Tamura J. K., Palmer-Hill F., Warrener P., Grakoui A., Rice C. M., Feinstone S. M., and Collett M. S. (1993) Hepatitis-C virus Ns3 protein polynucleotide-stimulated nucleoside triphosphatase and comparison with the related pestivirus and flavivirus enzymes. J. Virol. 67, 6152–6158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dürr H., Körner C., Müller M., Hickmann V., and Hopfner K. P. (2005) X-ray structures of the Sulfolobus solfataricus SWI2/SNF2 ATPase core and its complex with DNA. Cell 121, 363–373 [DOI] [PubMed] [Google Scholar]
- 25.Luo D., Xu T., Watson R. P., Scherer-Becker D., Sampath A., Jahnke W., Yeong S. S., Wang C. H., Lim S. P., Strongin A., Vasudevan S. G., and Lescar J. (2008) Insights into RNA unwinding and ATP hydrolysis by the flavivirus NS3 protein. EMBO J. 27, 3209–3219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soultanas P., Dillingham M. S., Velankar S. S., and Wigley D. B. (1999) DNA binding mediates conformational changes and metal ion coordination in the active site of PcrA helicase. J. Mol. Biol. 290, 137–148 [DOI] [PubMed] [Google Scholar]
- 27.Simmonds P. (2013) The origin of hepatitis C virus. Curr. Top. Microbiol. Immunol. 369, 1–15 [DOI] [PubMed] [Google Scholar]
- 28.Tortorici M. A., Duquerroy S., Kwok J., Vonrhein C., Perez J., Lamp B., Bricogne G., Rümenapf T., Vachette P., and Rey F. A. (2015) X-ray structure of the pestivirus NS3 helicase and its conformation in solution. J. Virol. 89, 4356–4371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marukian S., Andrus L., Sheahan T. P., Jones C. T., Charles E. D., Ploss A., Rice C. M., and Dustin L. B. (2011) Hepatitis C virus induces interferon-lambda and interferon-stimulated genes in primary liver cultures. Hepatology 54, 1913–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belon C. A., and Frick D. N. (2008) Monitoring helicase activity with molecular beacons. Biotechniques 45, 433–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frick D. N., Rypma R. S., Lam A. M., and Gu B. (2004) The nonstructural protein 3 protease/helicase requires an intact protease domain to unwind duplex RNA efficiently. J. Biol. Chem. 279, 1269–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jennings T. A., Chen Y., Sikora D., Harrison M. K., Sikora B., Huang L., Jankowsky E., Fairman M. E., Cameron C. E., and Raney K. D. (2008) RNA unwinding activity of the hepatitis C virus NS3 helicase is modulated by the NS5B polymerase. Biochemistry 47, 1126–1135 [DOI] [PubMed] [Google Scholar]
- 33.Pang P. S., Jankowsky E., Planet P. J., and Pyle A. M. (2002) The hepatitis C viral NS3 protein is a processive DNA helicase with cofactor enhanced RNA unwinding. EMBO J. 21, 1168–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chono K., Katsumata K., Kontani T., Kobayashi M., Sudo K., Yokota T., Konno K., Shimizu Y., and Suzuki H. (2010) ASP2151 a novel helicase-primase inhibitor possesses antiviral activity against varicella-zoster virus and herpes simplex virus types 1 and 2. J. Antimicrob. Chemother. 65, 1733–1741 [DOI] [PubMed] [Google Scholar]
- 35.Crute J. J., Grygon C. A., Hargrave K. D., Simoneau B., Faucher A. M., Bolger G., Kibler P., Liuzzi M., and Cordingley M. G. (2002) Herpes simplex virus helicase-primase inhibitors are active in animal models of human disease. Nat. Med. 8, 386–391 [DOI] [PubMed] [Google Scholar]
- 36.Kleymann G., Fischer R., Betz U. A., Hendrix M., Bender W., Schneider U., Handke G., Eckenberg P., Hewlett G., Pevzner V., Baumeister J., Weber O., Henninger K., Keldenich J., Jensen A., Kolb J., Bach U., Popp A., Mäben J., Frappa I., Haebich D., Lockhoff O., and Rübsamen-Waigmann H. (2002) New helicase-primase inhibitors as drug candidates for the treatment of herpes simplex disease. Nat. Med. 8, 392–398 [DOI] [PubMed] [Google Scholar]
- 37.Salam K. A., and Akimitsu N. (2013) Hepatitis C virus NS3 inhibitors: current and future perspectives. Biomed. Res. Int. 2013, 467869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Knegtel R. M., Kuntz I. D., and Oshiro C. M. (1997) Molecular docking to ensembles of protein structures. J. Mol. Biol. 266, 424–440 [DOI] [PubMed] [Google Scholar]
- 39.Mossessova E., and Lima C. D. (2000) Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol. Cell 5, 865–876 [DOI] [PubMed] [Google Scholar]
- 40.Collaborative Computational Project, Number 4 (1994) The CCP4 suite: Programs for Protein Crystallography. Acta Crystallogr D. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 41.Otwinowski Z., and Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 42.Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., and Warren G. L. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 43.Jones T. A., Zou J. Y., Cowan S. W., and Kjeldgaard M. (1991) Improved methods for building protein models in electron-density maps and the location of errors in these models. Acta Crystallogr. A 47, 110–119 [DOI] [PubMed] [Google Scholar]
- 44.Kallner A. (1975) Determination of phosphate in serum and urine by a single step malachite-green method. Clin. Chim. Acta 59, 35–39 [DOI] [PubMed] [Google Scholar]
- 45.Lindenbach B. D., Evans M. J., Syder A. J., Wölk B., Tellinghuisen T. L., Liu C. C., Maruyama T., Hynes R. O., Burton D. R., McKeating J. A., and Rice C. M. (2005) Complete replication of hepatitis C virus in cell culture. Science 309, 623–626 [DOI] [PubMed] [Google Scholar]
- 46.Witteveldt J., Evans M. J., Bitzegeio J., Koutsoudakis G., Owsianka A. M., Angus A. G., Keck Z. Y., Foung S. K., Pietschmann T., Rice C. M., and Patel A. H. (2009) CD81 is dispensable for hepatitis C virus cell-to-cell transmission in hepatoma cells. J. Gen. Virol. 90, 48–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chung H. Y., Gu M., Buehler E., MacDonald M. R., and Rice C. M. (2014) Seed sequence-matched controls reveal limitations of small interfering RNA Knockdown in functional and structural studies of hepatitis c virus NS5A-MOBKL1B interaction. J. Virol. 88, 11022–11033 [DOI] [PMC free article] [PubMed] [Google Scholar]