

Sepsis is a systemic inflammatory response syndrome caused by severe microbial infection. Uncontrollable activation of both inflammatory and coagulation pathways may result in multiple organ failure, a leading cause of death in intensive care units even in developed countries.
An adhesive protein, von Willebrand factor (VWF), plays a pivotal role in hemostasis and thrombosis by mediating platelet adhesion and aggregation,1 but is also known to be an acute-phase reactant whose plasma concentration is significantly increased in response to inflammation.1,2 From the standpoint of the human defense system, VWF could, therefore, be a key protein serving as a functional link between inflammation and thrombosis.2 Indeed, recent mouse model studies demonstrated that proper functional regulation of VWF-dependent inflammatory responses by ADAMTS13 considerably suppressed disease progression in patients with brain stroke3,4 or myocardial infarction.5,6 However, the precise mechanisms and roles of VWF in inflammation remain poorly understood thus far. We, therefore, studied the physiological relevance of VWF-dependent inflammatory responses in a mouse model of experimental sepsis involving cecal ligation and puncture (CLP).
VWF gene-deleted (VWF−/−; knock-out: KO) mice with a C57BL/6 background were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). All animal experiments were approved by the Institutional Animal Care and Use Committee of Nara Medical University. CLP was performed according to the established protocol with minor modifications.7 Briefly, each male KO mouse or wild-type (WT)-C57/BL6 mouse (10–13 weeks of age) was anesthetized with isoflurane and its abdominal wall was opened through a 1-cm midline incision. The exposed cecum was ligated with 4-0 Sofsilk (Covidien, Mansfield, MA, USA), punctured with an 18G needle and gently pressed until a small drop of stool appeared. The cecum was returned to the peritoneal cavity and the body wall and skin incision were closed with 4-0 Sofsilk. Two hundred microliters of saline were injected into the peritoneal cavity before the final suture to avoid dehydration. In some experiments (n=22 for the Kaplan-Meier analysis), human VWF (20 μg/mouse), purified from cryoprecipitate as previously described,8 was injected into KO mice via the tail vein just after CLP (indicated as “KO+VWF” in Figure 1). Excess bleeding was not observed in any of the WT or KO mice during or after the CLP operation. A preliminary experiment determined the ligation position of the cecum, a major factor in mouse mortality using the CLP approach.7 For induction of mid-grade sepsis, which would achieve the survival of approximately half of WT mice, the cecum was ligated at half the distance between the distal pole and the base of the cecum. In some experiments (WT, KO or KO+VWF; n=5 each), blood was harvested by cardiac puncture 24 h after CLP. Total white blood cell (WBC) numbers and the WBC differential were counted manually by microscopic examination of a blood sample that was spread in a thin film on a glass slide. Simultaneously, the ligated cecum was removed, fixed, and stained with hematoxylin and eosin to evaluate neutrophil recruitment in cecal tissues. In other experiments (WT; n=10, KO; n=13), blood was harvested from the submandibular vein to keep mice alive for subsequent observations. Mouse survival rates were analyzed by the Kaplan-Meier method and log-rank test. WBC and neutrophil numbers were analyzed with the Bonfferoni test or Student t-test. P values <0.05 were considered to be statistically significant.
Figure 1.
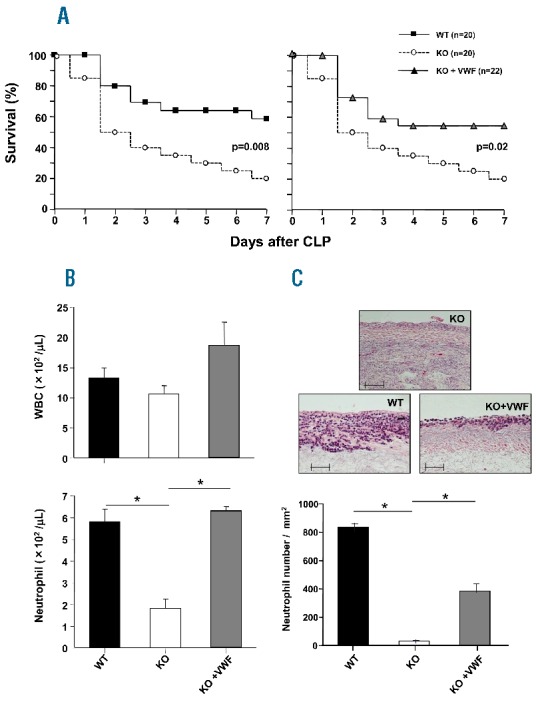
von Willebrand factor exerts beneficial effects in CLP-induced sepsis via recruitment of neutrophils to inflammatory sites. (A) Kaplan-Meier analysis of survival rates of WT (n=20: ■) or VWF-KO mice (n=20: ○) after CLP. The survival rate of WT mice 7 days after CLP was 60%. Note that KO mice died faster and had a significantly (P=0.008) lower survival rate (20.0%) than WT mice (left panel). Following bolus administration of human VWF (20 μg/mouse, n=22: ▲), this impaired survival rate in KO mice improved (to 54.5%) and became nearly indistinguishable from that of WT mice (see right panel), suggesting determinant effects of VWF in sepsis. (B) White blood cell and neutrophil numbers 24 h after CLP. These series of experiments (panels B and C), in which the recipient mice were sacrificed at day 1 (24 h) after CLP (n=5, each), were performed independently of the long-term observational experiments shown in panel (A). Each bar represents the mean + the standard error of the mean (SEM). KO mice demonstrated a significant (*P<0.05) decrease in neutrophil numbers (lower panel) compared with WT mice, while no significant difference between each group was seen in total WBC numbers (upper panel). Bolus injection of human VWF clearly restored neutrophil numbers to levels comparable to those in WT mice (*P<0.05). (C) Ligated ceca were harvested 24 h after CLP and subjected to hematoxylin and eosin staining. Images shown in the upper panel (original magnification, 200×; scale bar 100 μm) are representative of each three independent mouse samples. Note that ceca from KO mice showed necrotic tissue with poor neutrophil infiltration, whereas WT mice showed abundant neutrophil accumulation. Neutrophil recruitment was restored by bolus injection of human VWF. Statistical analyses corresponding to the images above are shown in the lower panel; each bar represents the average (+SEM) of 15 areas (each 1 mm2; five areas were randomly selected from three individual mouse samples). These analyses indicate that neutrophil numbers in cecal tissue are dramatically decreased in VWF-KO mice and are significantly (*P<0.05) restored by VWF administration.
Using the present CLP conditions, the survival rate of KO mice was significantly lower than that of WT animals (Figure 1A, left panel), but was restored by the administration of human VWF, indicating the crucial involvement of VWF in the pathological process of severe microbial infection (Figure 1A, right panel). Considering the significant decrease in neutrophils in peripheral blood as well as the reduced neutrophil infiltration in cecal tissue at 24 h after CLP in KO mice (Figure 1B,C), their lower survival rate most likely reflects defective leukokinetics associated with the absence of VWF. The cause of the neutrophil decrease in the blood of KO mice is not clear yet, but could be a reflection of the systemic status that overwhelms the bone marrow potential, as seen in severe or fatal sepsis in clinical settings. As a human defense mechanism, therefore, VWF may play a role in leukocyte recruitment at inflammatory loci in response to microbial infection. This scenario is consistent with a recent study demonstrating that the functional regulation of VWF by ADAMTS13 significantly reduced neutrophil accumulation at ischemic sites in a mouse model of myocardial infarction.6 In the current study, the retrospective analysis of neutrophil dynamics 24 h after CLP clearly indicated that the formation of a walled-off abscess in the peritoneal cavity was an absolute prerequisite for mouse survival, reflecting adequate neutrophil recruitment at the inflammatory focus (Figure 2A,B).
Figure 2.
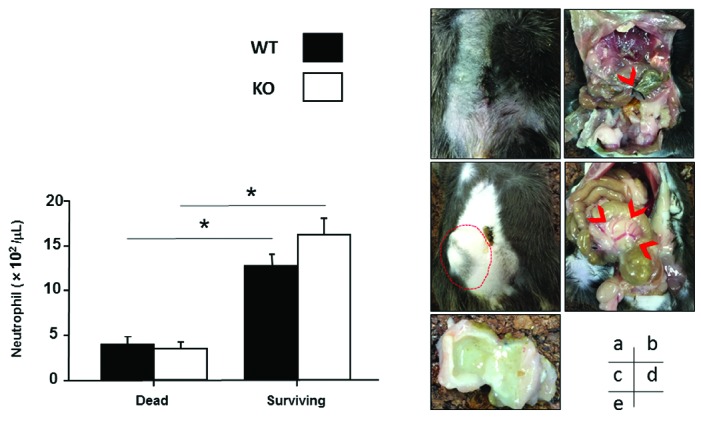
Retrospective analysis of neutrophil numbers 24 h after CLP, and the walled-off abscess seen in surviving mice. (A) Comparison of peripheral neutrophil numbers 24 h after CLP in dead mice and surviving mice. Peripheral blood was collected from the submandibular veins of WT or KO mice 24 h after CLP. Survival rates were evaluated after 6 days (see Figure 1A). Neutrophil numbers in the two groups (mice that died within 2 days vs. those that survived for 7 days after CLP) were then retrospectively compared in both WT (■; dead, n=5; surviving, n = 5) and KO (□; dead, n = 8; surviving, n = 5) mice. Surviving mice had significantly (*P<0.05) higher neutrophil numbers than dead mice at 24 h after CLP, regardless of genetic background. (B) Walled-off abscess formation in surviving mice. Mice that died within 2 days after CLP were dissected and the peritoneal cavity was observed. Mice that survived were sacrificed and dissected at day 7. Images displayed are representative of five independent mouse samples in each group. (a) No appreciable tumor formation is apparent externally in mice that died within 2 days after CLP. (b) Necrotic cecum at the distal position of ligation site (arrow head) in the peritoneal cavity of a mouse that died within 2 days after CLP. (c) Bulging abdominal wall of a surviving mouse (broken line). (d) Walled-off abscess formation in the peritoneal cavity of a surviving mouse (arrow head). (e) Cut surface of a walled-off abscess, in which pus was encapsulated, from a surviving mouse. Regardless of genetic background or VWF administration, walled-off abscess formation was observed in all surviving mice, whereas such abscess formation was not found in mice that died within 2 days after CLP.
It is, however, necessary to consider that VWF-KO mice not only have a lack of VWF, but also have impaired P-selectin functions due to the loss of intact Weibel-Palade bodies.9 Consequently, VWF-KO mice could exhibit reduced leukocyte rolling and adhesion on endothelial cell layers in vivo, resulting in impaired leukocyte recruitment. It is, therefore, unclear whether the impaired leukocyte recruitment in KO mice shown in the current study reflects VWF deficiency or the functional defect of endothelial P-selectin. In this regard, Petri et al.10 demonstrated that the function-blocking antibody of VWF significantly decreased neutrophil recruitment in WT-mice, albeit with intact P-selectin function as well as normal leukocyte rolling and adhesion on endothelium. In their study, the blockage of VWF function was shown to reduce neutrophil extravasation significantly, which could be essentially mediated by the VWF-platelet glycoprotein Ib interaction.
In addition, the lower survival rate (Figure 1A) and impaired leukocyte recruitment (Figure 1B,C) observed in VWF-KO mice were significantly restored by the administration of human VWF in the present study. Although, so far, human VWF reactivity to mouse platelets or ADAMTS13 is uncertain or reduced, our results in the human VWF infusion study strongly suggest that VWF is critically involved in neutrophil recruitment during inflammatory responses. Thus, VWF likely contributes to adequate leukocyte recruitment at inflammatory loci in response to severe microbial infection, possibly by supporting leukocyte extravasation from the microcirculation where blood flow creates high shear stress.1 Neutrophils that accumulate at the site of inflammation could encapsulate microbes and prevent their systemic dissemination.
In addition to neutrophil recruitment, the beneficial effects of VWF in sepsis may be explained by the recently proposed concept of “immunothrombosis”.11 In response to microbial infection, both inflammatory and thrombotic pathways are triggered as innate defense systems. Locally generated thrombi could seal in infectious microbes and detrimental inflammatory mediators at locally inflamed sites by interrupting blood flow. Indeed, recent studies have shown that mice deficient in coagulation factors, such as tissue factor, factor V and fibrinogen, had significantly lower survival rates than WT mice with experimental sepsis,12,13 reflecting impaired thrombin or fibrin generation. In this regard, decreased factor VIII activity due to the absence of VWF in VWF-KO mice may partly contribute to impaired thrombin generation. In addition, defective VWF-mediated platelet aggregation in peripheral capillaries could promote microbial dissemination throughout the body, resulting in the lower survival rate observed in VWF-deficient mice in this sepsis model, in analogy with fibrinogen-deficient mice which may fail to prevent microbial expansion due to the absence of fibrin-net formation.13
Although VWF-mediated thrombotic or inflammatory responses are beneficial for microbial extinction, as discussed above, it is important to consider a possibility that such VWF-dependent reactions may, conversely, be disadvantageous in septic states. VWF-dependent platelet aggregation may hinder microcirculatory blood flow, which is crucial for the viability of tissues or organs in the context of microbial infection. Indeed, mouse mortality in CLP-induced sepsis critically depends on the position of the cecal ligation as this determines the extent of the ischemic lesion.7 This might explain the previous discrepant observation by Lerolle et al.14 that in a model of CLP-induced sepsis, VWF-KO mice survived significantly longer than WT ones. Their CLP protocol, in which the mouse cecum was ligated about 15 mm proximal to the cecal pole (just below the ileo-cecal valve), was much more severe than ours, inducing high-grade sepsis and resulting in a 0% survival rate of WT mice within 30 h after their CLP.14 Although alternative explanations are possible, the beneficial effects of VWF deficiency on microcirculatory blood flow might have far outweighed the disadvantage of defective neutrophil recruitment on mouse survival in their severer CLP situations. A proper balance of VWF function is, therefore, crucial in the context of severe microbial infection, as it is in the case of bleeding or pathological thrombosis.
Overall, our results point to VWF being critically involved in neutrophil recruitment against fatal microbial infection. Although the functional regulation of VWF has recently been proven beneficial in various diseases,3–6,15 VWF-dependent inflammatory responses should be carefully considered in the context of these clinical applications.
Acknowledgments
The authors would like to thank Ayuri Nakamura for her technical assistance.
Footnotes
Funding: this work was supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan (No. 19591129 to M. Sugimoto), the 6th Baxter Coagulation Research Fund (to SK), and the Takeda Science Foundation (to M. Sugimoto).
Information on authorship contributionsand financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org
References
- 1.Ruggeri ZM. Von Willebrand factor, platelets and endothelial cell interactions. J Thromb Haemost. 2003;1(7):1335–1342. [DOI] [PubMed] [Google Scholar]
- 2.Kremer Hovinga JA, Zeerleder S, Kessler P, et al. ADAMTS-13, von Willebrand factor and related parameters in severe sepsis and septic shock. J Thromb Haemost. 2007;5(11):2284–2290. [DOI] [PubMed] [Google Scholar]
- 3.Kleinschnitz C, De Meyer SF, Schwarz T, et al. Deficiency of von Willebrand factor protects mice from ischemic stroke. Blood. 2009; 113(15):3600–3603. [DOI] [PubMed] [Google Scholar]
- 4.Fujioka M, Hayakawa K, Mishima K, et al. ADAMTS13 gene deletion aggravates ischemic brain damage: a possible neuroprotective role of ADAMTS13 by ameliorating postischemic hypoperfusion. Blood. 2010;115(8):1650–1653. [DOI] [PubMed] [Google Scholar]
- 5.Doi M, Matsui H, Takeda H, et al. ADAMTS13 safeguards the myocardium in a mouse model of myocardial infarction. Thromb Haemost. 2012;108(6):1236–1238. [DOI] [PubMed] [Google Scholar]
- 6.De Meyer SF, Savchenco AS, Haas MS, et al. Protective anti-inflammatory effect of ADAMTS13 on myocardial ischemia/reperfusion injury in mice. Blood. 2012;120(26):5217–5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009; 4(1):31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sugimoto M, Mohri H, McClintock RA, Ruggeri ZM. Identification of discontinuous von Willebrand factor sequences involved in complex formation with botrocetin. A model for the regulation of von Willebrand factor binding to platelet glycoprotein Ib. J Biol Chem. 1991;266(27):18172–18178. [PubMed] [Google Scholar]
- 9.Denis CV, André P, Saffaripour S, Wagner DD. Defect in regulated secretion of P-selectin affects leukocyte recruitment in von Willebrand factor-deficient mice. Proc Natl Acad Sci USA. 2001; 98(7):4072–4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petri B, Broermann A, Li H, et al. von Willebrand factor promotes leukocytes extravasation. Blood. 2010;116(22):4712–4719. [DOI] [PubMed] [Google Scholar]
- 11.Englemann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013;13(1):34–45. [DOI] [PubMed] [Google Scholar]
- 12.Sun H, Wang X, Degan JL, Ginsburg D. Reduced thrombin generation increases host susceptibility to group A streptococcal infection. Blood. 2009;113(6): 1358–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo D, Szaba FM, Kummer LW, et al. Protective roles for fibrin, tissue factor, plasminogen activator inhibitor-1, and thrombin activatable fibrinolysis inhibitor, but not factor XI, during defense against the gram-negative bacterium Yersinia enterocolitica. J Immunol. 2011; 187(4):1866–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lerolle N, Dunois-Lardé C, Badirou I, et al. von Willebrand factor is a major determinant of ADAMTS-13 decrease during mouse sepsis induced by cecum ligation and puncture. J Thromb Haemost. 2009; 7(5):843–850. [DOI] [PubMed] [Google Scholar]
- 15.Matsui H, Takeda M, Soejima K, et al. Contribution of ADAMTS13 to the better cell engraftment efficacy in mouse model of bone marrow transplantation. Haematologica. 2014;99(10):e211–213. [DOI] [PMC free article] [PubMed] [Google Scholar]