

Abstract
HIV-1 Rev protein mediates the nuclear export of viral RNA genomes. To do so, Rev oligomerizes cooperatively onto an RNA motif, the Rev-response element (RRE), forming a complex that engages with the host nuclear export machinery. To better understand Rev oligomerization, we determined four crystal structures of Rev N-terminal domain dimers, which show that they can pivot about their dyad axis, giving crossing-angles of 90° to 140°. In parallel, we performed cryo-EM of helical Rev filaments. Filaments vary from 11 to 15 nm in width, reflecting variations in dimer crossing-angle. These structures contain additional density, indicating that C-terminal domains become partially ordered in the context of filaments. This conformational variability may be exploited in the assembly of RRE/Rev complexes. Our data also revealed a third interface between Revs which offers an explanation for how the arrangement of Rev subunits adapts to the ‘A’-shaped architecture of the RRE in export-active complexes.
eTOC In Brief
Binding of 8–12 HIV-1 Rev subunits to the RRE of viral transcripts mediates nuclear export. DiMattia et al. describe a novel Rev-Rev interface and the remarkably flexible ways in which Rev can assemble, and propose how this allows Rev to engage with the A-shaped RRE.
Introduction
Rev is an HIV-1 protein that regulates the nuclear export of intron-containing viral RNA transcripts that would otherwise be retained in the nucleus to be spliced or degraded (Luo and Reed, 1999; Bousquet-Antonelli et al., 2000; Zhou et al., 2000). This mechanism of overriding eukaroytic function is essential for HIV-1 replication: un-spliced mRNA transcripts must be exported for packaging as genomes into nascent virions, and along with other partially-spliced transcripts, they are translated into the majority of the viral proteins (Cullen, 2003). In order to achieve this, multiple copies of Rev (8–12 molecules) oligomerize onto a cognate ~350 nucleotide RNA region within an intron of the HIV genome, known as the Rev Response Element (RRE) (Daly et al., 1989; Malim et al., 1989; Cook et al., 1991; Malim and Cullen, 1991; Daly et al., 1993; Mann et al., 1994; Pollard and Malim, 1998; Cullen, 2003; Pond et al., 2009). Once this ribonucleoprotein (RNP) complex forms, it co-opts the Crm1 nuclear export pathway (which is typically used to export host protein cargoes and small RNAs – reviewed in Okamura et al., 2015) to translocate the Rev-RRE RNP through nuclear pores into the cytoplasm (Fischer et al., 1995; Fornerod et al., 1997; Pollard and Malim, 1998; Cullen, 2003; Yedavalli et al., 2004).
At a mere 116 amino acid residues, Rev is a small protein with multiple functional motifs. It has two domains: an N-terminal domain (NTD; res. 1–65) which is responsible for (1) Rev self-assembly into dimers/higher-order oligomers and (2) RRE-association via an arginine-rich motif (ARM) that doubles as a nuclear localization signal (NLS) (Malim and Cullen, 1991); and a C-terminal domain (CTD; res. 66–116) which has a nuclear export signal (NES) and is responsible for bridging the interaction between the RRE and Crm1/Ran-GTP (Fischer et al., 1995; Fornerod et al., 1997; Cullen, 2003).
Rev’s conformationally dynamic nature and its propensity to aggregate and form filaments in vitro (Wingfield et al., 1991) have hindered structural investigation. Thus, the organization of the HIV RNP export complex remains unknown, although there is a growing number of structures of its components. Early on it was revealed how a single Rev ARM interacts with the primary Rev-binding site within the RRE (Battiste et al., 1996). Fourteen years later, two crystal structures showed how Rev dimerizes through interfacing of identical hydrophobic surfaces in its NTD (termed the A-A and B-B interfaces) (DiMattia et al., 2010; Daugherty et al., 2010). The NTDs are α-helical hairpins whose RRE-associating ARMs are located at the distal ends of the dimer. The CTDs are unstructured (or absent) in all crystal structures to date. More recently, a SAXS model has been determined for the RRE (without Rev) (Fang et al., 2013), and a crystal structure for the RevNTD dimer bound to an RRE fragment (Jayaraman et al., 2014). The interactions between Crm1 and the Rev NES have also been characterized (Güttler et al., 2010; Booth et al., 2014).
Despite these advances, it remains unclear how the oligomerization of Rev subunits is matched to the architecture of the RRE – for a recent review of the current Rev/RRE structural models, see Rausch and Le Grice, 2015 – such oligomerization being required for nuclear export. In this study, we have investigated the structural basis of Rev oligomerization in the absence of RNA by determining the crystal structures of four additional Rev dimers and by performing cryo-EM and 3D reconstruction of in vitro-assembled helical Rev filaments. We observe wide variation in the crossing angles of Rev dimers, a previously uncharacterized Rev-Rev oligomerization interface, and partial ordering of the Rev CTD in the context of filaments. Taken together, these observations shed light on how Rev subunits may oligomerize in the context of Rev-RRE RNP formation.
Results
After determining a crystal structure for the wild-type RevNTD dimer by using a Fab fragment as a crystallization chaperone (Stahl et al., 2010; DiMattia et al., 2010), we engineered a variant of this complex with a single-chain antibody variable fragment (scFv) instead of the full Fab molecule. The intent here was to improve resolution and to further explore Rev-Rev interactions, including the variable Rev dimer crossing angles previously observed (DiMattia et al., 2010; Daugherty et al., 2010). The scFv-Rev complex permitted tighter crystal packing and higher resolution X-ray diffraction data (up to 2.3 Å) (Table 1). Crystals of the scFv-Rev complex grew in a variety of reservoir conditions that gave rise to data sets in four different space groups. This allowed for separate structure determination of each crystal form (see Table 1 for data collection and refinement statistics).
Table 1.
X-ray diffraction data collection and refinement statistics
| Data collection | ||||
| Space group | P21 | P212121 | I4 | P3212 |
| Beamlinea | Diamond I24 | Diamond I02 | Diamond I02 | ESRF ID23-EH2 |
| Unit cell param. | ||||
| a, b, c (Å) | 43.9, 81.9, 98.8 | 44.5, 87.1, 166.7 | 121.1, 121.1, 89.3 | 48.5, 48.5, 264.5 |
| α, β, γ (°) | 90.0, 101.1, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 120.0 |
| Resolution (Å) | 48.5−2.30 (2.4−2.3) | 50.0−2.90 (3.0−2.9) | 60.8−3.10 (3.2−3.1) | 50.0−4.30 (4.4−4.3) |
| Wavelength (Å) | 0.9779 | 0.9793 | 0.9793 | 0.8726 |
| Completeness (%) | 100.0 (100.0) | 99.5 (99.2) | 99.8 (97.6) | 98.3 (98.6) |
| Rsym (%) | 10.3 (84.6) | 20.1 (100.0) | 15.1 (90.1) | 5.5 (71.7) |
| I/σI | 11.5 (1.6) | 10.3 (1.1) | 15.1 (2.7) | 20.4 (1.1) |
| Mean multiplicity | 8.1 (5.0) | 13.3 (13.3) | 7.5 (7.6) | 3.1 (2.8) |
| Refinement | ||||
| No. of reflections used in refinement | 30,611 | 14,917 | 11,655 | 2,588 |
| Rwork/Rfree (%) | 18.9/23.0 | 24.6/27.3 | 29.1/30.0 | 32.8/32.4 |
| Reflects used for Rfree (|Fo| > 0) (%) | 5.0 | 5.0 | 5.0 | 5.0 |
| Ramachandran Plot (%) | ||||
| Most favored | 96.9 | 92.6 | 95.7 | – |
| Allowed | 3.1 | 6.9 | 3.6 | – |
| Outliers | 0.0 | 0.5 | 0.7 | – |
| No. of protein/water atoms | 4344/180 | 4381/0 | 4444/0 | 2157/0 |
| Average B-factor(Å2) | 58 | 70 | 72 | – |
| Wilson B-factor (Å2) | 50 | 81 | 80 | – |
| RMSDs | ||||
| Bond lengths (Å) | 0.010 | 0.010 | 0.008 | – |
| Bond angles () | 1.15 | 1.21 | 1.14 | – |
Data were collected at beamlines Diamond I02 and I24 at the Diamond Light Source (Didcot, UK) and at Beamline ID23-EH2 at ESRF (Grenoble, France).
Ramachandran plots were calculated with Molprobity (Davis et al., 2007).
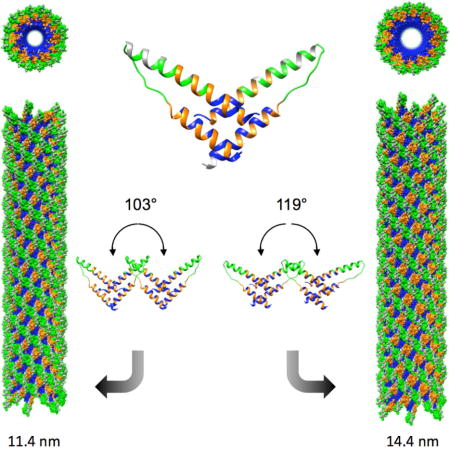
In parallel with the X-ray crystallography, filaments assembled from purified recombinant Rev were examined by cryo-electron microscopy. Two data sets were collected (see Experimental Procedures for details) from the same sample, with significantly different degrees of heterogeneity (manifesting as localized differences in width along their lengths) (Fig. 1A, B). This variability is greater in one set than the other. We posit that the filaments in the more homogeneous sample (according to filament diameter) had converged to an equilibrium conformation during storage at 4°C. This sample was checked for proteolytic degradation by SDS-PAGE and confirmed to be unaltered (data not shown). The averaged power spectrum from images in the more-homogeneous data set suggested a helical symmetry with a subunit rise and rotation of 21 Å and 22°, respectively, in addition to six-fold rotat ional symmetry (C6) (Fig. 1C). A final reconstruction was then generated to 8.3 Å resolution using the iterative real space reconstruction (IHRSR) method (Egelman, 2000) (with symmetry convergence to 21.2 Å and 22.1°) using 44,866 overlapping filament segments, each 300 pixels long (303 Å) (Fig. 1D).
Figure 1. Cryo-EM helical reconstruction of HIV Rev filaments, related to Fig. S4.

(A) Representative cryo-electron micrograph of HIV Rev filaments (scale bar = 50 nm).
(B) Cryo-electron micrograph depicting heterogeneous diameter of Rev filaments
(C) An averaged power spectrum, generated from 44,866 filament segments, each 512 pixels long (1.02 Å/px), is shown. The segments were windowed using a 50 pixel translation along the filament axis, diffraction pattern computed for each, and averaged. The Bessel orders are shown for the principal layer lines (n=−6, is at 1/55 Å in reciprocal space and n=+6, at 1/34 Å) as well as the meridional reflection (n=0, at 1/21 Å). Bessel order hand (−, left-handed; +, right-handed) was determined by fitting the crystal structure of individual Rev subunits into the EM density map.
(D) Reconstructed EM density map of HIV Rev filament. The asymmetric unit corresponds to one Rev dimer. The crystal structures of two Rev subunits were docked independently (using UCSF Chimera) and symmetrized with C6 and helical symmetry. Density for the Rev CTD (residues 66–116) is absent, either due to disorder or conformational heterogeneity.
(E) Extracted electron density corresponding to four Rev subunits, showing the presence of A-A, B-B, and C-C oligomerization interfaces. The density is contoured at thresholds of 1.5σ and 4σ to show the fit of α1 and α2 for each subunit.
(F) 2D representation of the helical assembly. The Rev filament displays left-handed and right-handed six-start helices (for which 2 and 3 starts are shown, respectively). Symmetry was imposed along the left-handed helix with an azimuthal rotation of−22.1° and 21.2 Å rise. The A-A, B-B, and C-C interfaces are colored blue, orange, and green, respectively.
(G) Surface representation of the Rev filament, colored as in (E).
(H) Transverse slice through EM reconstruction. The four punctate densities (marked by arrow) that repeat around the filament axis correspond to the four helices in one Rev dimer (scale bar = 10 nm).
(I) Transverse slice (same z-slice as in (G)) through map calculated from docked Rev NTD dimers into EM reconstruction.
Pivoting of Rev subunits at the oligomerization interfaces
The scFv-Rev crystal structures exhibit crossing angles of the RevNTD dimer ranging from ~90° to ~140°. They were measured after superposing the fiv e wild-type Rev dimer structures reported to date (PDB ID: 2X7L and presented herein), using one of the subunits for registration (Fig. 2A). We then asked: do interacting Rev subunits hinge about a specific set of residues or by a less specific “sliding” of one subunit across the other? Either scenario is plausible, given the predominantly hydrophobic interaction surface. To answer this question, we devised a calculable parameter for the Rev residues interfacing at oligomerization surfaces that we call spatial variance (SV). With all views of a given oligomerization interface superposed (after aligning the subunits), SV is defined as the degree to which a residue “moves” when comparing the different conformations of the opposing subunit. This measure is attractive in that if the Rev dimer subunits do move by lateral sliding, the SV values of the interfacing residues should all be roughly the same. However, this was not observed. At the A-A dimer interface, some residues had low SV values (between 0 and 2 Å2): V16, I19, K20, Y23, S56 and L60 (Fig. 2B) and the remaining had large SV values (between 2 and 12 Å2), from which we conclude that Rev dimers pivot via a ball-and-socket mechanism (Fig. 2B). Examination of this region confirmed that the aforementioned residues form a non-polar “socket” occupied by L64 of the opposing subunit (i.e. the “ball”). The mutation of L64 to Ala had no effect on the global conformation as judged by far-UV CD (Fig. S1) and near UV-CD (not shown) nor on its ability to form dimers (Fig. S1) yet completely abrogated the formation of filaments under conditions in which wild-type Rev polymerizes (Fig. S2). This suggests that L64 is a key residue for A-A interface formation and/or that dimer mobility is a requirement for ordered filament formation. For a systematic comparison of the residues interacting at the dimer interfaces across all of the observed views (with different dyad crossing angles), see Fig. S3.
Figure 2. Crossing angle variability of Rev NTD dimers, related to Fig. S1–S3.
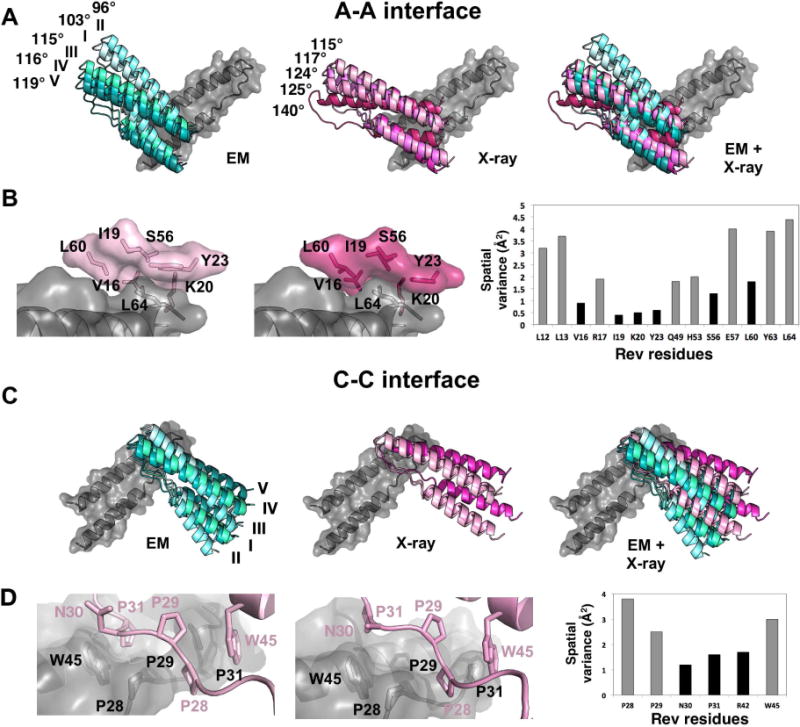
(A) Depicted are RevNTD dimer structures centered around the A-A interface. They are clustered by structural technique: determined by EM (cyan; docked into maps), X-ray crystallography (magenta), and an overlay of all. The crossing angles of the A-A dimers are labeled. The structures are overlapped on the gray subunits.
(B) Close-up of A-A dimerization interfaces. L64 of one subunit (grey) pivots within a hydrophobic pocket of the opposing subunit (shown in pink). The two panels show the interface for the dimers with most acute and most obtuse crossing angles. The spatial variance chart shows the results with the least “mobility” between different views of the respective interface (SV < 2; shown in black).
(C) Depicted are RevNTD dimers centered around the C-C interface, presented and colored as in panel A. Given the geometry of Rev oligomerization and the symmetry present in the observed conditions (helical filaments or crystal lattices), the crossing angles of C-C interfaces are equal to those of their respective A-A interface. The structures are overlapped on the gray subunits.
(D) Close-up of C-C dimerization interfaces. The spatial variance chart and C-C interface panel are depicted as in (B). The pivot point for this interface in N30 and P31.
Overall architecture of HIV-1 Rev filaments
HIV-1 Rev filaments are hollow tubes, with diameters that vary – in the less homogeneous data set — from ~11 to ~15 nm, average ~13 nm. These properties of the filaments are consistent with the findings of earlier studies at lower resolution (Watts et al., 1998). The asymmetric unit of the structure revealed V-shaped rod-like densities consistent with the α-helical hairpins visualized in RevNTD dimer crystal structures. Two hairpins representing a RevNTD dimer derived from the highest resolution crystal structure presented herein (see Fig. 3) were docked into the cryo-EM density and propagated according to the helical symmetry of the filament (see Experimental Procedures). The resolution of the reconstruction (8.3-Å, see Fig. S4) was sufficient to determine the hand of the helical filament, using the crystal structure of the RevNTD dimer. Its structure can be seen in the context of docked RevNTD molecules (Fig. 1D) as well as the fit of four RevNTD polypeptides into a portion of the map (Fig. 1E). The fit is further validated by visual comparison of corresponding z-slices of the EM map (Fig. 1H) and a map generated from the docked Rev subunits (Fig. 1I). The motif of four punctate densities, which is repeated six times around the filament cross-section, represents cross-cutting of the four α-helices in a Rev dimer (Fig. 1H, I). As expected, Rev subunits assemble via the homomeric mating of hydrophobic A-A and B-B interfaces, colored blue and orange, respectively (Fig. 1G) (DiMattia et al., 2010; Daugherty et al., 2010). However, a third, previously uncharacterized Rev-Rev interaction was observed which we term the C-C interface (green) (Fig. 1G). The repeating A-A and B-B interfaces form a hydrophobic core at an inner radius, while the “closed ends” of adjacent helical hairpins form the C-C interface. A 2D representation of this interaction network between Rev subunits in the filaments is shown in Fig. 1F.
Figure 3. The Rev C-C Interface, related to Fig. S5–S6.
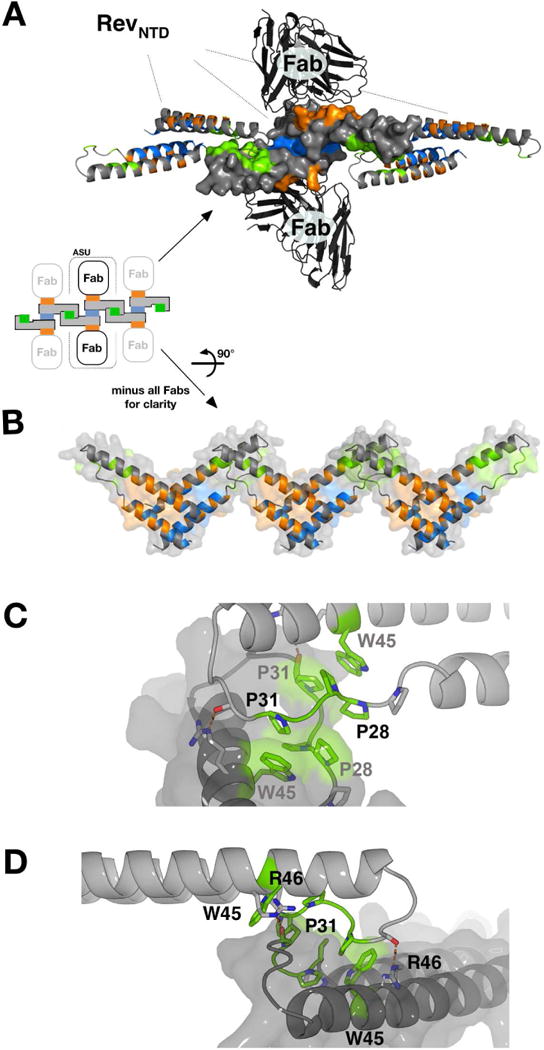
(A) Shown is the scFv-Rev P21 structure (PDB: 5DHV), with a Rev dimer at center (surface representation), formed through A-A interface (blue) and bound to two scFv molecules via B oligomerization surfaces (orange). The Rev dimer contacts two adjacent Rev dimers (ribbon representation) laterally via the C-C interface (green). Crystal packing of scFv-Rev lattices exhibits C-C interface in three of four space groups observed (see Fig S1).
(B) Rev dimers in (A) rotated 90°. The C-C interface bridges adjacent Re v dimers formed through the A-A oligomerization interfaces.
(C) The poly-proline loop (between α1 and α2) and W45 comprise the major contacts of the C-C interface. P28, P29, and P31 are involved, but P27 is not). The rigid poly-proline loops hook into each other, supported by stacking interactions between W45 and P31. The interface is further stabilized by two hydrogen bonds between R46 and backbone carbonyl moieties of the apposed subunit.
(D) Same as (C) but viewed from a different angle.
A third Rev-Rev oligomerization interface
After observing the C-C interface in the Rev filaments, we re-examined the crystal lattice packing of the scFv-Rev structures and found that in three out of four, the same interface forms as a lattice contact (Fig. 3A). As in the filaments, adjacent dimers interact laterally via the proline-rich loops of proximal helical hairpins (Fig. 3B). This interaction is homotypic, as in the A-A and B-B interactions, but differs in that it does not form a flat hydrophobic interface. Instead, W45 and the proline-rich loop (predominantly P28, P29, P31) hook into the groove formed by the same region on the opposing subunit (Fig 3C, D). Proline-proline interactions are common stabilizers of protein-protein interfaces, due to their relative rigidity compared to other exposed (i.e. non-globular) sequences (Kay et al., 2000). Further, the C-C interface has a stacking interaction between W45 and P31, a type of interaction which has been shown to be relatively strong despite the absence of any classical hydrogen bonding (Biedermannova et al., 2008). Supporting the importance of this region in forming the C-C interaction, the SV of the interface-participating residues N30 and P31 are the lowest (Fig. 2D). This suggests that the pivot point of the interaction localizes to this region (Fig. 2C). One hydrogen bond pair is predicted to form between R46 and backbone carbonyl moieties of the opposing subunit (Fig. 3D). Proline-rich sequences (such as the C-C interface) are known to form weaker protein-protein interactions than those formed by globular domains. It has been suggested that such weaker binding is advantageous in situations where a uniquely-defined complex is not the priority, but rather the recruitment of proteins in such a way that subsequent interactions are more probable, e.g. during cytoskeletal rearrangements and signaling cascades (Kay et al., 2000).
Biophysical analysis of Rev C-C Interface mutants
In order to better characterize the role of the C-C interface in Rev oligomerization in vitro, we generated three single mutation variants of Rev involving residues at this interface: P28A, P31A, and W45L. Protein expression levels and purification of Rev mutants proceeded similar to that of wild-type protein. Protein folding by dialysis against the buffer containing 100 mM sodium chloride resulted in little or no aggregation – the W45L mutant exhibiting the most aggregation. Analysis by sedimentation velocity (Fig. S5) indicates that the wild-type protein is physically homogeneous with a sedimentation coefficient (s) of 4.9, corresponding to a mass of 54 kDa indicating a tetrameric association as previously also reported by Nalin at al., (1990). The P28A mutant gave a similar result; however, the overlaid velocity profile indicates < 10% fast moving species presumed to be small aggregates. The presence of aggregates is more prominent in the P31A mutant (~ 20%) but the main species still corresponds to protein with limited association (s = 4.3). The W45L mutant is distinctly different from the others, apart from the small amount of aggregated protein (~ 15%), the low s value of 1.4 corresponds to dimeric protein as separately determined by sedimentation equilibrium (Fig. S6).
Rev wild-type, Rev P28A, Rev P31A and Rev W45L were folded and polymerized under identical conditions. As expected, Rev wild-type polymerized into long, hollow filaments (Fig. 4A). Rev P28A formed filaments very similar to those formed by Rev wild-type but much shorter (Fig. 4B). Rev P31A polymerized into filaments much narrower, without a lumen and apparently less well ordered than those formed by Rev wild-type (Fig. 4C). Much of the Rev P31A was in the form of small amorphous aggregates (not shown). Rev P31A was also examined by cryo-EM but judged unsuitable for further analysis (not shown). Rev W45L did not form any filaments and most of the protein appeared to be in the form of amorphous aggregates (Fig. 4D).
Figure 4. Negative-stain electron microscopy and circular dichroism of Rev C-C interface mutants.
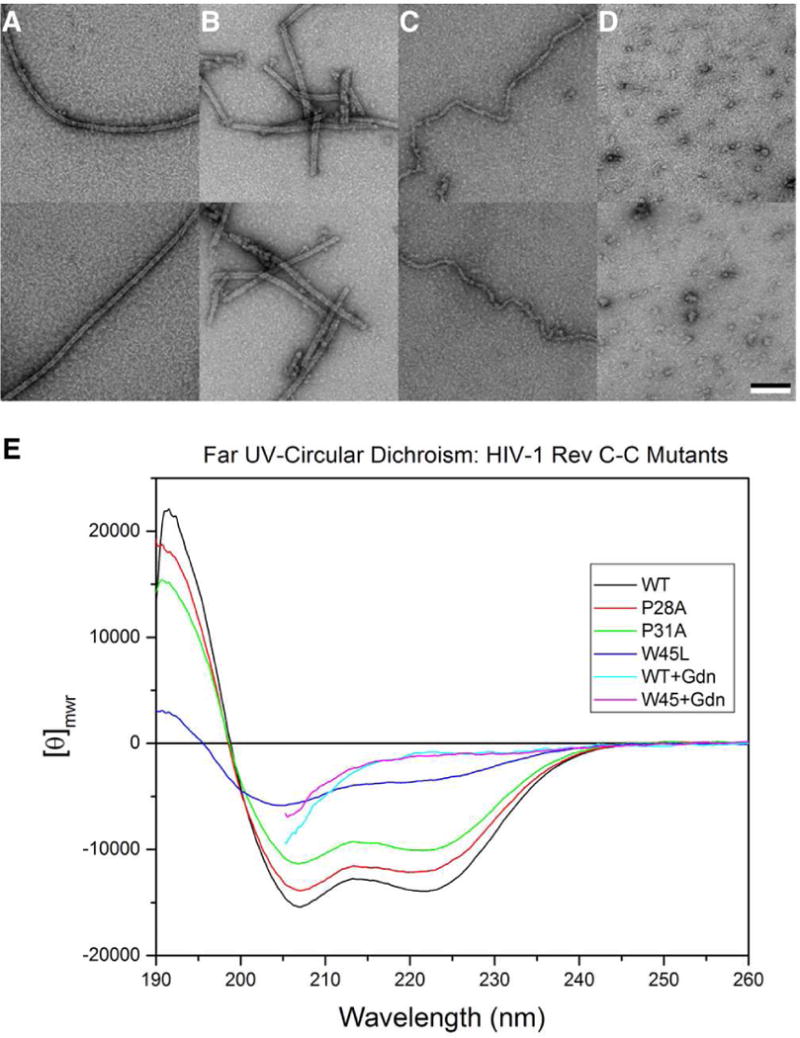
Shown are images of folded and polymerized Rev proteins.
(A) Rev wild-type, (B) Rev P28A, (C) Rev P31A, and (D) Rev W45L. Lower row, same as upper row but recorded in a different area on the grid. Bar = 100 nm.
(E) The far UV spectra shown for WT and P mutants have typical helical signatures whereas W45 mutant is conformationally perturbed but not necessarily unfolded. The unfolded spectra for WT and W45L are shown – spectra cannot extent further due to 4M Gdn-HCl. WT and two P mutants appear to associate into a tetramer. In the case of P31, especially, there is also some aggregation.
Polymorphism of Rev filaments
In order to characterize the different polymorphic structures of Rev filaments present in the less homogeneous data set, two-dimensional classification was performed on the cryo-EM images. The data were partitioned into five width classes, termed I–V (Fig. 5). The high resolution EM structure of Rev filaments presented above was low-pass filtered to 40-Å resolution and used as a starting reference for reconstruction of the images in each size class. The resulting reconstructions (Maps I–V, corresponding to the width classes) are shown as re-projections and 3D isosurfaces in Figs. 6A, B. The maps had resolutions in the range 9–11 Å; to facilitate comparison, all were filtered to 12-Å resolution. The diameters of the reconstructed filaments ranged from 11.4 to 14.4 nm. Rev dimer models were docked into these reconstructions and manually adjusted to optimize the fit, and thence to find the crossing angles at the A-A, B-B, and C-C interfaces. The appropriate helical symmetry was applied to each docked Rev dimer to generate a helical model of RevNTD for each polymorph (Fig. 6C). The variation between Rev subunit crossing angle and filament width is shown in Fig. 2A and 2C.
Figure 5. 2D classification of heterogeneous Rev filaments.

Principal components analysis and hierarchical classification were used to sort images of Rev filament segments into five bins. Shown are 2D-averages of each bin with protein density dark. The bins are designated I-V, and constitute the bins used for generation of the polymorphic reconstructions of Rev filaments in Fig. 6. For filament widths, see Figure 6.
Figure 6. Rev filament polymorphism.
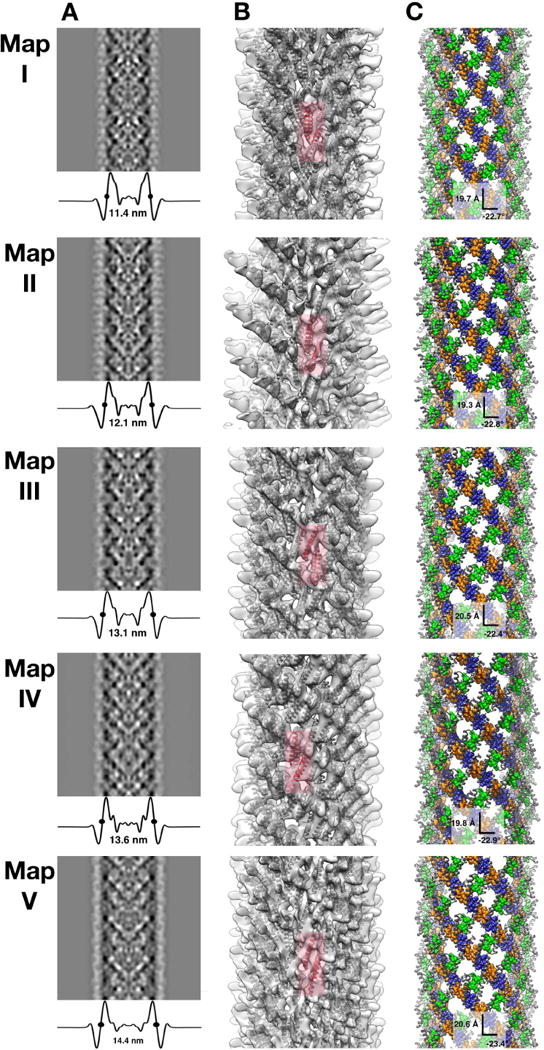
Maps I–V representing different polymorphic states of HIV Rev filaments are presented in each row. All of the maps were low pass-filtered to 12 Å resolution.
(A) Re-projections of each density map (protein is dark) with transverse profiles documenting the differences in filament diameter. In each case. the half-height diameter is marked with two dots.
(B) Isosurface representations, translucent to show docked Rev NTD subunits (gray). A representative A-A dimer is shown in red.
(C) Surface representation of each filament, with A-A, B-B and C-C interfaces colored blue, orange, and green, respectively. The helical step (rise and rotation) is labeled for each reconstruction.
HIV-1 Rev filament reconstructions offer a first glimpse of Rev CTD
After the RevNTD was docked into the cryo-EM maps, residual density was observed in each case, adjacent to the ordered termini of the docked dimers (Fig. 7). This density is on the outside of the filament, consistent with the decoration of filaments with a Fab molecule whose epitope is residues 94–116 of the CTD (Zhuang et al., 2014). Real-space difference maps were calculated between the experimental cryo-EM maps and maps generated from helically-propagated RevNTD dimers. The positive difference densities depicted in Fig. 7 are thresholded at 3σ and segmented based on their proximity to nearby termini. There is some variability in the shapes of the CTD-related densities in the different reconstructions. However, although somewhat weaker than the NTD-related density it is well above the level of background noise. We attribute these aspects to some variability in the degree of folding involved and/or some mobility in its association with the underlying filament.
Figure 7. Positive difference density attributed to Rev CTD, related to Fig. S7.
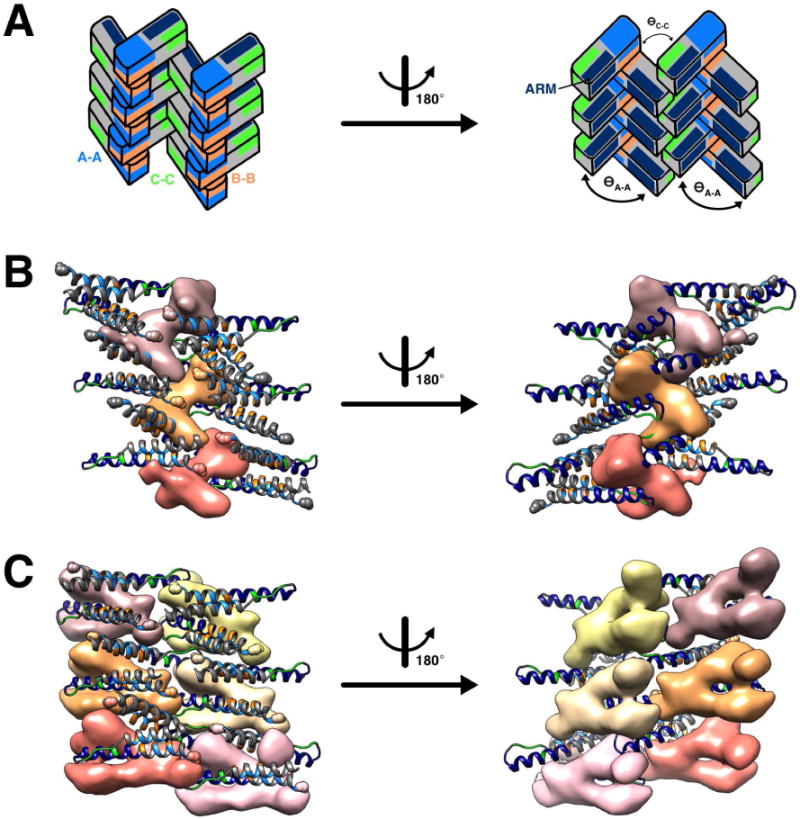
(A) Six dimers were extracted from the filament structure to show the adjacent positive difference densities. These cartoons depict the orientation of the Rev subunits in the panels below. The A-A, B-B, and C-C interfaces are shown in blue, orange, and green, respectively, and the ARM is in navy blue. The left and right cartoons are views of the filament interior and exterior, respectively.
(B) Positive difference density is shown (three asymmetric units; contoured at 3σ and with any floating density masked out) in pink, tan and salmon for filaments with acute A-A crossing angles (ӨA-A = ~100°). The density projects out between Rev subunits in a bi-lobed manner. The ordered C-termini from two Rev NTDs (small spheres) co-localize to form an apparent CTD dimerization domain.
(C) Positive difference density is shown, in a similar manner to (B), but for filaments with larger A-A crossing angles (ӨA-A = ~118°). Here, more CTD-attributed density appears to be ordered, forming a U-shaped domain from distinct Rev NTDs.
Two further arguments support the proposition that the CTD is at least partially folded in Rev filaments. Previously, we reported from Raman spectroscopy of Rev filaments that 20–25% of the polypeptide is β-sheet (Watts et al., 1998). Since the NTD is entirely 〈-helical, in filaments as in crystals, the β-sheet content must be part of the CTD. A second indication, albeit less direct, that at least part of the CTD becomes folded comes from the requirement of the CTD for filament assembly. Rev constructs 1–69 and 1–93, lacking all or approximately half of the CTD, do not form filaments (Fig. S7). While we cannot specify exactly how much of the CTD becomes folded in filaments, the case is strong that: the cryo-EM reconstructions reveal peripheral density that is attributable to the CTD; the CTD is at least partly folded; and the folded portion consists of β-sheet.
Discussion
HIV-1 Rev has proven to be a rather difficult biochemical entity to work with, due to its propensity to aggregate and form filaments in vitro. On the other hand, its oligomerization and RNA-binding properties are known to play essential roles in assembly of the Rev-RRE complex (Mann et al., 1994; Edgcomb et al., 2008; Pond et al., 2009; Daugherty et al., 2010a, 2010b). Aiming to achieve a better understanding of Rev-Rev interactions, we have used X-ray crystallography and cryo-EM to investigate helical filaments of Rev and to determine the inter-molecular interactions in Rev-containing co-crystals. Our principal findings may be summarized as follows: (1) a wide variation (~50°) in the Rev dimer crossing angles; (2) the existence of a third, heretofore unrecognized Rev-Rev oligomerization interface; and (3) partial ordering of the RevCTD in the context of Rev filaments. These results should inform studies exploring how Rev may interact with the RRE to form export-active complexes.
The Rev NTD forms an α-helical hairpin, with two opposing hydrophobic surfaces that can bind other Rev molecules. The existence of two such surfaces was inferred from a genetic and biochemical screen (Jain and Belasco, 2001), long before the crystal structure of a RevNTD dimer was first determined (DiMattia et al., 2010). The molecular details of one surface – that of the A-A interface-were explained by that structure. In it, the surface that engages in the B-B interface was occluded by Fab molecules. Another crystal structure revealed the structural determinants of the B-B interface (Daugherty et al., 2010). These two interfaces have been central elements in current models for Rev-RRE association. Our data now indicate the existence of a third Rev-Rev interaction, the C-C interface, involving residues P28, P29, and P31 (of the proline-rich loop) and W45 – all of which are highly conserved in HIV-1 isolates (Daugherty et al., 2010b). While this region has been thought to be important for maintaining the integrity of the hairpin (and hence of the A-A and B-B surfaces), we posit that it also promotes formation of the C-C interface.
The structures of Rev dimers that we have observed show a wide distribution of dimer crossing-angles (FIg. 2). From these structures we have identified the specific residues about which the dimer subunits pivot: L64 and P31 for the A-A and C-C interfaces, respectively (We do not have sufficient independent renderings of the B-B interface to do such a calculation). We have conservatively mutated these residues to Ala and tested the mutant proteins for their ability to fold properly and to polymerize into filaments. L64A retains the proper fold, but does not form filaments (Figs. S1, S2). With P31A, the helical hairpin fold may be slightly disrupted, and no filaments form (Fig. 4). The implications of these data are two-fold: (1) L64 and P31 are critical residues for the engagement of these oligomeric interactions. As such, they may be “hot spots” to focus upon for the development of anti-Rev oligomerization inhibitors, and (2) mutation of these residues to Ala may reduce the ability of adjacent Rev dimers to pivot against one another, thought to be an important adaptive feature for Rev assembly on the RRE.
To further probe the structural significance of the C-C interface, a number of limited mutations were made. We chose the highly conserved residues P28 and W45 (in addition to P31 described above) and made conservative variants with no charge alteration or increase in volume: P28A, P31A and W45L. The prolines 28 and 31 form part of the rigid poly-proline hairpin loop connecting the N and C-terminal helices of the Rev monomer. This loop is also stabilized by the stacking of W45 with P28 (Figs. 3C, D). Oligomerization and secondary structure were least perturbed by the P28A variant, compared to P31A and W45L. Presumably, increasing the conformational flexibility of residue 28 and thereby slightly tweaking the C-C interface does not compromise the fold of the Rev monomer. On the other hand, the mutation of P31 or W45 has a significant effect on Rev oligomerization and also perturbs the structure of Rev (Fig. 4E). Maintaining the structural integrity of the C-C interface appears to be crucial for mediating higher order Rev assembly. However, limiting the interpretability of these mutants are the apparent slight-to-moderate disruption of the Rev monomer fold that they appear to cause (the proximity of the C-C interface to the hinge of the helical hairpin hampered our ability to control these phenomena independently).
What is the justification for interpreting the inter-molecular interfaces observed in Rev filaments assembled in vitro in the context of Rev-RRE formation? As discussed above, our mutagenesis data support the role of the C-C interaction in higher order Rev oligomerization — a process wherein the A-A and B-B interfaces have been characterized, structurally and biochemically, in the context of Rev oligomerization both with and without the RRE. Further, the near-ubiquity with which we observe the CC interface in variously determined structures and its consistent juxtaposition to the A-A and B-B interfaces, suggest that the C-C interface is unlikely to be artifactual. In this context, it is noteworthy that Daugherty et al. (2008) found that the mutant W45A (W45 is located in ARM) encurred a 100-fold reduction in the affinity of oligomeric Rev for the RRE (despite the fact that that the W45A-ARM had been shown to bind Stem loop IIB with the same affinity as the wildtype ARM (Tan et al., 1993). This suggests that the mutation, similar to W45L, interferes with a Rev-Rev oligomerization interface rather than an RNA-binding surface.
Thus, we favor a Rev-RRE association model whereby two strands of Rev dimers connected via A-A and B-B interfaces can engage the RRE and also be linked via C-C interfaces. Adaptive dimer crossing angles would permit some subunits to engage the RNA while others form C-C contacts. This model reconciles the disparate features of the Rev-RRE assembly models that have thus far been reported (reviewed by Rausch et al. 2015). It has been proposed that the RRE adopts an “A”-shaped structure (Fang et al., 2013) and guides Rev oligomerization for RNP formation (Hope et al., 1990; Huang et al., 1991; Jain and Belasco, 2001; Malim and Cullen, 1991; Mann et al., 1994; Pollard and Malim, 1998). Remaining intact in the model are the well-characterized initial high-affinity interaction between Rev and stem loop IIB of the RRE (Cook et al., 1991; Heaphy et al., 1991; Huang et al., 1991; Kjems et al., 1991; Malim and Cullen, 1991; Iwai et al., 1992; Tiley et al., 1992) and a secondary Rev-RRE interaction with stem loop IA (Daugherty et al., 2008). The RRE is thought to present multiple Rev binding sites (downstream of the IIB and IA interactions) along Stem Loop I in an arrangement compatible with the subunit orientations defined by the Rev-Rev oligomerization interfaces (Jain and Belasco, 2001; Daugherty et al., 2008). Additionally, stem loop I appears to double back and engage the Stem loop IIABC junction via a tertiary interaction (Bai et al., 2014). (The situation is further complicated by the observation that the RRE is capable of adopting two different structures (Sherpa et al., 2015)).
Our current model hypothesizes that Rev dimers may polymerize along both legs of the “A”-shaped RRE rather than just the one, using the C-C interface as a molecular bridge between the two Rev tracks (Fig. 8A). Such an arrangement would enhance the specificity of Rev oligomerization onto the RRE, as the distance between the legs of the “A” is well-matched to that of two Rev dimers interacting laterally via a C-C interface. In this scenario, the Rev dimer crossing angles would need to be far more acute than those reported previously (120°, 140°) (DiMattia et al., 2010; Daugherty et al., 2010b) and presented herein (96°–125°). Given pot ential variability in the crossing angles of adjacently oligomerized Rev dimers, it is plausible that Rev may adopt the necessary conformations to bind the RRE in this way. In support of this, a recent crystal structure of a Rev dimer bound to a stem loop II-ABC junction had a crossing angle of ~50° (Jayarama n et al., 2014). There is also evidence that after Rev binds to the stem loop IIB and stem loop IA sites, additional Rev dimers propagate along Stem loop I via the A-A and B-B oligomerization interfaces (Daugherty et al., 2008).
Figure 8. Bilateral model for Rev-RRE RNP formation.
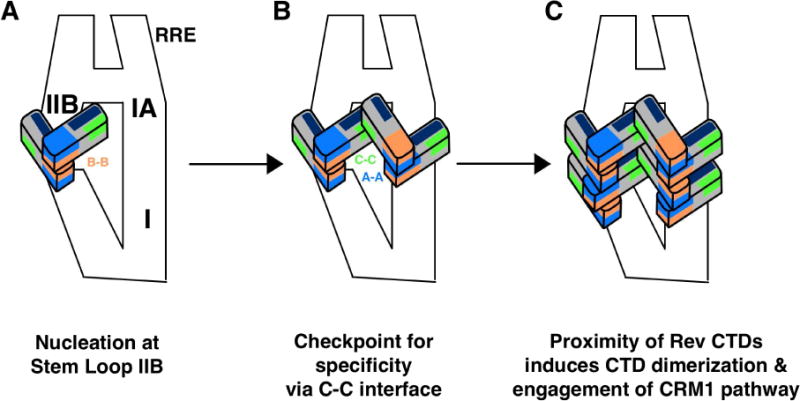
We propose an updated model for RNP formation that uses the C-C interface to help explain previously reported data.
(A) Rev nucleates at the Stem loop IIB site, whereb
(B) two additional Rev subunits are recruited across the RRE at Stem loop IA. The interaction of these two dimers via their C-C interface may be the specificity checkpoint characterized by Bai et al., 2014.
(C) Finally, additional Rev subunits may oligomerize onto the RRE along Stem loop I. The proximity of Rev subunits in this bilateral manner may induce organization and dimerization of the CTD and subsequently, engagement of the CRM1 pathway. A-A, B-B, and C-C interfaces are colored blue, orange, and green, respectively, and ARMs are colored in navy blue.
With a view to understanding the link between cooperative oligomerization of Rev on the RRE and nuclear export, the apparent disorder of the CTD has been enigmatic. Our observation of partial ordering of the CTD in the context of Rev filaments raises the possibility that some ordering may also take place when Rev associates with the RRE (Fig. 7). For example, the C-C interface may help to orient recruited Rev dimers such that laterally adjacent CTDs may engage each other (Fig. 8). Of note, a recent SHAPE-Seq and SAXS study of the Rev-RRE assembly pathway showed that after initial binding of a Rev dimer to stem loop IIb and a subsequent second dimer across the “A” at stem loop Ia, a “four-Rev” specificity checkpoint is reached prior to an induced-fit model for addition of more Rev molecules (Bai et al., 2014). We suggest that this checkpoint may be the formation of the C-C interface between the first two RRE-bound Rev dimers (Fig. 8B). This bilateral model may allow the recruitment of Rev dimers in a way that allows their CTDs to recruit a CRM1 dimer via their NES motifs for formation of the export-competent RNP (Fig. 8C) (Booth et al., 2014). Consistent with this is a report demonstrating that Rev oligomerization on the RRE functions to recruit at least two CTDs in order to mediate nuclear export of the unspliced and partially spliced viral RNAs (Hoffman et al., 2012).
In summary, our studies bring new structural insights into the oligomerization properties of HIV-1 Rev, in terms of the conformational adaptability with which Rev oligomers form, the interfaces between these assemblies, and a first view of the elusive CTD, in a partially ordered state. All of these properties are likely to be important for Rev’s ability to oligomerize onto the RRE and effect nuclear export of viral genomes. Further studies to determine the structure of the Rev-RRE nuclear export-competent complex are needed to expand our understanding of HIV-1 Rev’s key role in the nuclear export of viral mRNA.
Experimental Procedures
Protein expression and purification
Wild-type HIV-1 Rev was purified as previously described (Wingfield et al., 1991). Genes corresponding to Rev P28A, Rev P31A and Rev W45L were synthesized by GeneScript. Protein expression, purification, folding and polymerization were essentially as described previously for the wild-type protein. The purified proteins from the S-Sepharose column (in 2 M urea and ~ 1 M sodium chloride) were dialyzed either against 50 mM sodium phosphate pH 6.8, 100 mM sodium chloride and 1 mM DTT for analytical centrifugation, or against buffers promoting filament formation (first 50 mM sodium phosphate pH 7.0, 600 mM ammonium sulfate, 150 mM sodium chloride, 50 mM sodium citrate, 1 mM dithiothreitol, and 1 mM EDTA and then 50 mM sodium phosphate pH 7.0, 150 mM sodium chloride, 50 mM sodium citrate, 1 mM dithiothreitol, and 1 mM EDTA) for electron microscopy.
scFv-Rev complexes were prepared by mixing scFv with several-fold molar excess of wild-type Rev, followed by purification by Ni-sepharose chromatography, utilizing the C-terminal His tag present on the heavy chain of the scFv. As final polishing of the scFv-Rev complexes could not be achieved by gel filtration due to the strong adsorption of Rev to gel-filtration matrices, the sample was centrifuged at 100,000 g for 2 h to remove any protein aggregates. Final yields of the complexes were typically 0.5–1.0 mL at 5.0 mg/mL.
Crystallization and diffraction data collection
Purified scFv-Rev was concentrated to 5.0 mg mL−1 and crystallization trials were set up at 21° C in sitting drops containing 100 nL protein and 100 nL precipitant solution equilibrated against 95 uL reservoirs in 96-well plates (Walter et al., 2005). Crystals of scFv-Rev were initially grown in 20% PEG 3350, a variety of salts (200 mM sodium sulfate, sodium bromide, or ammonium phosphate dibasic), and pH ranging from 6.5–8.5. Optimized crystals based on the initial hit condition – varying protein-to-precipitant drop ratios and reservoir concentrations – were cryoprotected by a quick pass through reservoir solution supplemented with 30% v/v ethylene glycol before flash cooling in a cold (100 K) stream of nitrogen gas.
Five unique diffraction data sets of scFv-Rev were recorded, each from a single crystal. A total of 1800 frames were collected for each crystal with an oscillation angle of 0.1° on a Pilatus P6M detector at the Diamond Light Source, Beamline I24 (microfocus; Didcot, UK). The crystal lattices for each data set belonged to a distinct space group. The crystal that diffracted to the highest resolution was grown in 20% PEG 3350 and 200 mM sodium sulfate, from which diffraction data were collected to 2.3 Å resolution and subsequently processed using XDS and XSCALE (Kabsch, 2010).
Determination and refinement of crystal structures
Initial phase information for the scFv-Rev crystal structures was obtained by molecular replacement. Automated searches were done with Phaser (McCoy, 2007), using a search model of one variable Fab fragment bound to one Rev monomer, extracted from the original HIV-1 Rev structure determined in complex with Fabs (PDB 2X7L; DiMattia et al., 2010). Different reservoir conditions gave rise to crystals belonging to four different space groups. The molecular replacement solutions were rigid body-refined in Phenix (Adams et al., 2010) with the immunoglobulin domains and Rev treated independently. Positional refinement and group B-factor refinement (one group per residue) were carried out iteratively in both Phenix and AutoBuster (Blanc et al., 2004). The Molprobity server (Davis et al., 2007) and the validation tools in Coot informed the quality of the structure refinement process. Refinement statistics are given in Table 1, and final refined coordinates and structure factors have been deposited with the PDB with accession codes 5DHV, 5DHX, 5DHY, and 5DHZ.
Negative-stain electron microscopy
Following folding/polymerization the Rev proteins were diluted to 0.2 mg/ml with the final dialysis buffer and applied to carbon-coated grids glow-discharged (Fischione) immediately prior to use in a plasma of 25% oxygen, 75% argon. Grids were rinsed twice with distilled water and negatively stained with 1% uranyl acetate. In the case of Rev W45L, which did not polymerize, the protein was also diluted to 0.05 mg/ml and the grids were negatively stained with 1% uranyl acetate, 0.5 mM β-octyl glucoside. Micrographs were recorded on a CCD at 35,000x nominal magnification with an FEI CM120 electron microscope.
Cryo-electron microscopy
Suspensions of Rev filaments were applied to C-flat holey carbon grids, blotted to a thin film, and vitrified by plunge-freezing in liquid ethane. In earlier experiments, specimens were recorded on Kodak SO-163 photographic films observed with a CM200-FEG electron microscope (Philips/FEI) at an acceleration voltage of 120 kV (see Watts et al., 1998 for additional details). These data encompass the more heterogeneous filaments that were used for image classification and analysis of the filament polymorphism. Separately, a higher resolution, more homogeneous data set was collected with a Polara electron microscope (FEI) operated at an acceleration voltage of 300 kV. These data were recorded in low dose mode using acquisition package SerialEM (Mastronarde, 2005) with a K2 Summit direct electron detector (Gatan) operated in super-resolution movie mode with dose-fractionation. The nominal magnification was 20,000×, which corresponds to a calibrated pixel size of 2.02 Å/physical pixel, or 1.01 Å/pixel with super-resolution mode. Super-resolution mode was used to obtain images with a larger field of view and consequently longer, uninterrupted filaments. The parallel beam intensity was set to be 10 e−/pixel/s for proper electron counting. During a 9.9 s exposure, 33 300-ms frames were collected, resulting in a total electron dose of 25 e−/Å2. The frames were aligned using the program UCSF drift correction (Li et al., 2013). After motion correction, frames 3–33 were averaged to yield 93 processed micrographs that were subsequently binned two-fold, giving 2.02 Å/pixel.
Image Processing
The BSOFT software suite was used for all steps of image processing (Heymann et al., 2007). The defocus values were estimated using the program bctf and found to range from −0.99 to −3.09 m. Filaments were manually splined using bshow. Initially, boxed segments with dimensions 512 × 512 pixels were extracted from the filaments with 50-pixel offsets between them. Each segment was normalized and background-subtracted, phase-flipped, padded with zeroes to size 4096 × 4096 pixels, and the modulus squared of its Fourier transform was computed. The resulting spectra were averaged, showing an unambiguous pattern of layer-lines with reflections at (55 Å) −1, (34 Å) −1, and a meridional reflection at (21 Å)−1. Therefore, the axial rise per asymmetric unit in the filament was 21 Å and indexing of the layer lines yielded an azimuthal rotation per subunit of 22° (see Fig. 1C). The Bessel orders of the observed layer lines were multiples of 6, suggestive of C6 rotational symmetry.
Once the helical symmetry parameters were determined, the filaments were again boxed into overlapping segments using bshow and bfil with dimensions 300 × 300 pixels (303 × 303 Å). These images were CTF-corrected by phase-flipping in the initial stages and in later stages by phase and baseline compensation to enhance high-frequency components. An initial reference map was generated as a solid cylinder with diameter roughly equal to that of the filaments. Origins and orientations were determined by projection matching using borient in a restrained global search with 2–10° steps, using side-view projections only (plus v iews up to 10° away from equator) and then refined to a final step size of 0.5°, using brefine. Two de nsity maps were reconstructed from half data sets, using breconstruct, and then masked with a tight soft mask of the helical shell. These maps were then used to estimate the resolution by Fourier shell correlation at a cutoff of 0.3, using bresolve. The two maps were then averaged. At the end of each iteration of refinement, a real-space search of updated helical parameters was performed using bhelix, and the resulting helical parameters were used to symmetrize the reference map for the next iteration, as described in the IHRSR procedure (Egelman, 2000). This process was repeated until convergence of helical parameters was reached.
To calculate difference maps, the maps were adjusted to the same reciprocal-space amplitudes by use of bampweigh and then low-pass-filtered to 12 Å resolution and rescaled to the same real-space intensity average (0) and standard deviation (1), using bfilter. Finally, the adjusted maps were subtracted with bop to generate the difference map.
The more heterogeneous data collected on the CM-200 was similarly boxed, ctf parameters determined, and phases flipped as described above. However, the segments were classified by singular value decomposition (SVD), followed by principal components analysis and hierarchical classification using EMAN1 (Ludtke, et al., 1999). The final reconstruction above was filtered to 40 Å resolution and used as an initial reference for each 2D class in BSOFT, and the reconstruction process was carried out as described above for the initial map.
Structural analysis
The crossing angles of the Rev dimer structures were determined by calculating the dot product of the vectors that align with α2 helix (containing RRE-associating motif). Rev models were docked into the EM reconstructions using UCSF Chimera. In the higher-resolution map, the Fit in Map command was sufficient for docking of individual Rev subunits (one polypeptide at a time). For fitting of the lower-resolution polymorphic reconstructs, structural constraints were imposed based on the interacting residues in the crystal structures of the A-A, B-B, and C-C interfaces.
Analytical Ultracentrifugation
All samples were concentrated to about 1 mg/ml (~ 0.6 OD at 280 nm) except the W45L mutant, which was at 1.6 mg/ml (0.3 OD at 280 nm). A Beckman Optima XLI centrifuge with absorption optics, an An-60 Ti rotor, and standard double-sector centerpiece cells were used. For sedimentation velocity analyses samples were run for 3 h at 40,000 rpm at 10°C with absorbance scans every 8 min. Data analysis was done using DCDT+ 2.4.3 (Philo, 2006). For sedimentation equilibrium of Rev W45L, 18,000 rpm at 10°C was used with data sampling to 16 h followed by 45,000 rpm for 4 h for baseline correction.
Circular Dichroism
Spectra were collected at 20°C using a Jasco-715 spectrometer. The samples were (~ 1mg/ml) in 50 mM sodium phosphate, pH 6.8 containing 0.1M NaCl and 1 mM DTT. For each sample, four accumulations were collected between 180 nm and 260 nm, using a 0.02-cm path-length cell. Scanning was done at a speed of 20 nm/min with a 0.1-nm data pitch. After baseline subtraction, the raw data were converted to molar ellipticities and smoothed with Jasco software.
Supplementary Material
Highlights.
Rev NTD α-helical hairpins dimerize with widely varying crossing-angles
The unfolded CTDs become partially ordered when Rev assembles into helical filaments
Cryo-EM of filaments and crystal structures reveal a novel inter-molecular interface
This interface and variations in crossing-angle may help organize Rev-RRE complexes
Acknowledgments
We thank Stephen J. Stahl, Ira Palmer, and Joshua Kaufman (NIH/NIAMS) for support with preparation of reagents and Dennis Winkler (NIH/NIAMS) for installation and calibration of SerialEM, and the staff at Diamond Light Source (Didcot, UK) beamlines I02 and I24 and European Synchrotron Radiation Facility (Grenoble, France) beamline ED23-EH2 for support in data collection. This work was supported in part by the Intramural Research Programs of the National Institute for Arthritis and Skin Diseases, the National Institutes of Health (NIH) Intramural Targeted Antiviral Program, and the NIH-Oxford Scholars Program. D.I.S. is supported by the UK Medical Research Council (G100099) and J.M.G. by SPINE2COMPLEXES (LSHGST-2006-031220). The Wellcome Trust is acknowledged for providing administrative support (Grant 075491/Z/09).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions: P.T.W., J.M.G., D.I.S., A.C.S. designed research; M.A.D., N.R.W., N.C., R.H. performed research; J.B.H. contributed new software; M.A.D., N.R.W., J.B.H., P.T.W., J.M.G., D.I.S., and A.C.S. analyzed data; and M.A.D., N.R.W., P.T.W., J.M.G., D.I.S. and A.C.S. wrote the paper.
Accession Numbers: The PDB accession numbers for the atomic coordinates and structure factors for the scFV-Rev crystal structures are 5DHV, 5DHX, 5DHY, and 5DHZ. The EMDB accession number for the 8.3-Å cryo-EM helical reconstruction of a Rev filament is 6439.
References
- Bai Y, Tambe A, Zhou K, Doudna JA. RNA-guided assembly of Rev-RRE nuclear export complexes. eLife. 2014;3:e03656. doi: 10.7554/eLife.03656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battiste JL, Mao H, Rao NS, Tan R, Muhandiram DR, Kay LE, Frankel AD, Williamson JR. Alpha helix-RNA major groove recognition in an HIV-1 rev peptide-RRE RNA complex. Science. 1996;273:1547–1551. doi: 10.1126/science.273.5281.1547. [DOI] [PubMed] [Google Scholar]
- Biedermannova L, E Riley K, Berka K, Hobza P, Vondrasek J. Another role of proline: stabilizing interactions in proteins and protein complexes concerning proline and tryptophane. Phys Chem Chem Phys. 2008;10:6350–9. doi: 10.1039/b805087b. [DOI] [PubMed] [Google Scholar]
- Blanc E, Roversi P, Vonrhein C, Flensburg C, Lea SM, Bricogne G. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallographica Section D: Biol Cryst. 2004;60:2210–21. doi: 10.1107/S0907444904016427. [DOI] [PubMed] [Google Scholar]
- Booth DS, Cheng Y, Frankel AD. The export receptor Crm1 forms a dimer to promote nuclear export of HIV RNA. eLife. 2014;3:e04121. doi: 10.7554/eLife.04121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousquet-Antonelli C, Presutti C, Tollervey D. Identification of a regulated pathway for nuclear pre-mRNA turnover. Cell. 2000;102:765–775. doi: 10.1016/s0092-8674(00)00065-9. [DOI] [PubMed] [Google Scholar]
- Cook KS, Fisk GJ, Hauber J, Usman N, Daly TJ, Rusche JR. Characterization of HIV-1 REV protein: binding stoichiometry and minimal RNA substrate. Nucl Acids Res. 1991;19:1577–1583. doi: 10.1093/nar/19.7.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen BR. Nuclear mRNA export: insights from virology. Trends Biochem Sci. 2003;28:419–424. doi: 10.1016/S0968-0004(03)00142-7. [DOI] [PubMed] [Google Scholar]
- Daly TJ, Cook KS, Gray GS, Malone TE, Rusche JR. Specific binding of HIV-1 recombinant Rev protein to the Rev-responsive element in vitro. Nature. 1989;342:816–9. doi: 10.1038/342816a0. [DOI] [PubMed] [Google Scholar]
- Daly TJ, Doten RC, Rennert P, Auer M, Jaksche H, Donner A, Fisk G, Rusche JR. Biochemical characterization of binding of multiple HIV-1 Rev monomeric proteins to the Rev responsive element. Biochemistry. 1993;39:10497–505. doi: 10.1021/bi00090a028. [DOI] [PubMed] [Google Scholar]
- Daugherty MD, D’Orso I, Frankel AD. A solution to limited genomic capacity: using adaptable binding surfaces to assemble the functional HIV Rev oligomer on RNA. Mol Cell. 2008;31:824–34. doi: 10.1016/j.molcel.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty MD, Booth DS, Jayaraman B, Cheng Y, Frankel AD. HIV Rev response element directs assembly of the Rev homooligomer into discrete asymmetric complexes. Proc Nat’l Acad Sci USA. 2010a;107:12481–6. doi: 10.1073/pnas.1007022107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty MD, Liu B, Frankel AD. Structural basis for cooperative RNA binding and export complex assembly by HIV Rev. Nature Struct Mol Biol. 2010b;17:1337–42. doi: 10.1038/nsmb.1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. Molprobity: all-atom contacts and structure validation for proteins and nucleic acids. Nucl Acids Res. 2007;35:W375–83. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMattia MA, Watts NR, Stahl SJ, Rader C, Wingfield PT, Stuart DI, Steven AC, Grimes JM. Implications of the HIV-1 Rev dimer structure at 3.2 Å resolution for multimeric binding to the Rev response element. Proc Nat’l Acad Sci USA. 2010;107:5810–5814. doi: 10.1073/pnas.0914946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgcomb SP, Aschrafi A, Kompfner E, Williamson JR, Gerace L, Hennig M. Protein structure and oligomerization are important for the formation of export-competent HIV-1 Rev-RRE complexes. Prot Sci. 2008;17:420–430. doi: 10.1110/ps.073246608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelman EH. A robust algorithm for the reconstruction of helical filaments using single-particle methods. Ultramicroscopy. 2000;85:225–234. doi: 10.1016/s0304-3991(00)00062-0. [DOI] [PubMed] [Google Scholar]
- Fang X, Wang J, O’Carroll IP, Mitchell M, Zuo X, Wang Y, Yu P, Liu Y, Rausch JW, Dyba MA, Kjems J, Schwieters CD, Seifert S, Winans RE, Watts NR, Stahl SJ, Wingfield PT, Byrd Ra, Le Grice SF, Rein A, Wang YX. An unusual topological structure of the HIV-1 Rev response element. Cell. 2013;155:594–605. doi: 10.1016/j.cell.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer U, Huber J, Boelens WC, Mattaj IW, Lührmann R. The HIV-1 Rev activation domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell. 1995;82:475–483. doi: 10.1016/0092-8674(95)90436-0. [DOI] [PubMed] [Google Scholar]
- Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90:1051–1060. doi: 10.1016/s0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- Güttler T, Madl T, Neumann P, Deichsel D, Corsini L, Monecke T, Ficner R, Sattler M, Gorlich D. NES consensus redefined by structures of PKI-type and Rev-type nuclear export signals bound to CRM1. Nature Struct Mol Biol. 2010;17:1367–1376. doi: 10.1038/nsmb.1931. [DOI] [PubMed] [Google Scholar]
- Heaphy S, Finch JT, Gait MJ, Karn J, Singh M. Human immunodeficiency virus type 1 regulator of virion expression, rev, forms nucleoprotein filaments after binding to a purine-rich “bubble” located within the rev-responsive region of viral mRNAs. Proc Nat’l Acad Sci USA. 1991;88:7366–7370. doi: 10.1073/pnas.88.16.7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heymann JB, Belnap DM. Bsoft: Image processing and molecular modeling for electron microscopy. J Struct Biol. 2007;157:3–18. doi: 10.1016/j.jsb.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Hoffman D, Schwarck D, Banning C, Brenner M, Mariyanna L, Krepstakies M, Schindler M, Millar DP, Hauber J. Formation of Trans-Activation Competent HIV-1 Rev:RRE Complexes Requires the Recruitment of Multiple Protein Activation Domains. PLoS ONE. 2012;7:e38305. doi: 10.1371/journal.pone.0038305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope TJ, McDonald D, Huang XJ, Low J, Parslow TG. Mutational analysis of the human immunodeficiency virus type 1 Rev transactivator: essential residues near the amino terminus. J Virol. 1990;64:5360–5366. doi: 10.1128/jvi.64.11.5360-5366.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Hope TJ, Bond BL, McDonald D, Grahl K, Parslow TG. Minimal Rev-response element for Type 1 human immunodeficiency virus. J Virol. 1991;65:2131–2134. doi: 10.1128/jvi.65.4.2131-2134.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai S, Pritchard C, Mann DA, Karn J, Gait MJ. Recognition of the high affinity binding site in rev-response element RNA by the human immunodeficiency virus type-1 rev protein. Nucl Acids Res. 1992;20:6465–6472. doi: 10.1093/nar/20.24.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain C, Belasco JG. Structural model for the cooperative assembly of HIV-1 Rev multimers on the RRE as deduced from analysis of assembly-defective mutants. Mol Cell. 2001;7:603–14. doi: 10.1016/s1097-2765(01)00207-6. [DOI] [PubMed] [Google Scholar]
- Jayaraman B, Crosby DC, Homer C, Ribeiro I, Mavor D, Frankel AD. RNA-directed remodelling of the HIV-1 Rev protein orchestrates assembly of the Rev-Rev response element complex. eLife. 2014;3:e04120. doi: 10.7554/eLife.04120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Cryst D: Biol Cryst. 2010;66:125–32. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay BK, Williamson MP, Sudol M. The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000;14:231–241. [PubMed] [Google Scholar]
- Kjems J, Sharp PA. The basic domain of Rev from human immunodeficiency virus type 1 specifically blocks the entry of U4/U6.U5 small nuclear ribonucleoprotein in spliceosome assembly. J Virol. 1993;67:4769–4776. doi: 10.1128/jvi.67.8.4769-4776.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nature Meth. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo MJ, Reed R. Splicing is required for rapid and efficient mRNA export in metazoans. Proc Nat’l Acad Sci USA. 1999;96:14937–14942. doi: 10.1073/pnas.96.26.14937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- Malim MH, Cullen BR. HIV-1 structural gene expression requires the binding of multiple Rev monomers to the viral RRE: implications for HIV-1 latency. Cell. 1991;65:241–248. doi: 10.1016/0092-8674(91)90158-u. [DOI] [PubMed] [Google Scholar]
- Malim MH, Hauber J, Le SY, Maizel JV, Cullen BR. The HIV-1 rev trans-activator acts through a structured target sequence to activate nuclear export of unspliced viral mRNA. Nature. 1989;338:254–257. doi: 10.1038/338254a0. [DOI] [PubMed] [Google Scholar]
- Mann DA, Mikaelian I, Zemmel RW, Green SM, Lowe AD, Kimura T, Singh M, Butler PJ, Gait MJ, Karn J. A molecular rheostat: Co-operative rev binding to stem I of the Rev-response element modulates human immunodeficiency virus type-1 late gene expression. J Mol Biol. 1994;241:193–207. doi: 10.1006/jmbi.1994.1488. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Cryst D: Biol Cryst. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalin CM, Purcell RD, Antelman D, Mueller D, Tomchak L, Wegrzynski B, McCartney E, Toome V, Kramer R, HSuO M-C. Purification and characterization of recombinant protein of human immunodeficiency virus type 1. Proc Nat’l Acad Sci USA. 1990;87:7593–7597. doi: 10.1073/pnas.87.19.7593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura M, Inose H, Masuda S. RNA export through the NPC in eukayotes. Genes. 2015;6:124–49. doi: 10.3390/genes6010124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philo JS. Improved methods for fitting sedimentation coefficient distributions derived by time-derivative techniques. Anal Biochem. 2006;354:238–246. doi: 10.1016/j.ab.2006.04.053. [DOI] [PubMed] [Google Scholar]
- Pollard VW, Malim MH. The HIV-1 Rev Protein. Ann Rev Microbiol. 1998;52:491–532. doi: 10.1146/annurev.micro.52.1.491. [DOI] [PubMed] [Google Scholar]
- Pond SJ, Ridgeway WK, Robertson R, Wang J, Millar DP. HIV-1 Rev protein assembles on viral RNA one molecule at a time. Proc Nat’l Acad Sci USA. 2009;106:1404–1408. doi: 10.1073/pnas.0807388106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch JW, Le Grice SFJ. HIV Rev Assembly on the Rev Response Element (RRE): A Structural Perspective. Viruses. 2015;7:3053–3075. doi: 10.3390/v7062760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherpa S, Rausch JW, LeGrice SFJ, Hammaskjold M-L, Rekosh D. The HIV-1 Rev response element (RRE) adopts alternative conformations that promote different rates of virus replication. Nucl Acids Res. 2015;43:4676–4686. doi: 10.1093/nar/gkv313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl SJ, Watts NR, Rader C, DiMattia MA, Mage RG, Palmer I, Kaufman JD, Grimes JM, Stuart DI, Steven AC, Wingfield PT. Generation and characterization of a chimeric rabbit/human Fab for cocrystallization of HIV-1 Rev. J Mol Biol. 2010;397:697–708. doi: 10.1016/j.jmb.2010.01.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan R, Chen L, Buettner JA, Hudson D, Frankel AD. RNA Recognition by an Isolated alpha Helix. Cell. 1993;73:1031–1040. doi: 10.1016/0092-8674(93)90280-4. [DOI] [PubMed] [Google Scholar]
- Tiley LS, Malim MH, Twary HK, Stockley PG, Cullen BR. Identification of a high-affinity RNA-binding site for the human immunodeficiency virus type 1 Rev protein. Proc Nat’l Acad Sci USA. 1992;89:758–762. doi: 10.1073/pnas.89.2.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter TS, Diprose JM, Mayo CJ, Siebold C, Pickford MG, Carter L, Sutton GC, Berrow NS, Brown J, Berry IM, Stewart-Jones GB, Grimes JM, Stammers DK, Esnouf RM, Jones EY, Owens RJ, Stuart DI, Harlos K. A procedure for setting up high-throughput nanolitre crystallization experiments. Crystallization workflow for initial screening, automated storage, imaging, and optimization. Acta Cryst D: Biol Cryst. 2005;61:651–7. doi: 10.1107/S0907444905007808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts NR, Misra M, Wingfield PT, Stahl SJ, Cheng N, Trus BL, Steven AC, Williams RW. Three-dimensional structure of HIV-1 Rev protein filaments. J Struct Biol. 1998;121:41–52. doi: 10.1006/jsbi.1998.3964. [DOI] [PubMed] [Google Scholar]
- Wingfield PT, Stahl SJ, Payton MA, Venkatesan S, Misra M, Steven AC. HIV-1 Rev expressed in recombinant Escherichia coli: purification, polymerization, and conformational properties. Biochemistry. 1991;30:7527–34. doi: 10.1021/bi00244a023. [DOI] [PubMed] [Google Scholar]
- Yedavalli VS, Neuveut C, Chi YH, Kleiman L, Jeang KT. Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell. 2004;119:381–392. doi: 10.1016/j.cell.2004.09.029. [DOI] [PubMed] [Google Scholar]
- Zhuang X, Stahl SJ, Watts NR, DiMattia MA, Steven AC, Wingfield PT. A cell-penetrating antibody fragment against HIV-1 Rev has high antiviral activity: characterization of the paratope. J Biol Chem. 2014;29:20222–33. doi: 10.1074/jbc.M114.581090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Luo MJ, Straesser K, Katahira J, Hurt E, Reed R. The protein Aly links pre-messenger-RNA splicing to nuclear export in metazoans. Nature. 2000;407:401–405. doi: 10.1038/35030160. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.