

SUMMARY
Persistent accumulation of misfolded proteins causes endoplasmic reticulum (ER) stress, a prominent feature in many neurodegenerative diseases including amyotrophic lateral sclerosis (ALS). Here we report the identification of homeodomain interacting protein kinase 2 (HIPK2) as the essential link that promotes ER stress-induced cell death via the IRE1α-ASK1-JNK pathway. ER stress, induced by tunicamycin or SOD1G93A, activates HIPK2 by phosphorylating highly conserved serine and threonine residues (S359/T360) within the activation loop of the HIPK2 kinase domain. In SOD1G93A mice, loss of HIPK2 delays disease onset, reduces cell death in spinal motor neurons, mitigates glial pathology, and improves survival. Remarkably, HIPK2 activation positively correlates with TDP-43 proteinopathy in NEFH-tTA/tetO-hTDP-43ΔNLS mice, sporadic ALS and C9ORF72 ALS, and blocking HIPK2 kinase activity protects motor neurons from TDP-43 cytotoxicity. These results reveal a previously unrecognized role of HIPK2 activation in ER stress-mediated neurodegeneration, and its potential role as a biomarker and therapeutic target for ALS.
INTRODUCTION
An important hallmark of neurodegenerative diseases is the accumulation of abnormally folded proteins that lead to neuronal dysfunction and cell death. Accumulation of misfolded proteins in the endoplasmic reticulum (ER) activates highly conserved signal transduction pathways collectively known as the unfolded protein response (UPR). Under ER stress, UPR up-regulates genes to assist in protein folding and degradation, however, prolonged ER stress can activate irreversible signaling pathways leading to cell death (Kim et al., 2008; Oakes and Papa, 2015). Several lines of evidence indicate that ER stress plays a critical role in the pathogenesis of amyotrophic lateral sclerosis (ALS), an adult-onset neurodegenerative disease that affects upper and lower motor neurons (Boillee et al., 2006; Pasinelli and Brown, 2006). The discovery of genetic mutations in familial ALS (FALS) reveals novel mechanisms that are causally linked to disease pathogenesis. Among the “ALS disease genes”, four account for the majority of FALS cases, including SOD1, TDP-43 (TARDBP or TAR-DNA-binding protein-43), FUS/TLS (fused in sarcoma/translocation in liposarcoma or FUS), and GGGGCC hexanucleotide expansions in C9ORF72 gene. Characterizations of these mutations suggest that dysfunctions in RNA metabolism and protein homeostasis via the ubiquitin-proteasome pathways might contribute to pathogenesis and disease progression in ALS (Lee et al., 2012; Ling et al., 2013).
Several studies have investigated ER stress in relationship to neurodegeneration in ALS. For instance, misfolded mutant SOD1 protein has a high propensity to form insoluble aggregates in neuronal cytoplasm, where they trigger ER stress, disrupt axonal transport and increase mitochondrial damage (Atkin et al., 2006; Bruijn et al., 1998; Israelson et al., 2010; Kiskinis et al., 2014). Furthermore, emerging evidence indicates that the hexanucleotide expansion in C9ORF72 gene can generate non-ATG-initiated translation (RAN) dipeptides, which increases ER stress-induced cell death (Almeida et al., 2013; Ash et al., 2013; Donnelly et al., 2013; Mori et al., 2013). Finally, all C9ORF72-ALS (C9-ALS) patients and >95% SALS patients show TDP-43 proteinopathy characterized by cytoplasmic accumulations of ubiquitinated and phosphorylated TDP-43 protein aggregates in neurons and glia (Arai et al., 2006; Neumann et al., 2006). Consistent with these results, ER stress has been reported in the spinal cord tissues of SALS patients (Atkin et al., 2008). These results underscore the critical role of ER stress activation as an important mechanism that contributes to pathogenesis and disease progression in FALS and SALS.
Despite these findings, it remains unclear how misfolded proteins promote neurodegeneration. There are three major downstream targets of ER stress response, including two protein kinases IRE1α (inositol requiring enzyme 1α) and PERK (eukaryotic translation initiation factor 2 alpha kinase), and a transmembrane transcription factor ATF6 (Kim et al., 2008; Ron and Walter, 2007). The presence of misfolded proteins activates these downstream targets to promote adaptive ER stress response and increase protein folding capacity by transcriptionally up-regulating chaperones to unfold the misfolded proteins and restore ER homeostasis. However, when the accumulation of misfolded proteins becomes chronic and overwhelming, ER stress can activate destructive signaling pathways to promote cell death (Lin et al., 2007). Several groups have shown that activation of PERK correlates with disease progression in the mutant SOD1 model of ALS (Hetz et al., 2009; Kikuchi et al., 2006; Saxena et al., 2009; Wang et al., 2011). For instance, activation of PERK has been identified in a subset of motor neurons that are vulnerable to misfolded SOD1G93A proteins (Saxena et al., 2009). Interestingly, haploinsufficiency of PERK causes an accelerated disease onset and shortens lifespan in SOD1G85R mice, suggesting that PERK activation may protect against SOD1G85R-induced neurodegeneration (Wang et al., 2011).
Unlike PERK, much less is known about the role of IRE1α in ER stress-mediated neurodegeneration. IRE1α is an ER transmembrane protein that possesses both kinase and endoribonuclease (RNase) properties. Upon ER stress, the luminal domain of IRE1α detects the presence of unfolded proteins, causes IRE1α to oligomerize, and trans-phosphorylates the kinase domain of IRE1α. IRE1α kinase activates its RNase activity to promote X-Binding Protein (Xbp1) mRNA splicing and up-regulation of genes important for protein quality control and microRNAs that regulate apoptosis (Calfon et al., 2002; Han et al., 2009; Yoshida et al., 2001). In addition, activation of IRE1α can promote ASK1 (apoptosis signal-regulating kinase 1) and JNK-dependent cell death (Nishitoh et al., 2008; Ron and Walter, 2007). However, the mechanism that connects these IRE1α downstream targets to neurodegeneration remains poorly characterized.
Here we show that homeodomain interacting protein kinase 2 (HIPK2), previously implicated in developmental programmed cell death (Wiggins et al., 2004; Zhang et al., 2007), is an essential link that connects the IRE1α pathway to JNK activation and neuronal cell death during persistent and irreversible ER stress. HIPK2 activation precedes disease onset in SOD1G93A and NEFH-tTA/tetO-hTDP-43ΔNLS mice, and loss of HIPK2 reduces apoptosis in spinal motor neurons, mitigates glial pathology, and improves survival in SOD1G93A mice. Furthermore, HIPK2 activation positively correlates with TDP-43 proteinopathy in SALS and C9-ALS, while blocking HIPK2 kinase activity protects motor neurons from TDP-43 cytotoxicity. These results reveal a previously unrecognized role of HIPK2 in protein misfolding-mediated neurodegeneration and its potential role as a biomarker and therapeutic target for ALS.
RESULTS
HIPK2 is required for JNK activation during tunicamycin-induced ER stress
To characterize the role of HIPK2 in cell death, we treated wild type and Hipk2−/− mouse embryonic fibroblasts (MEFs) with several stress conditions, including staurosporine, anisomycin, and tunicamycin, which induced cell death either by blocking global protein synthesis or by inducing ER stress via blocking protein glycosylation and transport in the Golgi apparatus. Whereas wild type and Hipk2−/− MEFs were equally vulnerable to cell death induced by protein synthesis inhibitors staurosporine and anisomycin (Figure S1A–B), Hipk2−/− MEFs showed considerable resistance to tunicamycin-induced ER stress-related cell death detected by either the tetrazolium dye MTT or by the release of lactate dehydrogenase (LDH)(Figure S1C–D). Similarly, Hipk2−/− MEFs were resistant to ER stress induced by brefeldin A (BFA), which blocks Golgi trafficking by inhibiting small GTP-binding protein ADP ribosylation factor (Figure S1E). Finally, HEK293 cells treated with Hipk2 siRNA, but not control siRNA, also showed resistance to tunicamycin toxicity, whereas over-expression of HIPK2 restored the sensitivity to tunicamycin (Figure S1F).
To determine if loss of HIPK2 would similarly protect neurons from ER stress-induced cell death, we adopted an in vivo ER stress model using a single intraperitoneal injection of tunicamycin (1 mg/kg) in 3-month-old wild type and Hipk2−/− mice. Four days after the injection, brains and spinal cords from these mice were collected for histological analyses. Consistent with the results in MEFs, tunicamycin induced a marked increase of cell death in cortical neurons in wild type mice, detected by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), progressive increase in activated caspase 3, and ~30% reduction in NeuN+ cortical neurons (Figure 1A–F, M–N, Y). In contrast, the same treatment caused fewer apoptotic neurons in Hipk2−/− mutants, with no reduction in NeuN+ neuron number (Figure 1G–N). The protective effects of HIPK2 loss-of-function could also be detected in the spinal motor neurons, where Hipk2−/− mutants showed significantly fewer TUNEL+ spinal motor neurons and better preservation of neuron numbers (Figure 1O–X). Consistent with these data, cultured cortical neurons from embryonic day 15.5 (E15.5) Hipk2−/− mutants were more resistant to tunicamycin-induced cell death, characterized by the reduced number of activated caspase 3+ neurons (Figure S2A–I). Conversely, overexpressing HIPK2 in wild type cortical neurons enhanced tunicamycin-induced cell death (Figure S2J–S). These results show that HIPK2 is necessary and sufficient to promote cell death during ER stress.
Figure 1. Cortical and spinal motor neurons in Hipk2−/− mice are more resistant to tunicamycin-induced neurodegeneration.
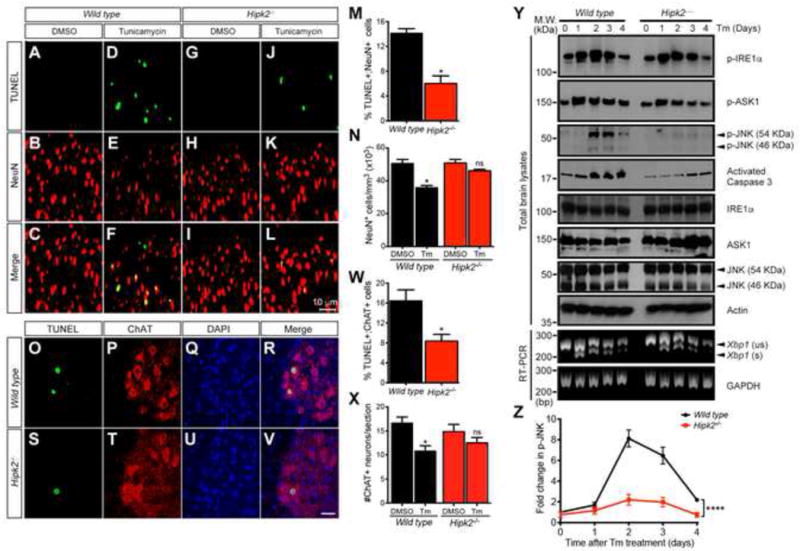
(A–L) Three-month-old wild type and Hipk2−/− mice treated with tunicamycin show TUNEL+ apoptotic neurons in sensorimotor cortex.
(M–N) Number of TUNEL+ and NeuN+ cortical neurons in the sensorimotor cortex of wild type and Hipk2−/− mutants. * p < 0.05, two-tailed unpaired Student’s t test.
(O–V) Wild type and Hipk2−/− mice injected with tunicamycin show TUNEL+ motor neurons in spinal cord.
(W–X) Number of TUNEL+ motor neurons in the cervical spinal cord of wild type and Hipk2−/− mice. * p < 0.05, two-tailed unpaired Student’s t test.
(Y) Protein lysates and RNA samples were prepared from the cerebral cortex of wild type and Hipk2−/− mice at 1, 2, 3 or 4 days after tunicamycin injection to characterize IRE1α-ASK1-JNK activation and Xbp1 splicing.
(Z) Quantification of the time-dependent p-JNK levels induced by tunicamycin. **** p < 0.0001, two-way ANOVA.
Among the three distinct signaling pathways downstream of ER stress, IRE1α is known to promote Xbp1 splicing, which converts XBP1 into a functionally active transcription factor that activates target genes to reduce ER stress response (Ron and Walter, 2007). If ER stress persists, prolonged activation of IRE1α irreversibly alters cell fate and activates cell death through ASK1 and JNK-dependent mechanisms that are poorly characterized. Since previous results indicate that HIPK2 can interact with JNK (Hofmann et al., 2003), we asked whether loss of HIPK2 might affect tunicamycin-induced activation of IRE1α, ASK1 and JNK. Using phosphorylation-specific antibodies for activated IRE1α, ASK1 and JNK, we observed that tunicamyin treatment induced IRE1α and ASK1 activation, and progressive increase in activated caspase 3 in wild type brain tissues preceding JNK activation (Figure 1Y). Interestingly, loss of HIPK2 almost completely blocked caspase 3 cleavage and JNK activation in Hipk2−/− brain tissues without affecting IRE1α or ASK1 activation or Xbp1 mRNA splicing (Figure 1Z). These results support the specific role of HIPK2 in promoting JNK activation under tunicamycin-induced ER stress.
ER stress promotes HIPK2 activation via site-specific phosphorylation
To investigate the role of HIPK2 in ER stress, we used in vitro kinase assays and mass spectrometry to determine whether tunicamycin-induced ER stress activates HIPK2 by phosphorylating specific amino acid(s) within the activation loop of HIPK2 (Figure 2A)(Shang et al., 2013). Consistent with the results in Figure 1, tunicamycin induced sequential phosphorylation of ASK1, HIPK2 and JNK in HEK293 cells, with HIPK2 phosphorylation, shown by the incorporation of γ-P32-ATP, occurred after ASK1 and before JNK activation (Figure 2B–C). To further identify specific amino acids in HIPK2 that become phosphorylated by ER stress, we expressed GFP-tagged HIPK2 in HEK293 cells, immunoprecipitated GFP-HIPK2 with magnetic beads conjugated anti-GFP antibody, performed on-bead digestion with trypsin, and analyzed the samples using liquid chromatography tandem MS (LC-MS/MS). In these experiments, we consistently observed mixed spectra with strong evidence of phosphorylation at either T360 or Y361 site (Figure 2D), suggesting that the phosphorylation at these two sites was mutually exclusive. Consistent with the western blot data, quantitation of MS peak intensities showed that tunicamycin treatment produced a similar increase in phosphorylation at both sites (Figure 2D). Phosphorylation was also detected in S359, although the signal was not as robust or consistent compared to T360 or Y361, perhaps due to steric hindrance from a nearby α-helix structure as predicted by the Phyre2 protein structure program (Figure S3A–C)(Kelley and Sternberg, 2009). To investigate how phosphorylation of these residues affects ER stress, we introduced point mutations in HIPK2 on S359 (HIPK2-S359A), T360 (HIPK2-T360A) and Y361 (HIPK2-Y361F), and used in vitro kinase assays to determine whether mutations in these amino acids affected the ability of HIPK2 to incorporate γ-P32-ATP. Surprisingly, tunicamycin-induced γ-P32-ATP incorporation in HIPK2 was not affected in HIPK2-Y361F, but was drastically decreased in HIPK2-T360A and HIPK2-S359A (Figure 2E). These results validate the essential role of T360 and S359 in ER stress-induced HIPK2 phosphorylation, and suggest that ER stress may induce HIPK2 phosphorylation that begins either simultaneously or sequentially at T360 and S359 to promote HIPK2 activation (Figure 2N). In contrast, loss of phosphorylation at the Y361 is not required for HIPK2 activation under ER stress.
Figure 2. ER stress promotes sequential activation of the ASK1-HIPK2-JNK pathway by phosphorylating HIPK2 at S359/T360.
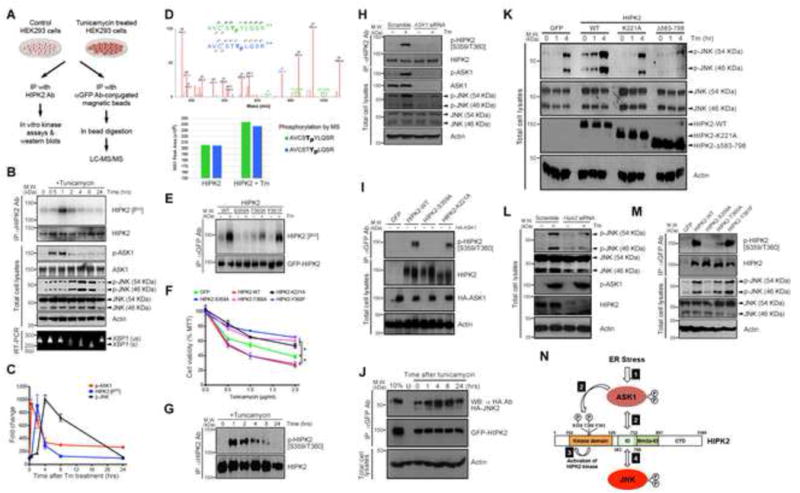
(A) Experimental procedures using in vitro kinase assays and liquid chromatography tandem mass spectrometry (LC-MS/MS) to characterize HIPK2 phosphorylation.
(B–C) Protein lysates from HEK293 cells treated with tunicamycin were used for in vitro kinase assays to detect HIPK2 activation measured by incorporation of γ-P32-ATP, and to detect p-ASK1 and p-JNK.
(D) (Top) Representative LC-MS/MS spectrum of a singly phosphorylated HIPK2 tryptic peptide spanning the S359, T360 and Y361 residues shows overlapping ion series corresponding to phosphorylation at T360 and Y361, highlighted in blue and green, respectively. All assigned ion peaks are marked in red. (bottom) Relative phosphorylation of HIPK2 at the two sites with and without tunicamycin treatment was compared by calculating peak areas of extracted ion chromatographs.
(E) Protein lysates from HEK293 cells expressing HIPK2-WT, HIPK2-S359A, HIPK2-T360A or HIPK2-Y361F were used in in vitro kinase assays to detect γ-P32-ATP incorporation in HIPK2 after tunicamycin treatment.
(F) HEK293 cells expressing HIPK2-WT, HIPK2-S359A, HIPK2-T360A and HIPK2 kinase-dead HIPK2-K221A were treated with tunicamycin. Cell viability was measured using MTT assay. Two-way ANOVA, * p < 0.05.
(G) Western blot using epitope-specific p-HIPK2 [S359/T360] Ab detects phosphorylated HIPK2 in tunicamycin-treated HEK293 cells.
(H) HEK293 cells were treated with scramble siRNA or ASK1 siRNA and tunicamycin to determine the effect on JNK phosphorylation.
(I) HEK293 cells co-expressing ASK1 and HIPK2-WT, HIPK2-S359A or kinase-dead HIPK2 (HIPK2-K221A) to determine the effect of ASK1 on HIPK2 phosphorylation.
(J) HEK293 cells expressing GFP-HIPK2 and HA-JNK2 treated with tunicamycin were used in co-immunoprecipitation assays to demonstrate HIPK2-JNK protein complex formation. “U” for untransfected HEK293 cells.
(K) HEK293 cells expressing different HIPK2 constructs were treated with tunicamycin to characterize endogenous JNK activation under ER stress.
(L) HEK293 cells were treated with control or Hipk2 siRNA and subsequently treated with tunicamycin to determine the effect on JNK phosphorylation.
(M) Lysates from HEK293 cells expressing HIPK2-WT, HIPK2-S359A, HIPK2-T360A, or HIPK2-Y361F and treated with tunicamycin were used to characterize the effect of HIPK2 phosphorylation on S359 and T360 in JNK phosphorylation.
(N) Diagram showing ER stress promotes a cascade of protein kinase activation, including (1) ASK1 activation, (2) ASK1 interaction with HIPK2 and ASK1-mediated site-specific phosphorylation of HIPK2, (3) HIPK2 kinase activation, and (4) HIPK2-JNK protein complex formation and JNK activation.
To characterize how HIPK2 phosphorylation might affect cellular response to ER stress, we expressed wild type HIPK2 (HIPK2-WT), HIPK2-S359A, HIPK2-T360A or HIPK2-Y361F in HEK293 cells, and treated these cells with tunicamycin. Both HIPK2-WT and HIPK2-Y361F promoted cell death in tunicamycin-treated HEK293 cells. In contrast, HIPK2-S359A and HIPK2-T360A protected cells from tunicamycin-induced cell death with similar efficiency (Figure 2F). Interestingly, the protective effects of HIPK2-S359A and HIPK2-T360A expression were similar to that of kinase-dead HIPK2-K221A mutant, supporting the idea that HIPK2 phosphorylation on S359/T360 might be coupled to HIPK2 kinase activation. To further characterize the role of HIPK2 phosphorylation in ER stress, we generated antibodies that recognized phosphorylated S359 and T360 residues in HIPK2. This epitope specific HIPK2 antibody (hereafter named p-HIPK2 [S359/T360] Ab) detected no HIPK2 phosphorylation in untreated cells. Upon tunicamycin treatment, however, p-HIPK2 [S359/T360] Ab detected an increase in phosphorylated HIPK2 with kinetics similar to those revealed by in vitro kinase assays (Figure 2G). In addition, p-HIPK2 [S359/T360] Ab detected phosphorylated HIPK2 in brain tissues from tunicamycin-treated wild type mice, but not Hipk2−/− mice (Figure S3D), supporting the specificity of this antibody.
HIPK2 acts downstream of ASK1 and upstream of JNK during ER stress
The sequential activation of ASK1, HIPK2 and JNK in response to ER stress raised the possibility that ASK1 may activate HIPK2 by phosphorylating S359 and T360, which then activates HIPK2 kinase activity and promotes JNK activation. To further delineate the relationship between ASK1, HIPK2 and JNK during ER stress, we expressed a series of HIPK2 deletion mutants in HEK293 cells and used co-immunoprecipitation to detect the interaction between HIPK2 and endogenous ASK1 after tunicamycin treatment. These results showed that removing the domain between amino acids 583 and 798 (HIPK2Δ583–798) completely abolished HIPK2-ASK1 interaction (Figure S4A), whereas over-expressing ASK1 promoted phosphorylation in wild type HIPK2, but not HIPK2Δ583–798 (Figure S4B). We then used siRNA to knock down ASK1 in HEK293 cells, which resulted in a near-complete loss of tunicaymcin-induced HIPK2-S359/T360 phosphorylation (Figure 2H). In contrast, over-expression of ASK1 enhanced S359/T360 phosphorylation in wild type HIPK2 and kinase-dead mutant HIPK2-K221A, but not in HIPK2-S359A (Figure 2I). These results suggest that S359/T360 phosphorylation in HIPK2 precedes HIPK2 kinase activation during ER stress, and that both are required for HIPK2 activation and its ability to promote the downstream cell death pathway.
To characterize the role of HIPK2 in ER stress-induced JNK activation, we performed co-immunoprecipitation using protein lysates from HEK293 cells expressing GFP-tagged HIPK2 and HA-tagged JNK2 and showed that tunicamycin induced a robust, time-dependent increase in protein complex formation between HIPK2 and JNK2 (Figure 2J). The protein complex formation between HIPK2 and JNK2 was significantly reduced in HIPK2 kinase-dead mutant, and almost completely abolished in HIPK2Δ583–798 (Figure 2K). Interestingly, acute knock down of endogenous HIPK2 using siRNA in HEK293 cells resulted in reduced JNK activation in response to tunicamycin without affecting ASK1 activation (Figure 2L), consistent with ASK1 acting upstream of HIPK2. In addition, over-expression of HIPK2-S359A and HIPK2-T360A, but not HIPK2-Y361F, blocked JNK activation in response to tunicamycin treatment (Figure 2M), suggesting that HIPK2 phosphorylation at S359/T360 is required to promote ER stress-induced JNK activation. Finally, Hipk2−/− MEF cells treated with tunicamycin also showed a significant reduction in p-JNK and p-c-Jun levels (Figure S5A–B). Similar to HEK293 cells, overexpression of HIPK2 in wild type MEF cells promoted tunicamyin-induced cell death. The pro-apoptotic effect of HIPK2 was almost completely blocked in Jnk1−/−;Jnk2−/− MEF cells (Figure S5C). Collectively, these results suggest that ER stress activates a cascade of signaling events, including ASK1-mediated phosphorylation of HIPK2 on S359/T360, which cooperatively activates HIPK2 kinase and HIPK2-JNK complex formation to promote cell death (Figure 2N).
HIPK2 promotes JNK activation and neuronal cell death in SOD1G93A mice
Previous studies indicate that the accumulation of misfolded mutant SOD1G93A proteins activate unfolded protein responses that contribute to neurodegeneration (Atkin et al., 2006; Nishitoh et al., 2008). To ask whether HIPK2 activation might promote neurodegeneration in this model, we used immunofluorescent microscopy with anti-HIPK2 antibody and showed that HIPK2 protein was expressed at low levels in wild type spinal motor neurons at postnatal day (P) 90 (Figure 3A–D, arrowheads). In contrast, spinal motor neurons in SOD1G93A littermates showed a significant increase in HIPK2 expression (Figure 3E–H, arrowheads). Importantly, spinal motor neurons in SOD1G93A mice with higher HIPK2 expression also showed prominent accumulation of misfolded SOD1G93A proteins, detected by conformation-specific C4F6 antibody (Bosco et al., 2010)(Figure 3E–H, arrow). Furthermore, HIPK2 protein signal intensity in spinal motor neurons in SOD1G93A mice showed an age-dependent increase that correlated with relative abundance of misfolded SOD1G93A proteins (Figure 3I).
Figure 3. Loss of HIPK2 blocks JNK activation and neuronal cell death in SOD1G93A model of ALS.
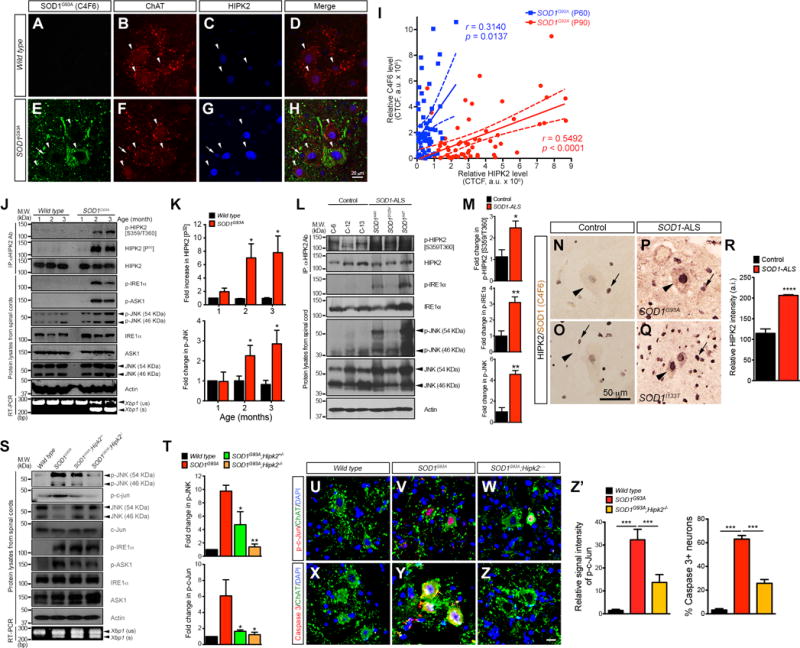
(A–H) Cervical spinal cord from P90 wild type and SOD1G93A were stained using C4F6 and HIPK2 antibodies.
(I) HIPK2 signal intensity in the spinal motor neurons of SOD1G93A mice at P60 and P90 is plotted as a function of the signal intensity for misfolded SOD1G93A proteins (C4F6 intensity). Pearson correlation coefficients, with 95% confidence and the p values.
(J) Spinal cord tissues from wild type and SOD1G93A mice at 1-, 2- and 3-month-old were used in immunoprecipitation-in vitro kinase assays for HIPK2 activation, and to detect the presence of p-IRE1α, p-ASK1, and p-JNK and Xbp1 splicing.
(K) Quantification of HIPK2 [P32] and p-JNK in wild type and SOD1G93A mice.
(L) Lysates from the cervical spinal cord of 3 SOD1-ALS patients and 3 age-matched controls were analyzed in western blots to detect the presence of IRE1α, HIPK2 and JNK phosphorylation.
(M) Relative signal intensity of p-HIPK2 [S359/T360], p-IRE1α and p-JNK. Two-tailed unpaired Student’s t test, * p < 0.05 and ** p < 0.01.
(N–R) Two-color immunohistochemistry for misfolded SOD1 proteins (detected by C4F6, brown color) and HIPK2 (blue color) was performed in spinal motor neurons of control and ALS patients with SOD1G93A or SOD1I133T mutation. Arrowheads in L–O indicate spinal motor neurons, whereas arrows indicate glial cells. The intensity of HIPK2 in motor neurons was quantified using NIH ImageJ.
(S) Spinal cord tissues from P90 wild type, SOD1G93A, SOD1G93A;Hipk2−/− and SOD1G93A;Hipk2−/− mice were used to detect activation of ER stress markers p-IRE1α, p-ASK1 and Xbp1 mRNA splicing.
(T) Quantification of p-JNK and p-c-Jun signal intensity. Two-tailed unpaired Student’s t test, * p < 0.05, ** p < 0.01.
(U–W) Confocal images of p-c-Jun in ChAT+ motor neurons in the cervical spinal cord of P120 wild type, SOD1G93A and SOD1G93A;Hipk2−/− mice.
(X–Z) Confocal images of activated caspase 3+ motor neurons in the cervical spinal cord of P120 wild type, SOD1G93A and SOD1G93A;Hipk2−/− mice.
(Z′) Quantification of p-c-Jun+ motor neurons and caspase 3+ neurons. Student’s t test, *** p < 0.001.
To investigate whether HIPK2 was activated as disease progressed in SOD1G93A mice, we used immunoprecipitation-in vitro kinase (IP-IVK) assays and showed that HIPK2 proteins in SOD1G93A mice exhibited a marked increase in γ-P32-ATP incorporation at P60 and P90 (Figure 3J–K). Consistent with these results, HIPK2 [S359/T360] antibody also detected a progressive increase in phosphorylated HIPK2 in the spinal cord tissues of SOD1G93A mice (Figure 3J). Similar to HIPK2, phosphorylation of IRE1α, ASK1, and JNK, as well as splicing of Xbp1 mRNA were all concurrently up-regulated in the spinal cord of SOD1G93A mice (Figure 3J–K). These results show that ER stress activation precedes disease onset and becomes more severe as disease progresses in SOD1G93A mice. To further characterize the role of HIPK2 in ALS, we used western blots and show that the activation of IRE1α-HIPK2-JNK pathway was indeed much more robust in spinal cord tissues from three FALS cases with mutations in the SOD1 gene (SOD1A4V, SOD1D125V and SOD1A4T)(Figure 3L–M & Table S1). Furthermore, we developed two-color immunostains for HIPK2 and misfolded SOD1 proteins and showed that spinal motor neurons in five patients with SOD1 mutations, including SOD1A4T, SOD1G93A, SOD1I133T, SOD1E100G and SOD1V14M, contained prominent misfolded SOD1 mutant proteins in the cytoplasm (brown color) and ~2-fold increase in HIPK2 immunoreactivity in the nucleus (blue color)(Figure 3P–R, arrowheads). In contrast, control motor neurons showed very low level of HIPK2 proteins in the nucleus and no detectable misfolded SOD1 proteins in the cytoplasm (Figure 3N–O, arrowheads).
To investigate whether HIPK2 activation might contribute to the pathogenesis of ALS caused by SOD1 mutations, we asked if loss of HIPK2 could block JNK activation in SOD1G93A mice. In support of this idea, western blots showed that p-JNK level was reduced by ~50% in the spinal cord of SOD1G93A;Hipk2+/− and >80% in SOD1G93A;Hipk2−/− mice (Figure 3S–T). Furthermore, phosphorylation of c-Jun, a major target of JNK kinase activity, also showed a drastic reduction in both SOD1G93A;Hipk2+/− and SOD1G93A;Hipk2−/− spinal cords. Consistent with these results, motor neurons in SOD1G93A;Hipk2−/− spinal cords showed much reduced p-c-Jun and activated caspase 3 (Figure 3U–Z′). Despite these changes, loss of HIPK2 had no detectable effects on the activation of IRE1α or ASK1, nor did it affect the splicing of Xbp1 mRNA (Figure 3S).
Loss of HIPK2 attenuates neurodegeneration in SOD1G93A mice
The reduced JNK activation and cell death phenotypes in spinal motor neurons in SOD1G93A;Hipk2−/− mice suggest that loss of HIPK2 might mitigate neurodegeneration in SOD1G93A mice. To test this, we quantified the number of choline acetyltransferase (ChAT)-positive motor neurons in the lumbar spinal cords of wild type, SOD1G93A, and SOD1G93A;Hipk2−/− mice at P90 and P120. Unlike the progressive reduction in motor neurons in SOD1G93A mice, SOD1G93A;Hipk2−/− mice showed significant preservation of motor neurons at P90 and P120 (Figure 4A–F, M). To determine whether loss of HIPK2 also affected glial pathology, we quantified the number of GFAP+ astrocytes and Iba1+ microglia in wild type, SOD1G93A and SOD1G93A;Hipk2−/− mice at P90 and P120. Whereas SOD1G93A mice showed an age-dependent progressive increase in astrogliosis and microgliosis in the spinal cord, both were significantly reduced in SOD1G93A;Hipk2−/− mice (Figure 4G–L, N).
Figure 4. HIPK2 deletion in SOD1G93A mice attenuates neurodegeneration, delays disease onset, prolongs survival and improves motor functions.
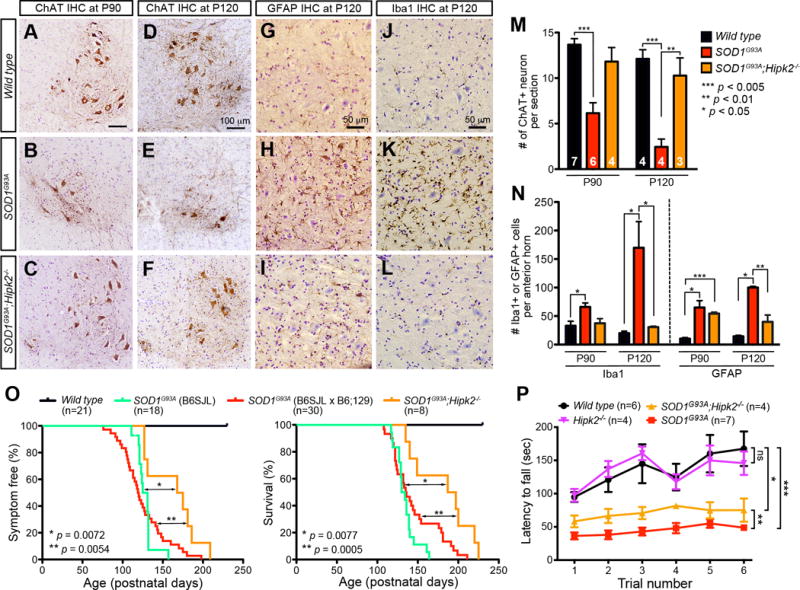
(A–F) Immunostaining of ChAT+ neurons in the cervical spinal cord of wild type, SOD1G93A and SOD1G93A;Hipk2−/− mice at P90 and P120.
(G–L) Immunostaining for GFAP+ astrocytes and Iba1+ microglia in wild type, SOD1G93A and SOD1G93A;Hipk2−/− mice at P120.
(M–N) Number of ChAT+ motor neurons, GFAP+ astrocytes and Iba1+ microglia in wild type, SOD1G93A and SOD1G93A;Hipk2−/− mice at P90 and P120. Two-tailed unpaired Student’s t test. *** p < 0.005, ** p < 0.01, and * p < 0.05.
(O) Kaplan–Meier analyses of disease onset and survival in wild type, SOD1G93A mice (B6SJL), SOD1G93A (B6SJL × B6;129) and SOD1G93A;Hipk2−/− mice.
(P) Rotarod testing in P120 wild type, Hipk2−/−, SOD1G93A and SOD1G93A;Hipk2−/− mice. Two-way ANOVA, * p = 0.04, ** p = 0.0082, *** p = 0.002, ns, not significant.
Because Hipk2−/− mice were maintained in B6;129 background and SOD1G93A mice were in B6;SJL background, the mating between these two mouse lines generated progenies with a mixed genetic background (B6SJL × B6;129), which slightly affected disease onset and survival curves compared with SOD1G93A in B6:SJL background (Gurney et al., 1994). Despite this minor difference, SOD1G93A (B6SJL × B6;129) mice had a median disease onset of 119.5 days and median survival 135.5 days, similar to SOD1G93A (B6;SJL). Compared to SOD1G93A littermates (B6SJL × B6;129) and SOD1G93A mice in B6:SJL background, SOD1G93A;Hipk2−/− mice exhibited a significant delay in disease onset (median 171.5 days, p = 0.0054) and better survival (median 192 days, p = 0.0005)(Figure 4O). Our previous results indicate that 1–2 months old Hipk2−/− mice exhibit poor motor coordination on Rotarod tests due to partial loss of ventral midbrain dopaminergic neurons (Zhang et al., 2007). However, as Hipk2−/− mice reached P120, their performance on Rotarod tests improved and was similar to that in wild type mice (Figure 4P). Compared to SOD1G93A mice, SOD1G93A;Hipk2−/− mice showed better performance on Rotarod tests at P120 (p = 0.0082). However, SOD1G93A;Hipk2−/− mice performed less well compared to wild type or Hipk2−/− mice (p = 0.04)(Figure 4P). Together, these results indicate that loss of HIPK2 significantly attenuates neurodegeneration in SOD1G93A mice.
ER stress and HIPK2 activation in TDP-43, but not FUS, ALS Model
To determine whether ER stress is also a prominent disease mechanism shared among other mouse ALS models, we characterized the state of IRE1α-HIPK2-JNK activation in FUS-R521C mice that express the most common human FUS mutation FUS-R521C and the bi-transgenic, tetracycline-inducible NEFH-tTA/tetO-hTDP-43ΔNLS mice that express truncated human TDP-43 lacking the nuclear localization signal (Qiu et al., 2014; Walker et al., 2015). In our previous studies, we show that ~60% of FUS-R521C transgenic mice develop ALS-like symptoms before P90 due to severe dendritic and synaptic defects (Qiu et al., 2014), whereas removing doxycycline from the drinking water for NEFH-tTA/tetO-hTDP-43ΔNLS mice induces progressive ALS-like symptoms with accumulation of soluble and insoluble hTDP-43ΔNLS proteins in the cytoplasm of cortical and spinal motor neurons (Walker et al., 2015). Unlike SOD1G93A mice, FUS-R521C mice that reached end-stage disease at P90 showed no evidence of activation in IRE1α, HIPK2 or JNK (Figure 5A–B). In contrast, expressing hTDP-43ΔNLS in NEFH-tTA/tetO-hTDP-43ΔNLS mice led to a progressive increase in p-IRE1α, p-HIPK2 [S359/T360] and p-JNK that reached its maximum at 11 weeks after induction (Figure 5C–D). Consistent with the western blot results, spinal motor neurons in NEFH-tTA/tetO-hTDP-43ΔNLS mice showed a significant increase in HIPK2 proteins 10 weeks after induced expression of hTDP-43ΔNLS (Figure 5E–K). Together, these results identify ER stress and IRE1α-HIPK2-JNK pathway as a common disease mechanism shared by SOD1G93A and NEFH-tTA/tetO-hTDP-43ΔNLS mice, but not FUS-R521C mice.
Figure 5. Activation of IRE1α-HIPK2-JNK in SOD1G93A and NEFH-tTA/tetO-hTDP-43ΔNLS, but not FUS-R521C, ALS models.
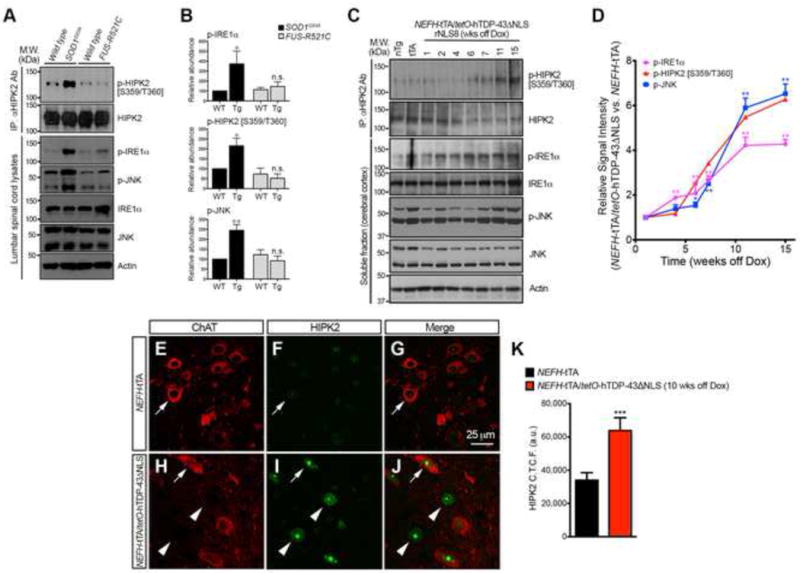
(A–B) Spinal cord lysates from SOD1G93A, FUS-R521C and wild type littermates were used in western blots to compare the levels of p-IRE1α, p-HIPK2 [S359/T360] and p-JNK. Student’s t test, * p < 0.05 and ** p < 0.01.
(C–D) RIPA soluble lysates are prepared from the cortex non-transgenic control (nTg), NEFH-tTA mice (tTA), and NEFH-tTA/tetO-hTDP-43ΔNLS mice that are “off doxycycline” for 1, 2, 4, 6, 7, 11 and 15 weeks to detect activated and total IRE1α, HIPK2 and JNK. Two-tailed unpaired Student’s t test, * p < 0.05, ** p < 0.01.
(E–K) Confocal images of HIPK2 in ChAT+ motor neurons in NEFH-tTA mice and NEFH-tTA/tetO-hTDP-43ΔNLS (rNLS8) mice that are “off doxycycline” for 10 weeks. Arrows indicate ChAT+ neurons, and arrowheads ChAT− neurons. Relative signal intensity of HIPK2 was quantified by NIH ImageJ. Two-tailed unpaired Student’s t test, *** p < 0.001.
HIPK2 activation correlates with TDP-43 proteinopathy in SALS and C9-ALS
Given the results from tunicamycin-induced ER stress, SOD1G93A and NEFH-tTA/tetO-hTDP-43ΔNLS mouse models, we reasoned that the activation of HIPK2-JNK pathway may have a broader role in the pathogenesis of ALS. Since TDP-43 protein misfolding and aggregation is a common pathological feature in SALS and C9-ALS (Brettschneider et al., 2014; Mackenzie et al., 2014), we asked whether misfolded TDP-43 proteins might trigger ER stress and HIPK2 activation to promote disease initiation and progression. To test this, we prepared protein lysates from the spinal cord of 28 cases of SALS and 16 cases of C9-ALS using published protocols (Neumann et al., 2006). In addition, we also prepared 13 spinal cord and 10 frontal cortex samples from cases with no history of neurodegenerative disease, and samples from the frontal cortex of 7 cases with Alzheimer’s disease (Table S1).
We used western blots to quantify the relative abundance of p-HIPK2 [S359/T360], p-JNK, p-c-Jun, ubiquitinated TDP-43 and TDP-43 phosphorylated on S409/410 residues (p-TDP-43 [S409/410]). Whereas control cases showed very low or no detectable p-HIPK2 [S359/T360], p-TDP-43 [S409/410], ubiquinated TDP-43 or p-JNK, the majority of SALS cases showed a marked increase in all these proteins (Figure 6A–F)(Figure S6). Remarkably, the level of p-HIPK2 [S359/T360] in SALS cases positively correlated with p-TDP-43 [S409/410], ubiquitinated TDP-43, and p-JNK (Figure 6G–I). Similar to SALS cases, samples from C9-ALS cases also showed significant increases in p-HIPK2 [S359/T360], p-TDP-43 [S409/410], ubiquitinated TDP-43 and p-JNK (Figure 6C–F)(Figure S7), and the level of p-HIPK2 [S359/T360] positively correlated with ubiquitinated TDP-43 and p-JNK, but not with p-TDP-43 [S409/410] (Figure 6J–L). Consistent with these results, the remaining spinal motor neurons in SALS and C9-ALS cases showed a robust increase in HIPK2 protein levels by immunostaining (Figure 6MP). In contrast, samples from the frontal cortex of Alzheimer’s disease cases showed no evidence of TDP-43 proteinopathy or HIPK2 phosphorylation (Figure S8).
Figure 6. HIPK2-JNK activation positively correlates with TDP-43 proteinopathy in sporadic ALS and familial ALS with C9ORF72 mutations.
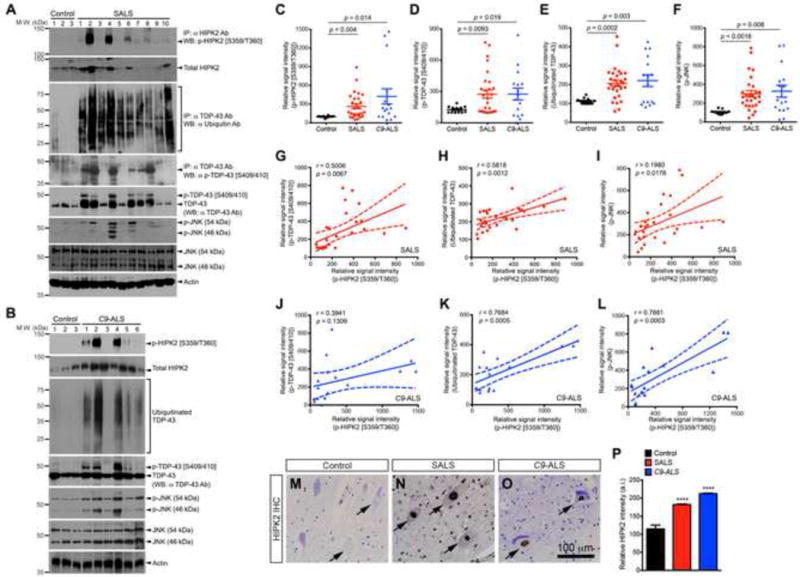
(A–B) Spinal cord tissues from SALS and C9-ALS cases were used in western blots to detect p-HIPK2 [S359/T360], ubiquinated TDP-43, p-TDP-43 [S409/410], and p-JNK.
(C–F) Relative abundance of p-HIPK2 [S359/T360], p-TDP-43 [S409/410], ubiquitinated TDP-43 and p-JNK. Two-tailed Student’s t test.
(G–L) Relative abundance of p-HIPK2 [S359/T360] is plotted as a function of the signal intensity of p-TDP-43 [S409/410], ubiquitinated TDP-43 or p-JNK in SALS cases (G–I) and C9-ALS (J–L). Pearson correlation coefficients and p values are indicated. Dash lines represent 95% confidence.
(M–P) Spinal cord tissues from control, SALS and C9-ALS cases were stained with HIPK2 antibody, and the intensity of HIPK2 quantified using NIH ImageJ.
HIPK2 kinase inhibitors attenuate cytotoxicity from tunicamycin, SOD1G93A and TDP-43 in spinal motor neurons
To determine the effects of HIPK2 kinase inhibitors in blocking ER stress-induced cell death, we used tunicamycin and SOD1G93A-induced cell death as models to screen a number of HIPK2 kinase inhibitors (Miduturu et al., 2011). Of the 5 HIPK2 kinase inhibitors screened (Table S2), A64 and JWD-065 showed the most robust effects in blocking HIPK2 phosphorylation on S359/T360 and JNK activation in HEK293 cells (Figure 7A–B), blocking tunicamycin-induced cell death with an EC50 of 0.403 μM and 0.641 μM, respectively (Figures 7C). Western blots and immunostaining for activated caspase 3 also showed that A64 blocked p-HIPK2 [S359/T360] in primary spinal motor neurons and protected >60% of these neurons from tunicamycin- and SOD1G93A-induced cell death (Figure 7D–L).
Figure 7. HIPK2 kinase inhibitors block S359/T360 phosphorylation and attenuate ER stress-induced cell death.
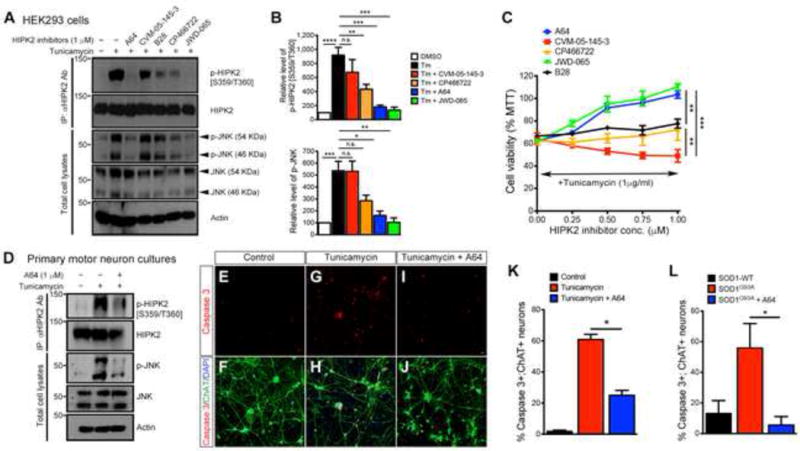
(A) HIPK2 kinase inhibitors (1 μM each) were tested in HEK293 cells for their ability to block HIPK2 phosphorylation and JNK phosphorylation.
(B) Quantification of p-HIPK2 [S359/T360] and p-JNK. Two-tailed unpaired Student’s t test; * p < 0.05, ** p < 0.01, *** p < 0.005 and **** p < 0.0001.
(C) HEK293 cells were treated with tunicamycin (1μg/ml) and HIPK2 inhibitors at the designated concentrations. Cell viability is determined using MTT assays. 2 way ANOVA; ** p < 0.01 and *** p < 0.005.
(D) Motor neurons were treated with DMSO, tunicamycin (1 μg/ml) or tunicamycin and HIPK2 inhibitor A64 (1μM). Protein lysates from these neurons were used to characterize HIPK2 and JNK phosphorylation.
(E–J) Cortical neurons were treated with DMSO, tunicamycin (1 μg/ml) or tunicamycin and HIPK2 inhibitor A64 (1 μM). Cell death was analyzed using antibodies for activated caspase 3.
(K) Quantificaiton of caspase 3+ neurons. Data represents mean ± SEM. Two-tailed unpaired Student’s t test; * p < 0.05.
(L) Motor neurons transfected with mutant SOD1G93A were treated with HIPK2 inhibitor A64, and cell death quantified using activated caspase 3 antibody.
To determine whether HIPK2 inhibitors can also protect neurons from cytotoxicity from TDP-43 proteinopathy, we transfected HEK293 cells with constructs expressing wild type human TDP-43 (Myc-hTDP-43-WT) or ALS-associated TDP-43 mutations, including hTDP-43G348C-Myc-His, mTDP-43A315T-Myc-His, mTDP-43A90V-Myc-His and mTDP-43M337V-Myc-His. Interestingly, overexpression of wild type and mutant TDP-43 led to marked increases in ubiquitinated TDP-43 and p-TDP-43 [S409/410](Figure 8AB), and robustly increased p-HIPK2 [S359/T360] and other ER stress effectors, including p-IRE1α and Xbp1 splicing. Co-expression of wild type HIPK2 with TDP-43 further enhanced cell death in HEK293 cells, whereas co-expression of HIPK2-S359A reduced TDP-43 cytotoxicity (Figure 8C). In contrast, HIPK2 inhibitor A64 mitigated the cytotoxicity caused by TDP-43 in HEK293 cells.
Figure 8. HIPK2 inhibitor A64 attenuates TDP-43 proteinopathy-induced cell death in HEK293 cells and spinal motor neurons.
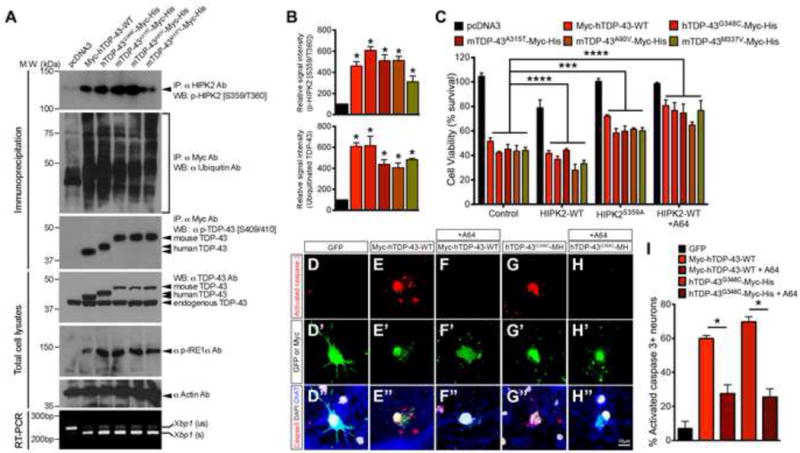
(A) Lysates from HEK293 cells expressing wild type or mutant TDP43 were used in western blots to determine TDP-43 ubiquitination, phosphorylation on TDP-43, IRE1α and HIPK2, and Xbp1 splicing.
(B) Quantification of p-HIPK2 [S359/T360] and ubiquitinated TDP-43. * p < 0.05, unpaired two-tailed Student’s t test.
(C) HEK293 cells were co-transfected with TDP-43 constructs, together with HIPK2-WT, HIPK2-S359A or HIPK2-WT treated with HIPK2 inhibitor A64 (1 μM). Two days after transfection, cells were harvested for MTT assays. Two way ANOVA, *** p < 0.001 and **** p < 0.0001.
(D–H) Activated caspase 3 in rat spinal motor neurons two days after transfection with GFP, Myc-hTDP-43-WT or TDP-43G348C-Myc-His, and treated with DMSO or HIPK2 inhibitor A64 (1 μM).
(I) Qunatification of apoptotic motor neurons. * p < 0.05, two-tailed Student’s t test.
Finally, to determine whether HIPK2 inhibitors also protect neurons from TDP-43 cytotoxicity, we expressed Myc-hTDP-43-WT or hTDP-43G348C-Myc-His in cultured spinal motor neurons. Similar to the results in HEK293 cells, Myc-hTDP-43-WT and hTDP-43G348C-Myc-His markedly increased activated caspase 3+ neurons (Figure 8D–E, G & I). In contrast, A64 treatment protected neurons from cell death induced by either wild type or mutant TDP-43 (Figure 8F, H & I). These results support that HIPK2 activation promotes cell death, whereas blocking HIPK2 may mitigate neurodegeneration, caused by TDP-43 proteinopathy.
DISCUSSION
HIPK2 Activation Links Persistent ER Stress to Cell Death
Chronic ER stress contributes to many human diseases, including diabetes, viral infections, cancer and neurodegeneration (Kim et al., 2008; Oakes and Papa, 2015). Indeed, several studies indicate that misfolded mutant SOD1 proteins can robustly activate adaptive UPR, including activation of ASK1 and PERK, Xbp1 mRNA splicing and transcriptional up-regulation of protein chaperone machinery (Walker and Atkin, 2011). These studies demonstrate that PERK activation and up-regulation of ER chaperones and members of the protein disulfide isomerase (PDI) family alleviate neurodegeneration. Similarly, removal of XBP1 or ASK1 prolongs survival of SOD1G93A mice (Hetz et al., 2009; Nishitoh et al., 2008). The implications of ER stress activation in the pathogenesis of ALS have been further supported by the findings that autosomal dominant mutations in the human ALS8 gene, VAPB, can potentiate ER stress response-mediated cell death (Suzuki et al., 2009). Collectively, these results support the conceptual framework that disruption of the homeostatic control of ER stress is critical to the pathophysiology and disease progression in ALS.
Despite the important role of ER stress in ALS, it remains unclear what dictates the decision between survival and cell death during ER stress response. The resistance of Hipk2−/− neurons to tunicamycin-induced cell death implicates HIPK2 as the key mediator of ASK1-JNK pro-apoptotic pathway during persistent ER stress response (Figure 1 and Figure S1). The mechanism by which ER stress activates HIPK2 involves epitope-specific phosphorylation of S359/T360 residues within the activation loop of HIPK2 kinase domain, which activates HIPK2 kinase activity and promotes HIPK2-JNK protein complex formation (Figure 2). Not only do these results solidify the evidence that HIPK2 links ER stress to pro-apoptotic JNK activation (Oakes and Papa, 2015; Ron and Walter, 2007), they also open up new opportunities to interrogate the state of ER stress response in a pathway-specific fashion. Indeed, using Xbp1 mRNA splicing and epitope-specific p-IRE1α, p-ASK1, p-HIPK2 [S359/T360] and p-JNK antibodies, we show that the pathway downstream of IRE1α is robustly up-regulated in the spinal cord of SOD1G93A mice as early as P60 when these mice are free of any neurological symptoms (Figure 3). Interestingly, loss of HIPK2 selectively blocks JNK activation without affecting Xbp1 splicing or the activation of IRE1α and ASK1 (Figure 3S–T). Moreover, loss of HIPK2 reduces neuronal cell death and attenuates neurodegeneration in SOD1G93A mice (Figures 3–4). These results place HIPK2 downstream of IRE1α and ASK1 in the ER stress pathway and underscore the specific role of HIPK2 in promoting neuronal cell death under ER stress conditions.
ER Stress and Mechanisms of Neurodegeneration in ALS
Neuropathological examination of the majority of SALS and C9-ALS cases has shown that TDP-43 proteinopathies, characterized by ubiquitination and phosphorylation of TDP-43, are key diagnostic features associated with both diseases. These results suggest that TDP-43 protein misfolding might induce ER stress response and thereby contribute to the pathogenesis of SALS and C9-ALS. In support of this, ER stress response has been reported in a small number of sporadic ALS cases and ALS-associated TDP-43 mutations can increase sensitivity to ER stress and induce XBP1 nuclear translocation (Atkin et al., 2008; Walker et al., 2013). One intriguing observation from our study is the lack of ER stress or HIPK2 activation in the FUS-R521C transgenic mouse model. Consistent with these results, histopathology and RNA-seq analyses in FUS-R521C mice show robust defects in RNA transcription/splicing and DNA damage repair, but no significant increase in ER stress response (Qiu et al., 2014). These results underscore the divergent role of ER stress-induced neurodegeneration in the pathogenesis of different subtypes of ALS.
How does ER stress contribute to the pathogenesis and disease progression in C9-ALS? Several recent studies provide important insights into the mechanisms that may contribute to ER stress in C9-ALS. First, it has been shown that the accumulation of dipeptide repeat proteins, encoded by the mRNA from C9orf72 hexanucleotide expansion via repeat associated non-ATG (RAN) translation, can induce ER stress and TDP-43 proteinopathy (Chew et al., 2015; Zhang et al., 2014). Second, neurons derived from C9-ALS patients show a marked increase of cell death upon tunicamycin treatment, suggesting that these neurons are much more vulnerable to ER stress (Haeusler et al., 2014). Finally, two recent studies show that C9orf72 mutations can also disrupt nucleocytoplasmic transport proteins, such as RanGAP (Freibaum et al., 2015; Zhang et al., 2015). These results raise the possibility that defects in the nucleocytoplasmic transport machinery may impair nuclear import of RNA binding proteins, such as TDP-43, leading to cytoplasmic accumulation of TDP-43 to trigger ER stress.
Unlike the recent advances in the C9-ALS disease mechanisms, much less is known about the relationship between ER stress and TDP-43 accumulation in SALS. By quantifying the relative abundance of p-HIPK2 [S359/T360] and p-JNK in a large number of sporadic ALS and C9-ALS cases, we demonstrate that HIPK2 and JNK activation are tightly coupled in disease and positively correlate with TDP-43 proteinopathy (Figure 6). In addition, these cases show robust activation of p-IRE1α and p-ASK1 (S.L., unpublished results), again supporting the idea that the entire ASK1-HIPK2-JNK pathway is activated in human ALS. Finally, similar to SOD1G93A mice and FALS patients with SOD1 mutations (Figure 3), many remaining spinal motor neurons in sporadic ALS and C9-ALS cases continue to show robust accumulation of HIPK2 in the nucleus (Figure 6). These results support the hypothesis that the pro-apoptotic ER stress pathway, involving ASK1, HIPK2 and JNK, is a prominent feature conserved in sporadic and familial ALS.
HIPK2 Activation as a Therapeutic Target and Biomarker for ALS
The prominent role of HIPK2 activation in ER stress-induced cell death raises the possibility that HIPK2 might be a feasible therapeutic target for ALS. This is supported by the results that HIPK2-S359A, HIPK2-T360A and kinase-dead HIPK2-K221A can mitigate tunicamycin-induced JNK activation and cell death in HEK293 cells (Figure 2). In addition, loss of HIPK2 attenuates neurodegenerative phenotypes in SOD1G93A mice (Figure 4). Furthermore, HIPK2 inhibitor A64 protects cytotoxicity caused by tunicamycin, SOD1G93A, and over-expression of wild type or mutant TDP-43 (Figures 7–8). Thus, the broad benefits of blocking HIPK2 activation in these conditions provide affirmative evidence that inhibition of HIPK2 kinase can protect neurons from ER stress-induced cell death in ALS.
By analyzing a single dominant pro-apoptotic pathway in the ER stress response, we reveal a robust and positive correlation between HIPK2-JNK activation and TDP-43 proteinopathy in a large number of sporadic ALS and C9-ALS cases. However, these results also reveal significant heterogeneity in the magnitude of HIPK2-JNK activation in ALS patients. Such heterogeneity most likely reflects the stage of disease, the number of remaining neurons with TDP-43 inclusions, and/or other complex and diverse pathogenic factors that contribute to the chronic loss of motor neurons via cell autonomous and non-cell autonomous mechanisms. Given the robust correlation between HIPK2 activation and TDP-43 proteinopathy, it is tempting to speculate that this correlation may serve as an important biomarker to (1) stratify ALS patients based on the magnitude of intrinsic ER stress response, and (2) to identify patients that are most likely to benefit from intervention using HIPK2 inhibitors. In addition, HIPK2-JNK activation may also serve as a pharmacodynamic indicator to monitor the treatment efficacy of blocking the ER stress response, or as a prognostic indicator of disease progression and recurrence.
EXPERIMENTAL PROCEDURES
Animals
Hipk2−/− (RRID: MGI_5008273 and RRID: MGI_3510466), SOD1G93A mice (RRID: MGI_3785391), FUS-R521C mice (JAX Stock #026406) and NEFH-tTA/tetO-hTDP-43ΔNLS (rNLS8) mice (JAX Stock #028412) have been previously described (Gurney et al., 1994; Qiu et al., 2014; Walker et al., 2015; Wiggins et al., 2004). Animal care was approved by the Institutional of Animal Care and Use Committee (IACUC) and followed the National Institute of Health guidelines.
Human Brain and Spinal Cord Tissues
Frozen and paraffin-embedded spinal cord tissues were procured from controls with no known neurodegenerative diseases, SALS patients, and FALS patients with SOD1 or C9ORF72 mutations. Frozen tissues from frontal cortex were obtained from controls and patients with Alzheimer’s disease. All cases were evaluated at the University of California San Francisco, University of Pennsylvania and Northwestern University, and tissues were collected with informed consents and institutional IRB approvals. Demographic information and clinical data of these cases are provided in Table S1.
HIPK2 Kinase Inhibitors
HIPK2 kinase inhibitors, A64, B28 and CP466722, were reported previously (Miduturu et al., 2011; Rainey et al., 2008), whereas CVM-05-145-3 and JWD-065 have not been published. The structure these inhibitors, their SMILES (Simplified Molecular-Input Line-Entry System) information, and EC50 to suppress tunicamycin-induced cell death are provided in Table S2.
Supplementary Material
Acknowledgments
We thank Linda Kwong, John Robinson and Theresa Schuck from the Center for Neurodegenerative Disease Research at the University of Pennsylvania for their assistance with NEFH-tTA/tetO-hTDP-43ΔNLS and human samples, Dr. Kevan Shokat for advice on HIPK2 kinase characterization, and members of the Huang lab for discussions. This work was supported by NIH F31 NS081905-02 (S.A.R.), U54 HG006097-02 (N.S.G.), 8P41GM103481 & 1S10OD016229-01 (A.L.B.), P50 AG023501 and P01 AG019724 (W.W.S.), OD010927 & OD011915 (E.J.H.), AG017586 (V.M-Y.L), AG013854 (E.H.B.), NS062796 (J.R.C.), Les Turner ALS Foundation (T.S.), and the Dept of Veterans Affairs Merit Review Awards I01-BX001108 and I01-RX002133 (E.J.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental information includes Extended Experimental Procedures, eight figures, and two tables can be found with this article on line.
AUTHOR CONTRIBUTIONS
S.L., Y.S. and E.J.H. designed the study. S.L., Y.S., S.R., A.U., A.A.T., R.A.P., K.H.L. and A.J. carried out experiments or contributed critical reagents and protocols, S.L., Y.S., A.U., A.A.T., and R.A.P. analyzed data and performed statistical analyses, J.W. and N.S.G. provided HIPK2 inhibitors and related SAR data, V.M-Y.L, and J.Q.T. contributed NEFH-tTA/tetO-hTDP-43ΔNLS tissues, W.W.S., T.S., E.H.B., V.M-Y.L, and J.Q.T. contributed human ALS samples. A.L.B., A.D.G., J.R.C. and E.J.H. provided supervisory oversights. S.L., Y.S. and E.J.H. wrote the manuscript, and all authors approved the manuscript.
References
- Almeida S, Gascon E, Tran H, Chou HJ, Gendron TF, Degroot S, Tapper AR, Sellier C, Charlet-Berguerand N, Karydas A, et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013;126:385–399. doi: 10.1007/s00401-013-1149-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW, 3rd, Rademakers R, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkin JD, Farg MA, Turner BJ, Tomas D, Lysaght JA, Nunan J, Rembach A, Nagley P, Beart PM, Cheema SS, et al. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J Biol Chem. 2006;281:30152–30165. doi: 10.1074/jbc.M603393200. [DOI] [PubMed] [Google Scholar]
- Atkin JD, Farg MA, Walker AK, McLean C, Tomas D, Horne MK. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis. 2008;30:400–407. doi: 10.1016/j.nbd.2008.02.009. [DOI] [PubMed] [Google Scholar]
- Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, Goolsby H, Fontaine BA, Lemay N, McKenna-Yasek D, et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13:1396–1403. doi: 10.1038/nn.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettschneider J, Arai K, Del Tredici K, Toledo JB, Robinson JL, Lee EB, Kuwabara S, Shibuya K, Irwin DJ, Fang L, et al. TDP-43 pathology and neuronal loss in amyotrophic lateral sclerosis spinal cord. Acta Neuropathol. 2014;128:423–437. doi: 10.1007/s00401-014-1299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes-Casey M, Lee CW, Jansen-West K, Kurti A, Murray ME, et al. Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science. 2015;348:1151–1154. doi: 10.1126/science.aaa9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80:415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525:129–133. doi: 10.1038/nature14974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507:195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, Backes BJ, Oakes SA, Papa FR. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, Martinez G, Cuervo AM, Brown RH, Glimcher LH. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009;23:2294–2306. doi: 10.1101/gad.1830709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann TG, Stollberg N, Schmitz ML, Will H. HIPK2 regulates transforming growth factor-beta-induced c-Jun NH(2)-terminal kinase activation and apoptosis in human hepatoma cells. Cancer Res. 2003;63:8271–8277. [PubMed] [Google Scholar]
- Israelson A, Arbel N, Da Cruz S, Ilieva H, Yamanaka K, Shoshan-Barmatz V, Cleveland DW. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron. 2010;67:575–587. doi: 10.1016/j.neuron.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Kikuchi H, Almer G, Yamashita S, Guegan C, Nagai M, Xu Z, Sosunov AA, McKhann GM, 2nd, Przedborski S. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc Natl Acad Sci U S A. 2006;103:6025–6030. doi: 10.1073/pnas.0509227103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nature reviews Drug discovery. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- Kiskinis E, Sandoe J, Williams LA, Boulting GL, Moccia R, Wainger BJ, Han S, Peng T, Thams S, Mikkilineni S, et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell. 2014;14:781–795. doi: 10.1016/j.stem.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, Lee VM, Trojanowski JQ. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci. 2012;13:38–50. doi: 10.1038/nrn3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Frick P, Neumann M. The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol. 2014;127:347–357. doi: 10.1007/s00401-013-1232-4. [DOI] [PubMed] [Google Scholar]
- Miduturu CV, Deng X, Kwiatkowski N, Yang W, Brault L, Filippakopoulos P, Chung E, Yang Q, Schwaller J, Knapp S, et al. High-throughput kinase profiling: a more efficient approach toward the discovery of new kinase inhibitors. Chem Biol. 2011;18:868–879. doi: 10.1016/j.chembiol.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Nishitoh H, Kadowaki H, Nagai A, Maruyama T, Yokota T, Fukutomi H, Noguchi T, Matsuzawa A, Takeda K, Ichijo H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008;22:1451–1464. doi: 10.1101/gad.1640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10:173–194. doi: 10.1146/annurev-pathol-012513-104649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- Qiu H, Lee S, Shang Y, Wang WY, Au KF, Kamiya S, Barmada SJ, Finkbeiner S, Lui H, Carlton CE, et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J Clin Invest. 2014;124:981–999. doi: 10.1172/JCI72723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey MD, Charlton ME, Stanton RV, Kastan MB. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res. 2008;68:7466–7474. doi: 10.1158/0008-5472.CAN-08-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009;12:627–636. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- Shang Y, Doan CN, Arnold TD, Lee S, Tang AA, Reichardt LF, Huang EJ. Transcriptional corepressors HIPK1 and HIPK2 control angiogenesis via TGF-beta-TAK1-dependent mechanism. PLoS Biol. 2013;11:e1001527. doi: 10.1371/journal.pbio.1001527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Kanekura K, Levine TP, Kohno K, Olkkonen VM, Aiso S, Matsuoka M. ALS-linked P56S-VAPB, an aggregated loss-of-function mutant of VAPB, predisposes motor neurons to ER stress-related death by inducing aggregation of co-expressed wild-type VAPB. J Neurochem. 2009;108:973–985. doi: 10.1111/j.1471-4159.2008.05857.x. [DOI] [PubMed] [Google Scholar]
- Walker AK, Atkin JD. Stress signaling from the endoplasmic reticulum: A central player in the pathogenesis of amyotrophic lateral sclerosis. IUBMB Life. 2011;63:754–763. doi: 10.1002/iub.520. [DOI] [PubMed] [Google Scholar]
- Walker AK, Soo KY, Sundaramoorthy V, Parakh S, Ma Y, Farg MA, Wallace RH, Crouch PJ, Turner BJ, Horne MK, et al. ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS One. 2013;8:e81170. doi: 10.1371/journal.pone.0081170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AK, Spiller KJ, Ge G, Zheng A, Xu Y, Zhou M, Tripathy K, Kwong LK, Trojanowski JQ, Lee VM. Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol. 2015;130:643–660. doi: 10.1007/s00401-015-1460-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Popko B, Roos RP. The unfolded protein response in familial amyotrophic lateral sclerosis. Hum Mol Genet. 2011;20:1008–1015. doi: 10.1093/hmg/ddq546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggins AK, Wei G, Doxakis E, Wong C, Tang AA, Zang K, Luo EJ, Neve RL, Reichardt LF, Huang EJ. Interaction of Brn3a and HIPK2 mediates transcriptional repression of sensory neuron survival. J Cell Biol. 2004;167:257–267. doi: 10.1083/jcb.200406131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Zhang J, Pho V, Bonasera SJ, Holtzman J, Tang AT, Hellmuth J, Tang S, Janak PH, Tecott LH, Huang EJ. Essential function of HIPK2 in TGFbeta-dependent survival of midbrain dopamine neurons. Nat Neurosci. 2007;10:77–86. doi: 10.1038/nn1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525:56–61. doi: 10.1038/nature14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Jansen-West K, Xu YF, Gendron TF, Bieniek KF, Lin WL, Sasaguri H, Caulfield T, Hubbard J, Daughrity L, et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014;128:505–524. doi: 10.1007/s00401-014-1336-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.