

1. Summary
The allosteric mechanism of the heterodimeric enzyme imidazole glycerol phosphate synthase was studied in detail with solution NMR spectroscopy and molecular dynamics simulations. We studied IGPS in complex with a series of allosteric activators corresponding to a large range of catalytic rate enhancements (26 – 4900 fold), in which ligand binding is entropically driven. Conformational flexibility on the millisecond timescale plays a crucial role in intersubunit communication. Carr-Purcell-Meiboom-Gill relaxation dispersion experiments probing Ile, Leu, and Val methyl groups reveal that the apo- and glutamine-mimicked complexes are static on the millisecond timescale. Domain-wide motions are stimulated in the presence of the allosteric activators. These studies, in conjunction with ligand titrations, demonstrate that the allosteric network is widely dispersed and varies with the identity of the effector. Further, we find that stronger allosteric ligands create more conformational flexibility on the millisecond timescale throughout HisF. This domain-wide loosening leads to maximum catalytic activity.
2. Introduction
The functional implications of direct interactions between enzyme and ligand due to electrostatic and hydrophobic contacts are often readily rationalized from structural and kinetic data,(Wolfenden et al., 2001) however an understanding of how these binding interactions are propagated to remote sites is often less clear. Nonetheless, an enzyme’s ability to leverage the free energy of effector ligand binding to coordinate structural and dynamical changes over long molecular-scale distances is a hallmark of allostery. Given the central role of allostery in a variety of biological processes, including its functional regulation of enzymatic reactions, allostery has been the object of scientific inquiry for decades(Koshland Jr. et al., 1966; Monod et al., 1965; Tsai et al., 2014) and several well-defined structural,(Daily et al., 2009; Demerdash et al., 2009; Fairman et al., 2011; Laskowski et al., 2009; Popovych et al., 2009) energetic,(Hilser, 2010; Hilser et al., 2012; Motlagh et al., 2012; Motlagh et al., 2014) and dynamic (Bahar et al., 2007; Bakan et al., 2009; Gunasekaran et al., 2004; Ming et al., 2005; Popovych et al., 2006; Rivalta et al., 2012; Rousseau et al., 2005; Tzeng et al., 2009) models exist to describe this phenomenon. Additionally, it has also recently been demonstrated that multiple allosteric models can be operative in a single enzyme.(Freiburger et al., 2011; Freiburger et al., 2014)
The glutamine amidotransferase imidazole glycerol phosphate synthase (IGPS) is an allosteric enzyme that lies at the branch point of purine and histidine biosynthetic pathways of bacteria, archea, plants, and fungi.(Chaudhuri et al., 2001; Myers et al., 2005; Sinha et al., 2004) IGPS from Thermatoga maritima is a 51 kDa heterodimeric complex of HisH (23 kDa) and HisF (28 kDa) enzymes that synchronizes two distinct catalytic reactions in active sites separated by over 25 Å.(Beismann-Driemeyer et al., 2001) HisH catalyzes the hydrolysis of glutamine (Gln) to yield glutamate and ammonia after which HisF utilizes the newly generated NH3 in the cyclization of its substrate, N′-[5′-phosphoribulosyl)formimino]-5-aminoimidazole-4-carboxamide-ribonucleotide (PRFAR) as shown in Figure 1. The mechanism of PRFAR cyclization to yield imidazole glycerol phosphate (IGP) and 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) has been described in detail. (Beismann-Driemeyer et al., 2001; Chaudhuri et al., 2001; Chaudhuri et al., 2003) PRFAR cyclization is dependent on successful production, sequestration, and translocation of NH3 that is generated nearly 30 Å from the HisF effector site, and the glutaminase and cyclization reactions are tightly coupled in a 1:1 stoichiometry (Chaudhuri et al., 2001) despite occurring in different enzyme subunits.
Figure 1.
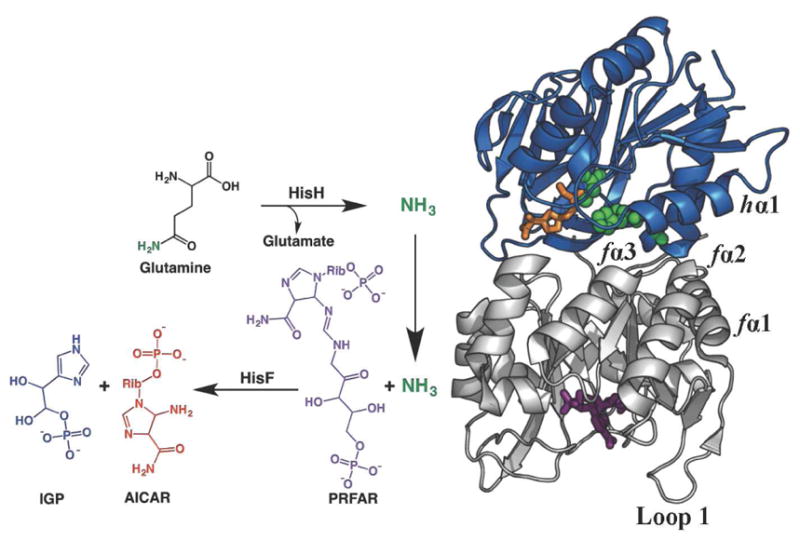
IGPS Reaction. The T. maritima IGPS complex contains two enzyme subunits; HisH (blue), which catalyzes the hydrolysis of glutamine (the Gln analogue acivicin is shown in orange sticks) near a conserved catalytic triad (green spheres) and HisF (gray), which catalyzes the cleavage and cyclization of the allosteric activator and substrate PRFAR (purple sticks). The inverted arrow depicts the path of NH3 travel and key secondary structural elements of IGPS are labeled.
In the absence of HisF-bound PRFAR, the rate of Gln hydrolysis in the HisH subunit is negligible (1.2 x 10−3 s−1)(Myers et al., 2003) but PRFAR binding stimulates the glutaminase subunit, enhancing this basal rate of NH3 production by 4900-fold. We have previously shown by nuclear magnetic resonance (NMR) spectroscopy that PRFAR binding allosterically activates a network of dynamic amino acid residues in IGPS. This network is significantly different and strengthened compared to apo IGPS.(Lipchock et al., 2010; Rivalta et al., 2012) Thus, binding of PRFAR to HisF converts the rigid apo enzyme to one that undergoes protein-wide conformational exchange motions on the millisecond timescale.(Lipchock et al., 2009, 2010) A subsequent computational analysis further demonstrated that molecular motions were involved in propagating allosteric information from the PRFAR binding site to the HisF/HisH interface.(Manley et al., 2013; Rivalta et al., 2012) In particular, communication among dynamic clusters determined from network analysis is more robust when PRFAR is bound compared to the loosely connected communities of the apo enzyme. The network of dynamic residues identified in NMR and computational studies represents a putative allosteric pathway involving α-helices within HisF (fα2, fα3) and HisH (hα1)1, extending from the PRFAR binding site to the dimer interface and beyond to the HisH glutaminase active site. Importantly, this work showed that PRFAR binding causes a merger of two separate community networks and the location of this merger is adjacent to the bond in PRFAR that is eventually cleaved upon reaction with NH3.(Rivalta et al., 2012) Furthermore, NMR and computational studies provided little evidence for large amplitude ligand-induced structural changes in IGPS, instead suggesting that ligand binding activates ms molecular motions that enable glutaminase chemistry to occur by allowing sampling of the active conformation with the appropriate dipolar stabilization of the developing oxyanion on the glutamine substrate.(Lipchock et al., 2010; Rivalta et al., 2012) The motions induced by PRFAR are widespread throughout the HisF subunit, however other ligands that bind to the PRFAR site were shown to activate glutaminase activity, albeit to a lesser amount than PRFAR. This observation prompted the need to investigate whether a single allosteric network or multiple delocalized pathways are responsible for allosteric information transfer.
In order to further elucidate pathways in this intricate allosteric network, we examined the effects of the binding of a series of small molecule allosteric activators on the conformational motions in IGPS. These allosteric ligands stimulate glutaminase activity in IGPS to varying degrees, with PRFAR being the most activating followed by the combination of IGP and AICAR, IGP alone, and AICAR alone.(Myers et al., 2003) The enhancement of glutamine catalysis over the basal level is 4900, 330, 110, and 26 fold for PRFAR, AICAR & IGP, IGP, and AICAR, respectively. Here, we detail the effects of the binding of these ligands on the structure and dynamics of IGPS. We studied binary complexes of IGPS with each of these effectors and ternary complexes that mimic the catalytic state of Gln- and PRFAR-bound IGPS by utilizing the covalently bound Gln analogue, acivicin.(Chittur et al., 2001) NMR analysis of the individual IGPS-ligand complexes highlight a widely dispersed and effector-dependent allosteric network in this enzyme and show that each of these ligands is capable of stimulating ms motions in the HisF domain and beyond, however the number and location of these dynamic residues is variable. Interestingly, the degree to which each allosteric ligand induces conformational flexibility is intertwined with its ability to activate the enzyme.
3. Results
Allosteric Ligand Binding is Entropically Driven
Isothermal titration calorimetry (ITC) experiments demonstrated that PRFAR binding is an endothermic and entropically driven process ΔH = 6.3 ± 0.04 kcal/mol and −TΔS = −14.5 ± 0.06 kcal/mol at 298 K.(Lipchock et al., 2010) Endothermic ITC binding profiles for the other activating ligands, IGP and AICAR, are also observed (Figure 2). The ITC-derived Kd for IGP is 0.5 mM and is within a factor of 3 of its apparent activation constant.(Myers et al., 2003) The unfavorable binding enthalpy of IGP (ΔH = 1.34 kcal/mol) is compensated by an entropic gain (−TΔS = −5.89 kcal/mol) that drives the spontaneous process. The binding of AICAR yields reproducible isotherms with minimal enthalpy change, complicating quantitation of the integrated heat. However, previous kinetic reports(Myers et al., 2005; Myers et al., 2003) as well as NMR data from this work conclusively show that AICAR binds to IGPS. The Kd for AICAR obtained from NMR lineshape analysis (Fig. S2) is 1.5 ± 0.2 mM, and based on this value and an estimated calorimetric ΔH ~ 0, −TΔS for AICAR binding is approximately −3.5 kcal/mol. The lineshape derived Kd value for AICAR is similar to its apparent activation constant, Kact (1.3 mM) obtained from kinetic data.(Myers et al., 2003) Interestingly, these ITC data show a correlation between degree of glutaminase activation by a particular allosteric ligand and the magnitude of the increase in TΔS.
Figure 2.

Ligand binding to IGPS by ITC. Changes in free energy (black), enthalpy (red), and entropy (blue) are shown for the formation of ternary complexes through binding of AICAR, IGP, or PRFAR to acivicin-conjugated IGPS. The catalytic rate enhancements caused by each allosteric ligand are shown under the ligand label. See also Figures S1, S2.
Allosteric Ligands Cause Small but Measurable Chemical Shift Changes
The NMR chemical shift parameter is exquisitely sensitive to the three-dimensional structure of proteins. To assess changes in the structure of IGPS caused by the formation of binary (IGPS and various effector ligands) and ternary (IGPS, effector ligand and the covalent Gln analogue, acivicin) complexes with AICAR, IGP, AICAR & IGP, and PRFAR, the chemical shifts of Ile, Leu, and Val (ILV) methyl groups as well as 1H-15N backbone amide resonances were monitored in a series of 1H-13C HMQC (Tugarinov et al., 2004; Velyvis et al., 2009) and 1H-15N TROSY(Pervushin et al., 1997) NMR spectra, respectively, during a titration with the ligands until IGPS was saturated. PRFAR and IGP cause the most substantial changes in HisF chemical shifts (Figure 3) yet the majority of shifts are less than 25 parts per billion (ppb). AICAR binding has an even smaller effect. Interestingly, somewhat larger chemical shift perturbations reappear when AICAR is titrated together with IGP.
Figure 3.

Ligand-induced chemical shift perturbations. Shown are changes in (A) AICAR, (B) IGP, (E) AICAR & IGP, and (F) PRFAR effector-bound ternary complexes relative to apo IGPS. Black lines represent 1.5σ from the 10% trimmed mean of the four combined data sets. Significant changes (above the black line) are mapped onto the HisF structure (C,D,G,H) (PDB 1GPW).(Douangamath et al., 2002) See also Figures S3–S5.
Combined chemical shift differences (Δδ1H13C) reporting the magnitudes of perturbations following formation of the IGPS ternary complexes were determined from , where Δδ1H and Δδ13C are the chemical shift differences between the effector-saturated and apo IGPS. Significant perturbations determined from the 10% trimmed mean of the Δδ1H13C values are mapped onto the HisF structure for each ternary complex (Figure 3), highlighting regions of IGPS that are most sensitive to the presence of allosteric ligands. Several residues in close proximity to the HisF binding pocket experience the largest chemical shift changes in the presence of these effectors. The fL50 methyl groups located in the HisF binding pocket and are within 3.5 Å of PRFAR, appear particularly sensitive to ligand binding, as these chemical shifts are perturbed in the presence of every effector tested with the exception of AICAR (Fig. S4). Further, effector binding poses obtained from 100 ns MD simulations detail the effect of HisF ligands on the fV48, fL50 and fI52 cluster at the fβ2 strand, as previous results have indicated that these hydrophobic contacts are part of the IGPS allosteric pathway originating at the HisF binding pocket. Here, PRFAR binding induces disruption of the fL48-fL50 interaction, which is present in the apo enzyme, in favor of a new fL50-fI52 contact (Fig. S4c), thereby altering hydrophobic interactions with the nearby Loop 1 and fα2 helix (Rivalta et al., 2012). The MD trajectories indicate that IGP is strongly bound to IGPS via the glycerol phosphate moiety and its motion does not interfere with those of the fβ2 hydrophobic residues (Fig. S4b). AICAR, however, is also mobile at the HisF active site, weakening the fL50-fI52 contact and imposing a configuration of these residues different from that of PRFAR. This outcome is consistent with NMR data (Fig. S4d) indicating that the fL50 resonance is not affected by the AICAR titration while it is sensitive to IGP, AICAR & IGP, and PRFAR titrations, which alter the hydrophobic fβ2 cluster upon binding.
These small chemical shift perturbations caused by the allosteric ligands extend beyond the HisF binding site, reaching the HisF/HisH interface in all cases. Even the weakest glutaminase activator, AICAR, perturbs ILV chemical shifts that are >15 Å away from the effector site. These data suggest the allosteric network of IGPS is widely dispersed and illuminate ligand-specific pathways that may be essential for propagation of the allosteric signal. Interestingly, IGP perturbs the chemical shifts of more residues than PRFAR, even though it is a weaker allosteric activator, indicating that chemical shift effects alone do not provide the entire picture of IGPS allostery (Figure 3). For example, the number residues that experience significant ligand-dependent chemical shift changes are 2, 36, 23, and 15 for AICAR, IGP, AICAR & IGP, and PRFAR, respectively. Thus the two most activating allosteric ligands perturb the chemical shifts of fewer residues than lesser activating ligands. However, the allosteric pathways outlined by these chemical shift data differ with each effector, suggesting that a potential ‘PRFAR-specific’ pathway is still the most preferable for allosteric communication.
To obtain additional insight into ligand-induced chemical shift changes, we calculated the synergistic chemical shift changes upon formation of the IGPS ternary complex. These shifts (Δδ) were determined by comparing the chemical shift change between the apo (δApo) and ternary (δTernary) complex versus the sum of the difference between apo and the two binary acivicin-bound (δAcivicin), and effector-bound (δEffector) complexes as given by ΔΔδ = ΔδTernary-Apo − (ΔδEffector-Apo + ΔδAcivicin-Apo). Thus, values of ΔΔδ that are larger than the defined cutoff value (1.5 times greater than the standard deviation of the 10% trimmed mean value) are indicated as ‘synergistic’ chemical shifts. This analysis shows that the PRFAR-bound IGPS enzyme undergoes a greater number (fV12, fV48, fL50, fV126, fL153, fI168, fL222, fV226, fV234, and fL237) of synergistic chemical shifts in HisF than do the other ligands (Fig. S5). Analysis of ΔΔδ for AICAR-bound IGPS shows only four ILV resonances with significant synergistic shifts, (fL2, fV12, fV79, and fL169). The next most activating ligand, IGP, produces synergistic shifts in the resonances of eight ILV residues (fL2, fL10, fV12, fL50δ1, fL50δ2, fL65, fV126, fL179, fL222) as does the simultaneous binding of both AICAR and IGP (fL10, fI44, fV69, fV79, fI93, fV125, fV134, and fV226) shown in Fig. S5. Formation of the PRFAR ternary complex induces synergistic perturbations in ten ILV residues, only half of which show significant ΔΔδ in the analysis of other effectors. Thus, synergistic changes in the chemical shifts of fV48, fL153, fI168, fV234, and fL237 are exclusive to PRFAR-bound IGPS, suggesting they or residues nearby may be particularly important for allostery in the formation of the most enzymatically active complex. In support of this, mutagenesis of residues fD98, fK99, hY138 and hK181, which are adjacent to these residues cause a varying degree of allosteric defects in IGPS, presumably through disruption of the interfacial salt bridges proposed to regulate NH3 transport to HisF.(List et al., 2012) Further, the significant number of ligand-dependent differences in this synergy, especially those found in comparison to the PRFAR-bound ternary complex, indicates there are likely multiple allosteric networks within IGPS. Thus, the best allosteric activator does not cause large chemical shifts changes in the most residues in comparison to other ligands; but PRFAR binding does result in the largest number of residues with synergistic chemical shift changes. Moreover, the number of synergistic chemical shift changes correlates well with the activating power to the allosteric ligands.
The Allosteric Signal Travels to the HisH Active Site
The allosteric signal resulting from ligand binding in HisF must travel more than 25 Å to the glutaminase active site of HisH. Previous work suggested that PRFAR, rather than cause large amplitude structural changes, induces millisecond motions that result in temporal sampling of the catalytically active HisH structure without large deviations from the most populated, inactive solution structure. It is possible that sampling of the active conformer occurs more readily when PRFAR is bound compared to the other allosteric effectors. Crystallographic(Chaudhuri et al., 2001; Chaudhuri et al., 2003; Douangamath et al., 2002), NMR(Lipchock et al., 2010), and molecular dynamics(Manley et al., 2013; Rivalta et al., 2012) studies suggest a rotation of the HisH active site hPro49-hGly50-hVal51-hGly52 (PGVG) loop, which lies adjacent to the HisH catalytic triad, is required for proper formation of the oxyanion hole during catalysis, although double-mutant studies suggest additional factors are essential for HisH catalysis.(List et al., 2012) To examine long-range perturbations at the glutaminase site caused by effector binding, we monitored the amide backbone chemical shifts of the hG50 and hG52 resonances of the PGVG loop during allosteric ligand titration into 2H,15N-labeled HisH-IGPS (Figure 4).
Figure 4.

Ligand-induced exchange broadening of hG50 and hG52 located in the HisH PGVG loop. (A) The ligand titration ranges from red (apo) to purple (effector-saturated) and percent decrease in peak intensity is shown in the inset. (B) One dimensional slices of the hG50 resonance showing the decrease in peak intensity from PRFAR binding. Relative peaks heights of the ligand-bound resonances are shown with arrows in the bottom panel.
Similar to our observations in HisF, we detected very small chemical shift changes in the hG50 and hG52 resonances during titration with PRFAR and no changes during titration with IGP or AICAR. However, a significant amount of exchange broadening is observed (Figure 4), indicative of enhanced ms motions at these residues. These ligand-induced motions at the HisH active site are consistent with MD simulations that showed greater flexibility in the PGVG loop residues in the presence of PRFAR.(Manley et al., 2013; Rivalta et al., 2012) The extent of the broadening is effector dependent for hG50 and hG52, where saturation with PRFAR, results in a 76% and 46% decrease in peak intensity relative to that in apo IGPS. The combination of AICAR & IGP has a greater effect than either ligand alone, most notably on hG50, which is broadened by 36%. IGP broadens hG50 by 28%, but has minimal effect on hG52. AICAR binding decreases the intensities of the hG50 and hG52 resonances by 19% and 28%, respectively. Interestingly, the degree of exchange broadening found in these HisH PGVG loop residues correlates with effector strength, especially for hG50. More importantly, these data demonstrate that all allosteric ligands can induce motional changes that span the dimer interface, reaching the HisH active site. Thus, these data indicate that PRFAR alters the motions at the HisH active site more significantly than all other activators and the degree to which motions are altered is loosely correlated with the effectiveness of allosteric activation.
Allosteric Ligands Induce Millisecond Motions in HisF
Formation of IGPS ternary complexes results in ligand-induced exchange broadening of HisF resonances in the 1H-15N TROSY HSQC spectra of each effector-bound IGPS complex (Figure 5). Binding of AICAR to a preformed complex of acivicin-IGPS causes broadening of four amide resonances, although two of these, fL10 and fL253, lie at the PRFAR binding site and at the HisF/HisH interface, respectively (Figure 5B). Ternary complexes containing IGP and AICAR & IGP show similar but amplified effects on the HSQC spectrum of HisF-IGPS, as 32 and 34 resonances are broadened beyond detection, respectively (Figure 5C,D). Both effectors induce broadening near the ligand site as well as the dimer interface. Additional broadening is observed in Loop 1, adjacent to the effector binding site, and the fα1 and fα3 helices. The combination of AICAR & IGP also broadens small clusters of resonances belonging to fα2, fα4 and fα6. Most strikingly, PRFAR binding causes 65 (25% of HisF) residues to be broadened beyond detection (Figure 5A). Clusters of broadened resonances in the PRFAR complex correspond to amino acids near the ligand binding site, Loop 1, the dimer interface, and smaller subsets of resonances from fα1-fα7 (Figure 5A,E).
Figure 5.

Ligand-induced exchange broadening in HisF. (A) Representative 1H-15N HSQC spectrum of apo IGPS (orange) and PRFAR-bound IGPS (purple). Residues that are significantly broadened in the presence of allosteric effectors are mapped onto the IGPS structure for the (B) AICAR, (C) IGP, (D) AICAR & IGP, and (E) PRFAR complexes. See also Figure S3.
A lesser degree of exchange broadening was observed in the 1H-13C TROSY HMQC spectra monitoring ILV residues of each effector-bound IGPS complex allowing additional investigation into these effects using NMR relaxation methods. To better quantitate ligand-induced millisecond motions, we compared ILV-based NMR CPMG relaxation dispersion experiments on binary and ternary complexes of AICAR-, IGP-, AICAR & IGP-, and PRFAR- bound HisF-IGPS.
Millisecond Motions Occurs in All Effector-Bound Complexes
A comparison of the millisecond motions in binary and ternary IGPS complexes show a number of similarities. Below, focus is restricted to the differences among the ternary complexes given its particular relevance to the active form of an allosteric enzyme.(Kimmel et al., 2000) A summary of the NMR results for all binary and ternary complexes is provided in Tables 1 – 6 in the Supporting Information.
Formation of each ternary complex results in changes in the ms motions at numerous amino acid sites. Figure 6 shows distinct CPMG relaxation dispersion profiles caused by the different allosteric activators for residues located throughout IGPS. Non-linear fitting of the dispersion curves results in unique rate constants (kex) for motion of each residue, with the PRFAR-bound ternary complex being the exception where the motion of all residues are better represented with a single kex value = 225 ± 30 s−1.
Figure 6.

Ligand-induced millisecond motions in IGPS. (A) Representative CPMG curves for effector-bound binary and (B) ternary complexes of IGPS for apo (black), AICAR (red), IGP (blue), AICAR & IGP (green), and PRFAR (purple) IGPS. See also Table S1 and Figure S8.
Formation of the ternary PRFAR complex induces millisecond motion in 36 ILV methyl groups, eight of which are unique to this ternary complex: fV12, fV17, fV48, fL63, fI116, fV140, fV190, and fL196. As noted above, all 36 methyl groups in the PRFAR ternary complex are well described by a single kex value, suggesting concerted motions in this most catalytically active ternary complex. All other binary and ternary complexes exhibit ms motions with large ranges of kex values (Figures 6, 7). A number of the motions detected in the PRFAR ternary complex result from the presence of both acivicin and PRFAR, and these motions are not detected in their respective binary complexes.
Figure 7.

Structural clustering of kex from NMR relaxation dispersion. (A) The magnitudes of kex are mapped onto the structure of the IGP (left, middle) and PRFAR (right) ternary complexes for all residues undergoing ms exchange. Other IGPS complexes are in SI Fig 7. (B) Distribution of kex values for the apo enzyme (left), IGP ternary complex (middle), and PRFAR ternary complex (right). Colors in each histogram correlate with the legend in (A). Optimal bin sizing for these data were determined using the protocol outlined in (Scott, 1979). IGP, PRFAR, and acivicin are represented with blue, purple, and orange sticks, respectively. See also Tables S2–S7 and Figure S6.
Similarly, millisecond motions occur for a unique subset of residues in IGPS ternary complexes containing AICAR, IGP, and AICAR & IGP when compared to their corresponding binary state (Tables S1 – S7). In the ternary complex fewer ILV methyl groups undergo conformational exchange in AICAR-, IGP-, and AICAR & IGP enzyme forms compared to those in the PRFAR complex. However, these motions remain interspersed throughout the HisF α8β8 barrel (Figure 8). PRFAR induced motions are widespread in IGPS whereas dynamic residues within the other ternary complexes are primarily located in the β-strands of the HisF barrel. No ms motions were detected for residues in the HisF helices α4, α5, α6, or α7 surrounding the β-barrel in the IGP and AICAR & IGP complexes, unlike those observed for PRFAR. Thus, the left side of the HisF subunit (as oriented in Figure 8) in which the ribonucleotide portion of PRFAR binds, appears more static in ternary complexes with weaker effectors. A lack of flexibility in this region of HisF becomes more evident in the AICAR ternary complex, where even fewer secondary structural elements contain dynamic ILV residues. Millisecond motions are absent from the α1, α4, α5, α6, α7, and α8 helices and the β4, β5, β6, and β7 strands, confining the 18 dynamic methyl groups to a small region of HisF when AICAR is bound. Interestingly, there is a linear correlation between the number of flexible residues induced by a particular ligand and the enhancement of kcat/Km for glutaminase activity (Figure 8A).
Figure 8.

Flexible residues and catalytic rate enhancement in IGPS. (A) Correlation of kcat/Km vs. the number of ILV residues that experience ms motions as determined from CPMG relaxation dispersion measurements for AICAR, IGP, AICAR and IGP, and PRFAR ternary complexes. (B–J) Flexible residues that show ligand induced ILV dispersion curves or amide exchange broadened (beyond detection) in each of the ternary complexes. (C, E, G, and I) Rotated views of B, D, F, H, respectively. Residues are shown as a surface rendering with each active site labeled. The allosteric ligands are shown as wire mesh. The HisH subunit is shown in cyan. See also Figures S7, S8.
Unique motions in the PRFAR ternary complex extend from the HisF ligand site to the HisF/HisH interface and are dispersed throughout the domain (Figure 8). The locations of uniquely flexible residues in the ternary complex containing AICAR & IGP are similar to those found with PRFAR, occurring near the HisF active site and dimer interface. However, flexible residues unique to the IGP- and AICAR-bound ternary complexes are concentrated exclusively near the HisF/HisH interface. Differences in the number and identity of flexible residues among the ternary complexes points to an allosteric network with multiple pathways of information relay between the cyclase to glutaminase active sites.
The concerted motions observed in the PRFAR ternary complex are not observed for any other allosteric ligand. Relaxation dispersion profiles measured for IGPS ternary complexes containing AICAR, IGP, and AICAR & IGP cannot be described by a single global process, and non-linear fits of these dispersion data yield a range of kex values, many of which are significantly higher than the exchange rate constant determined for the PRFAR complex. The fastest rates of conformational exchange are found in the IGP complex, with values of kex ranging from ~ 1000 – 6000 s−1. Exchange rates measured for ILV methyl groups in the AICAR & IGP-bound ternary complex are in a slower regime, but some still have a maximum kex value of 4900 s−1. Conformational exchange is slowest in AICAR ternary complex, occurring at a nearly identical rate to that of its corresponding binary complex (200 – 1500 s−1), with the exception of a single residue, fI44. These ligand-induced motions are due to conformational exchange in IGPS and not from binding and dissociation of the ligands themselves. Values of kon and koff for each ligand obtained by NMR lineshape analysis indicate that the rates of ligand binding and unbinding are too fast to contribute to the CPMG dispersion data. Thus, these data reflect motions within the enzyme itself.
The individually determined kex values mapped onto the structures of AICAR-, IGP-, and AICAR & IGP-bound IGPS reveal local clusters of similar exchange rates in all complexes. With the exception of the IGP ternary complex (Figure 7), most flexible residues experience motions slower than 1500 s−1 (Fig. S6). Smaller clusters of residues undergoing faster motions generally occur near the effector binding site and along the α-helices of the HisF barrel. These clusters, regardless of effector ligand and motional regime, universally form connective pathways from the HisF binding site to the dimer interface, which in some cases involve multiple routes. However, differences in the kinetic regimes of dynamic clusters could hinder efficient transfer of allosteric information.
Interestingly, nearly every flexible residue in the IGP ternary complex is shifted to a higher exchange timescale than that of the analogous PRFAR complex (Figure 7). Thus, the rate of conformational exchange is also not the sole determinant of activator efficiency, suggesting that a delicate balance exists between the number, location, and timescale of motions in HisF.
The ability to stimulate concerted conformational fluctuations appears to be unique to PRFAR, suggesting that non-concerted motions or motions faster than ~ 230 s−1 are less than optimal for catalytic activation and may partially explain the weakened activating power of IGP and AICAR. Although AICAR and IGP are allosteric activators of IGPS, there are significant gaps in the abilities of these effectors to enhance glutamine hydrolysis (4900-fold increase with PRFAR, 110-fold for IGP, 26-fold for AICAR). The differences between the conformational exchange rates measured for the PRFAR-bound ternary complex and those containing AICAR, IGP, or AICAR & IGP suggest that enhancement of ms motions alone are not enough to effectively relay allosteric information to the HisH active site. It appears that controlled, concerted motion in specific locations throughout the HisF domain provides the proper conformational fluctuations that are essential for efficient glutaminase activation, which is unique to the PRFAR-bound ternary complex.
4. Discussion
The enhancement of millisecond motions in IGPS is vital to communication between the HisF and HisH active sites. Numerous experimental studies of IGPS demonstrate a lack of structural change upon effector binding,(Chaudhuri et al., 2001; Chaudhuri et al., 2003; Douangamath et al., 2002; Lipchock et al., 2009, 2010; List et al., 2012; Rivalta et al., 2012; Vanwart et al., 2012) ruling out significant conformational changes as a means of relaying allosteric information. The X-ray crystal structures of apo and PRFAR-bound IGPS (PDB 1OX4 and 1OX5) reveal negligible changes in the structure due to PRFAR binding (rmsd = 0.41 Å).(Chaudhuri et al., 2003) Further, fluorescence quenching experiments examining solvent accessibility at the HisF/HisH interface yield the same results for apo, acivicin, binary PRFAR-bound, and ternary PRFAR-bound complexes, suggesting there are minimal differences in these structures.(Lipchock et al., 2010) However, a striking difference between apo IGPS and the allosterically activated enzymes is the ligand-enhanced ms motions. The extent of line broadening in the 1H-15N HSQC NMR spectrum of HisF-labeled IGPS during titrations with PRFAR indicate that this ligand causes a change in backbone amide motion on the millisecond timescale with over 25% of the amide resonances being broadened beyond detection. NMR relaxation experiments additionally demonstrate enhancement of conformational fluctuations in the 13CH3 groups of Ile, Leu, and Val residues within IGPS, when bound to PRFAR.(Lipchock et al., 2010)
To more fully understand the role of conformational fluctuations in IGPS allosteric signaling, we investigated the interactions of additional allosteric effectors, AICAR, IGP, and AICAR & IGP, with IGPS. These effectors provide a scale of chemical probes that are known to activate the glutaminase domain to different extents. Motions of the methyl groups of Ile, Leu, and Val residues within HisF were quantified in both binary and ternary complexes with AICAR, IGP, and AICAR & IGP with CPMG relaxation dispersion experiments. Based on these data, we have established a relationship between the glutaminase activation potential of allosteric ligands and the number of residues experiencing millisecond conformational flexibility. The increase in conformational flexibility caused by allosteric ligands also correlates with ITC measurements, which showed a trend in enthalpy/entropy compensation of AICAR, IGP, and PRFAR binding in which the more activating ligand is more enthalpically disfavored and more entropically favored (Figure 2).
Comparison of ms motions in AICAR-, IGP-, AICAR & IGP-, and PRFAR-bound ternary complexes reveals that better glutaminase activators cause a larger number of amide and ILV methyl groups to undergo millisecond conformational exchange (Figure 8). The location of these dynamic residues is also important as PRFAR, the native allosteric ligand, imparts the greatest degree of flexibility to the α-helices and β-strands of the α8β8 barrel at the center of HisF. AICAR & IGP and IGP alone are significantly weaker allosteric activators and although motions present in each of these ternary complexes are also spread around the HisF α8β8 barrel, they occur primarily on the β-strands. The weakest allosteric activator, AICAR, is capable of enhancing flexibility of only one half of the α8β8 barrel reminiscent of motions observed in apo and acivicin-bound IGPS. Millisecond motions are enhanced in β1, α2, β2, β3, and β8 of every ternary complex (Figure 8), suggesting that these regions of HisF are fundamental to relaying allosteric information. However, optimal allosteric signaling requires domain wide flexibility that includes additional structural elements within the α8β8 barrel, namely in the α-helices. Flexibility around the entire HisF barrel is presumably necessary for the highest level of glutaminase activation because it promotes fluctuations in the distances of the charged gate residues (fR5, fE46, fK99, and fE167), which are proposed to regulate passage of NH3 across the interface.
There have been a number of biochemical studies that have identified amino acids that are critical for relaying allosteric information between the PRFAR binding site and the glutaminase site in HisH (Amaro et al., 2007; Beismann-Driemeyer et al., 2001; Chaudhuri et al., 2003; Chittur et al., 2001; Klem et al., 1993). Many of those residues also show ms motions in the IGPS complexes studied here. There are four residues that comprise the salt bridge gate noted above. Of those, the amides of fK99 and fE167 are not assigned but adjacent residues fD98, fG96, fV100, and fV102 are assigned, as are fL169 and fL170. PRFAR stimulates ms motions for all of those gate residues or residues adjacent to the gate (fA3, fI6, fV48, fG96, fD98, fV100, fV102, fL169, and fL170). AICAR & IGP, IGP, and AICAR only activate ms motions in a subset of these residues. Notably, residues near fK99 are only flexible when PRFAR is bound.
Mutation of Loop 1 residue fK19 results in reduced allosteric communication in IGPS. fK19 is a critical component of the flexible region of Loop 1 that may interact with allosteric ligands in the cyclization site, consistent with significant increase in the normal 1:1 stoichiometry of glutamine hydrolysis:PRFAR cleavage in fK19 mutants.(Beismann-Driemeyer et al., 2001) The 1H-15N amide of fK19 is not assigned. Nonetheless, binding of PRFAR results in enhanced motions in fT21, fG20, fV18, fV17, and fV12 all located in Loop 1. The other ligands again only enhance ms motions in a subset of these residues. Thus, these ligands result in enhanced flexibility in amino acid residues that are known to be critically important for allostery.
The β-strands in IGPS are more conserved than are the α-helices (Fig. S8). The two weaker allosteric ligands, AICAR and IGP, primarily enhance motions in the conserved β-strands of the α8β8 barrel, while PRFAR and to a lesser extent AICAR & IGP, induce conformational flexibility throughout all secondary structural elements of HisF. Thus, ligand-induced motions within the highly conserved β-strands of HisF may be an intrinsic property of IGPS, regardless of effector, while additional flexibility in the α-helices of the HisF barrel more strongly influences the ability of allosteric ligands to activate T. maritima IGPS.
The extent of motions in a number of β-strands and several other localized regions of HisF in response to AICAR, IGP, AICAR & IGP, and PRFAR binding indicates that the allosteric network of IGPS is disperse. These data are consistent with several investigations of IGPS mutants, which have primarily concentrated on the HisF binding pocket and the HisF/HisH interface. Smaller numbers of mutants affecting the HisF α8β8 barrel were reported to disrupt glutaminase activity.(Beismann-Driemeyer et al., 2001; List et al., 2012)Our NMR chemical shift titrations and CPMG relaxation dispersion measurements indicate that numerous residues along the α8β8 barrel, such as fLeu50, fVal126, and fLeu222 undergo significant changes in chemical shifts and millisecond dynamics, lending support to the importance of mutational studies in the barrel region of HisF (Figs. S4,S5).
In addition to the effects of allosteric ligands on the HisF domain of IGPS, NMR line broadening also indicates ms motional changes in the HisH PGVG loop during chemical shift titrations, consistent with a dynamic mechanism involving loop rearrangement to the catalytically competent form of the enzyme.(Rivalta et al., 2012) The ability of allosteric effectors to induce broadening provides evidence that binding information in HisF is relayed across the dimer interface.
The obvious difference between PRFAR and the lesser activating ligands is the chemical linkage at the C7 position joining the AICAR portion to the uncyclized glycerol phosphate moiety. Interestingly, the site of bond cleavage and cyclization near C7 is positioned next to two HisF residues (G145 and T171) that only demonstrate flexibility when PRFAR is bound. The backbone amide of fG145 interacts with PRFAR through a bound water molecule and the hydroxyl side chain of fT171 is within H-bonding distance of N8 and the 2″ carbonyl of PRFAR. Simultaneous binding of AICAR and IGP only increase the flexibility of G145, whereas only T171 is flexible when IGP binds. Thus it is possible that these two residues ‘sense’ the presence of PRFAR and are the initiators of the concerted ms motions only observed in this complex.
The NMR data in this work are not consistent with a two-site population shift between inactive (apo) IGPS and an active structure. If this were the case, it would be expected that the ligand-induced shifts would follow a linear trend with PRFAR causing the largest shifts and the magnitude of other ligand-induced shifts would be commensurate with their activating power. This is not what is observed (Fig. S5). In some cases the shifts are linear and PRFAR does cause the largest change. But in the majority of cases the largest chemical shift perturbation is caused by IGP binding (Fig. S5). In addition, for a number of residues there is no linear trend among the ligands. Although the data are consistent with a ligand-induced effect it does not support a two-state T-to-R like transition between distinct structures.
The Rex parameter (Rex = papbΔωHΔωC) determined from relaxation dispersion experiments contains useful structural information about the lowly populated conformer in solution.(Beach et al., 2005; Grey et al., 2003; Massi et al., 2006) Previous studies (Beach et al., 2005; Boehr et al., 2006) have noted a correlation between Rex and Δω that provides supportive evidence for the so-called conformational selection model. In this work, we observe no correlation between the ligand induced chemical shift changes and Rex (Table S7). Moreover, the magnitude of Rex is not correlated with the activating power of the ligands. For example, on a per-residue basis, Rex for the IGP ligand is greater than that for PRFAR. These data seem to support a broadening of the ensemble of enzyme conformations while the average solution conformation does not change significantly. Within this ensemble it seems likely that multiple active states exist and PRFAR allows for more efficient sampling of these active conformations.
Concluding Remarks
We have used several small molecule effectors with varying abilities to activate the IGPS glutaminase domain to gain insight into the allosteric mechanism of this enzyme, specifically, the role that ms motions play in allosteric communication between the HisF and HisH active sites. Enhancement of ms motions in the highly conserved β1, β2, β3, and β8 strands of HisF are fundamental for allosteric signal transmission, but flexibility around the entire α8β8 barrel appears necessary for the most efficient glutaminase activation. Conformational fluctuations around the HisF barrel hint at an allosteric mechanism that relies on ‘unplugging’ the gated path at the HisF/HisH interface, allowing transportation of NH3 between domains. We also find that the number of ILV methyl groups undergoing conformational exchange in binary and ternary effector-bound IGPS complexes correlates with the activating power of the ligand. This conformational exchange is a concerted motional process in PRFAR-bound IGPS; however, residues in ternary complexes containing AICAR, IGP, and AICAR & IGP undergo asynchronous motions that generally occur much more quickly (> 1000 s−1). Thus, the concerted motion measured in the PRFAR-bound ternary complex sets it apart from other effectors and may contribute significantly to its high activating power. The results presented in this work reveal an intricate balance of contributions from the number, location, and rate of motions within HisF and outline important differences among these complexes that provide insight into the allosteric mechanism of IGPS.
5. Experimental NMR Procedures
Materials
5-Aminoimidazole-4-carboxamide ribotide (AICAR) and the glutamine analogue acivicin were purchased from Sigma (St. Louis, MO). D-erythro-imidazole glycerol phosphate (IGP) was purchased from Santa Cruz Biotechnology (Dallas, TX). Antibiotics used in protein expression and other analytical grade chemicals were purchased from AmericanBio (Natick, MA). PRFAR was synthesized as previously described.(Lipchock et al., 2010)
Acivicin labeling
Covalent modification of IGPS with the non-hydrolyzable Gln analogue acivicin was achieved by incubating IGPS with two molar equivalents of acivicin for 8–10 hours.(Chittur et al., 2001; Lipchock et al., 2010) A reduction in free thiol (Cys) content was confirmed with a DTNB assay. Excess acivicin was removed by dialysis against 10 mM HEPES (with 10 mM KCl, 0.5 mM EDTA) buffer at pH 7.3, and IGPS was subsequently concentrated using an Amicon centrifugal cell.
Isothermal Titration Calorimetry
ITC measurements were carried out using a MicroCal iTC 200 using 19 x 2.0 μL injections of ligand after an initial 0.2 μL injection, with 250 second spacing between injections. IGPS was prepared by dialysis against 2x 4.0 L of 50 mM HEPES (pH 7.6) with 10 mM KCl. Final IGPS concentration was measured by UV absorbance at 280 nm (ε = 29005 M−1cm−1). Experimental concentrations of IGPS were 0.15 mM (stoichiometrically modified with acivicin) while effector concentrations were 1.14 mM, 13 mM, and 20 mM for PRFAR, IGP, and AICAR, respectively.
NMR Titrations
NMR titrations were performed on a 14.1 T Varian Inova NMR spectrometer at 30 °C by collecting a series of 1H-13C TROSY HMQC and 1H-15N TROSY HSQC spectra with increasing ligand concentration. The 1H, 13C, carrier frequencies were centered at 0.75, 19.5 in the HMQC while the 1H and 15N carrier frequencies were set to the water resonance and 120 ppm, respectively. Saturation of HisF-IGPS was monitored by following resonance shifts until additions of ligand produced no further perturbation. PRFAR was titrated to a concentration of 0.9 mM, IGP to a concentration of 12 mM, and AICAR to a concentration of 14 mM. The NMR observed ligand saturation point is consistent with the expected values based on prior ITC experiments.
Methyl-TROSY Multiple Quantum (MQ) CPMG Dispersion Experiments
MQ CPMG experiments probing Ile, Leu, and Val methyl groups (13CH3) were performed on 14.1 T Varian Inova and 18.8 T Agilent NMR spectrometers at 30 °C. The methyl-TROSY CPMG pulse sequence was based on the report of Kay and coworkers(Korzhnev et al., 2004a; Korzhnev et al., 2004b). A constant relaxation delay period of 40 ms was used in the CPMG sequence, with varied τcp points of 0.0, 0.4167, 0.50, 0.625, 0.7682, 1.0, 1.4286, 2.0, 2.5, 3.333, 5.0, and 10.0 ms, with a recycle delay of 2.0 seconds.
Data Analysis and Processing
NMR spectra were processed with NMRPipe(Delaglio et al., 1995) and analyzed in SPARKY(Goddard et al.). Transverse relaxation rates (R2) were determined by measuring peak intensities of each ILV methyl resonance at multiple τcp delay points with a Perl-based exponential curve-fitting script. Relaxation dispersion curves were generated by plotting R2 vs. 1/τcp using RELAX(Bieri et al., 2011; d’Auvergne et al., 2008a, b) with the R2eff, NoRex, and MMQCR72 (full two-site Carver-Richards) models.(Morin et al., 2014) Relaxation dispersion data obtained at two static magnetic fields were fit simultaneously using the fast CPMG Equation and uncertainty values were obtained within RELAX from replicate spectra.
Supplementary Material
Highlights.
Allosteric ligand binding is correlated to structural dynamics in a model enzyme
A widely dispersed allosteric network has been identified
Catalytic rate enhancement varies with the degree of millisecond flexibility
Allosteric ligand binding is communicated over a 25-Å distance
eTOC Blurb.
Lisi et al. use NMR and computational methods to probe the allosteric influence of small molecule activators. Allosteric activator-induced changes in ms motions are responsible for enhancing the catalytic rate at a distant active site. Interestingly the most activating ligand results in the largest degree of motions.
Acknowledgments
This work was supported by NIH grant GM106121 to J.P.L. and V.S.B. We thank Kyle East for assistance with lineshape analysis.
Footnotes
We use lower case, italicized f and h when specifically referring to residues or secondary structure elements in the HisF or HisH subunits, respectively.
Author Contributions
J.P.L. and V.S.B. designed the research; G.P.L. and G.A.M. performed the research; G.P.L., G.A.M., and J.P.L. analyzed the data; G.P.L. and J.P.L. wrote the paper; I.R. and V.S.B. designed computational research; H.H. and I.R. performed computational work; I.R. and V.S.B. wrote computational work. ¶G.P.L. and G.A.M. contributed equally to this work.
Conflict of Interest
The authors declare no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amaro RE, Sethi A, Myers RS, Davisson VJ, Luthey-Schulten ZA. A Network of Conserved Interactions Regulates the Allosteric Signal in a Glutamine Amidotransferase. Biochemistry. 2007;46:2156–2173. doi: 10.1021/bi061708e. [DOI] [PubMed] [Google Scholar]
- Bahar I, Chennubhotla C, Tobi D. Intrinsic Dynamics of Enzymes in the Unbound State and Relation to Allosteric Regulation. Curr Opin Struct Biol. 2007;117:633–640. doi: 10.1016/j.sbi.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakan A, Bahar I. The Intrinsic Dynamics of Enzymes Plays a Dominant Role in Determining the Structural Changes Induced Upon Inhibitor Binding. Proc Natl Acad Sci USA. 2009;106:14349–14354. doi: 10.1073/pnas.0904214106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach H, Cole R, Gill M, Loria JP. Conservation of μs-ms Enzyme Motions in the Apo- and Substrate-Mimicked State. J Am Chem Soc. 2005;127:9167–9176. doi: 10.1021/ja0514949. [DOI] [PubMed] [Google Scholar]
- Beismann-Driemeyer S, Sterner R. Imidazole Glycerol Phosphate Synthase from Thermatoga maritima: Quaternary Structure, Steady-State Kinetics, and Reaction Mechanism of the Bienzyme Complex. J Biol Chem. 2001;276:20387–20396. doi: 10.1074/jbc.M102012200. [DOI] [PubMed] [Google Scholar]
- Bieri M, d’Auvergne EJ, Gooley PR. relaxGUI: A New Software for Fast and Simple NMR Relaxation Data Analysis and Calculation of ps-ns and us Motion of Proteins. J Biomol NMR. 2011;50:147–155. doi: 10.1007/s10858-011-9509-1. [DOI] [PubMed] [Google Scholar]
- Boehr DD, McElheny D, Dyson HJ, Wright PE. The Dynamic Energy Landscape of Dihydrofolate Reductase Catalysis. Science. 2006;313:1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- Chaudhuri BN, Lange SC, Myers RS, Chittur SV, Davisson VJ, Smith JL. Crystal Structure of Imidazole Glycerol Phosphate Synthase: A Tunnel Through the (Beta/Alpha)8 Barrel Joins Two Active Sites. Structure. 2001;9:987–997. [PubMed] [Google Scholar]
- Chaudhuri BN, Lange SC, Myers RS, Davisson VJ, Smith JL. Toward Understanding the Mechanism of the Complex Cyclization Reaction Catalyzed by Imidazole Glycerolphosphate Synthase: Crystal Structures of a Ternary Complex and the Free Enzyme. Biochemistry. 2003;42:7003–7012. doi: 10.1021/bi034320h. [DOI] [PubMed] [Google Scholar]
- Chittur SV, Klem TJ, Shafer CM, Davisson VJ. Mechanism for Acivicin Inactivation of Triad Glutamine Amidotransferases. Biochemistry. 2001;40:876–887. doi: 10.1021/bi0014047. [DOI] [PubMed] [Google Scholar]
- d’Auvergne EJ, Gooley PR. Optimisation of NMR Dynamic Models I. Minimisation Algorithms and Their Performance within the Model-Free Brownian Rotational Diffusion Spaces. J Biomol NMR. 2008a;40:107–119. doi: 10.1007/s10858-007-9214-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Auvergne EJ, Gooley PR. Optimisation of NMR Dynamic Models II. A New Methodology for the Dual Optimisation of the Model-Free Parameters and the Brownian Rotational Diffusion Tensor. J Biomol NMR. 2008b;40:121–133. doi: 10.1007/s10858-007-9213-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daily MD, Gray JJ. Allosteric Communication Occurs via Networks of Tertiary and Quaternary Motions in Proteins. PLoS Comput Biol. 2009;5:e1000293–e1100306. doi: 10.1371/journal.pcbi.1000293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A Multidimensional Spectral Processing System Based on UNIX Pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Demerdash ONA, Daily MD, Mitchell JC. Structure-Based Predictive Models of Allosteric Hot Spots. PLoS Comput Biol. 2009;5:e1000531–e1000555. doi: 10.1371/journal.pcbi.1000531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douangamath A, Wlaker M, Beismann-Driemeyer S, Vega-Fernandez MC, Sterner R, Wilmanns M. Structural Evidence for Ammonia Tunneling Across the (Beta/Alpha)8 Barrel of the Imidazole Glycerol Phosphate Synthase Bienzyme Complex. Structure. 2002;10:185–193. doi: 10.1016/s0969-2126(02)00702-5. [DOI] [PubMed] [Google Scholar]
- Fairman JW, Wijerathna SR, Ahmad MF, Xu H, Nakano R, Jha S, Prendergast J, Welin RM, Flodin S, Roos A, et al. Structural Basis for Allosteric Regulation of Human Ribonucleotide Reductase by Nucleotide-Induced Oligomerization. Nat Struct Mol Biol. 2011;18:316–322. doi: 10.1038/nsmb.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiburger LA, Baettig O, Sprules T, Berghuis AM, Auclair K, Mittermaier AK. Competing Allosteric Mechanisms Modulate Substrate Binding in a Dimeric Enzyme. Nat Struct Mol Biol. 2011;18:288–294. doi: 10.1038/nsmb.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiburger LA, Miletti T, Zhu S, Baettig O, Berghuis AM, Auclair K, Mittermaier AK. Substrate-Dependent Switching of the Allosteric Binding Mechanism of a Dimeric Enzyme. Nat Chem Biol. 2014;10:937–942. doi: 10.1038/nchembio.1626. [DOI] [PubMed] [Google Scholar]
- Goddard TD, Kneller DG. SPARKY 3. University of California; San Francisco: [Google Scholar]
- Grey MJ, Wang C, Palmer AG., III Disulfide Bond Isomerization in Basic Pancreatic Trypsin Inhibitor: Multisite Chemical Exchange Quantified by CPMG Relaxation Dispersion and Chemical Shift Modeling. J Am Chem Soc. 2003;125:14324–14335. doi: 10.1021/ja0367389. [DOI] [PubMed] [Google Scholar]
- Gunasekaran K, Ma B, Nussinov R. Is Allostery an Intrinsic Property of All Dynamic Proteins? Proteins: Structure, Function, and Bioinformatics. 2004;57:433–443. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- Hilser VJ. An Ensemble View of Allostery. Science. 2010;327:653–654. doi: 10.1126/science.1186121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilser VJ, Wrabl JO, Motlagh HN. Structural and Energetic Basis of Allostery. Annu Rev Biophys. 2012;41:585–609. doi: 10.1146/annurev-biophys-050511-102319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel JL, Reinhart GD. Reevaluation of the Accepted Allosteric Mechanism of Phosphofructokinase from Bacillus stearothermophilus. Proc Natl Acad Sci USA. 2000;97:3844–3849. doi: 10.1073/pnas.050588097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klem TJ, Davisson VJ. Imidazole Glycerol Phosphate Synthase: The Glutamine Amidotransferase in Histidine Biosynthesis. Biochemistry. 1993;32:5177–5186. doi: 10.1021/bi00070a029. [DOI] [PubMed] [Google Scholar]
- Korzhnev DM, Kloiber K, Kanelis V, Tugarinov V, Kay LE. Probing Slow Dynamics in High Molecular Weight Proteins by Methyl-Trosy NMR Spectroscopy: Application to a 723-Residue Enzyme. J Am Chem Soc. 2004a;126:3964–3973. doi: 10.1021/ja039587i. [DOI] [PubMed] [Google Scholar]
- Korzhnev DM, Kloiber K, Kay LE. Multiple-quantum relaxation dispersion NMR spectroscopy probing millisecond time-scale dynamics in proteins: theory and application. J Am Chem Soc. 2004b;126:7320–7329. doi: 10.1021/ja049968b. [DOI] [PubMed] [Google Scholar]
- Koshland DE, Jr, Nemethy G, Filmer D. Comparison of Experimental Binding Data and Theoretical Models in Proteins Containing Subunits. Biochemistry. 1966;5:365–368. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Gerick F, Thornton JM. The Structural Basis of Allosteric Regulation in Proteins. FEBS Lett. 2009;583:1692–1698. doi: 10.1016/j.febslet.2009.03.019. [DOI] [PubMed] [Google Scholar]
- Lipchock JM, Loria JP. Millisecond Dynamics in the Allosteric Enzyme Imidazole Glycerol Phosphate Synthase (IGPS) from Thermatoga Maritima. J Biomol NMR. 2009;45:73–84. doi: 10.1007/s10858-009-9337-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipchock JM, Loria JP. Nanometer Propagation of Millisecond Motions in V-Type Allostery. Structure. 2010;18:1596–1607. doi: 10.1016/j.str.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- List F, Vega MC, Razeto A, Hager MC, Sterner R, Wilmanns M. Catalysis Uncoupling in a Glutamine Amidotransferase Bienzyme by Unblocking the Glutaminase Active Site. Chemistry & Biology. 2012;19:1589–1599. doi: 10.1016/j.chembiol.2012.10.012. [DOI] [PubMed] [Google Scholar]
- Manley G, Rivalta I, Loria JP. Solution NMR and Computational Methods for Understanding Protein Allostery. J Phys Chem B. 2013;117:3063–3073. doi: 10.1021/jp312576v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massi F, Wang C, Palmer AG., III Solution NMR and Computer Simulation Studies of Active Site Loop Motion in Triosephosphate Isomerase. Biochemistry. 2006;45:10787–10794. doi: 10.1021/bi060764c. [DOI] [PubMed] [Google Scholar]
- Ming D, Wall ME. Allostery in a Coarse-Grained Model of Protein Dynamics. Phys Rev Lett. 2005;95:198103–198106. doi: 10.1103/PhysRevLett.95.198103. [DOI] [PubMed] [Google Scholar]
- Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: A plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- Morin S, Linnet T, Lescanne M, Schanda P, Thompson GS, Tollinger M, Teilum K, Gagne S, Marion D, Griesinger C, et al. relax: The Analysis of Biomolecular Kinetics and Thermodynamics using NMR Relaxation Dispersion Data. Bioinformatics. 2014;30:2219–2220. doi: 10.1093/bioinformatics/btu166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motlagh HN, Li J, Thompson EB, Hilser VJ. Interplay Between Allostery and Intrinsic Disorder in an Ensemble. Biochem Soc Trans. 2012;40:975–980. doi: 10.1042/BST20120163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motlagh HN, Wrabl JO, Li J, Hilser VJ. The Ensemble Nature of Allostery. Nature. 2014;508:331–339. doi: 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers RS, Amaro RE, Luthey-Schulten ZA, Davisson VJ. Reaction Coupling Through Interdomain Contacts in Imidazole Glycerol Phosphate Synthase. Biochemistry. 2005;44:11974–11985. doi: 10.1021/bi050706b. [DOI] [PubMed] [Google Scholar]
- Myers RS, Jensen JR, Deras IL, Smith JL, Davisson VJ. Substrate-Induced Changes in the Ammonia Channel for Imidazole Glycerol Phosphate Synthase. Biochemistry. 2003;42:7013–7022. doi: 10.1021/bi034314l. [DOI] [PubMed] [Google Scholar]
- Pervushin K, Riek R, Wider G, Wuthrich K. Attenuated T2 Relaxation by Mutual Cancellation of Dipole-Dipole Coupling and Chemical Shift Anisotropy Indicates an Avenue to NMR Structures of Very Large Biological Macromolecules in Solution. Proc Natl Acad Sci USA. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically Driven Protein Allostery. Nat Struct Mol Biol. 2006;13:831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovych N, Tzeng SR, Tonelli M, Ebright RH, Kalodimos CG. Structural Basis for cAMP-Mediated Allosteric Control of the Catabolite Activator Protein. Proc Natl Acad Sci U S A. 2009;106:6927–6932. doi: 10.1073/pnas.0900595106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivalta I, Sultan MM, Lee NS, Manley G, Loria JP, Batista VS. Allosteric Pathways in Imidazole Glycerol Phosphate Synthase. Proc Natl Acad Sci USA. 2012;109:E1428–E1436. doi: 10.1073/pnas.1120536109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau F, Schymkowitz JA. A Systems Biology Perspective on Protein Structural Dynamics and Signal Transduction. Curr Opin Struct Biol. 2005;15:23–30. doi: 10.1016/j.sbi.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Scott DW. On Optimal and Data-Based Histograms. Biometrika. 1979;66:605–610. [Google Scholar]
- Sinha SC, Chaudhuri BN, Burgner JW, Yakovleva G, Davisson VJ, Smith JL. Crystal Structure of Imidazole Glycerol-Phosphate Dehydratase: Duplication of an Unusual Fold. J Biol Chem. 2004;279:15491–15498. doi: 10.1074/jbc.M312733200. [DOI] [PubMed] [Google Scholar]
- Tsai CJ, Nussinov R. A Unified View of “How Allosery Works”. PLOS Comp Biol. 2014;10:1–12. doi: 10.1371/journal.pcbi.1003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugarinov V, Kay LE. An Isotope Labeling Strategy for Methyl TROSY Spectroscopy. J Biomol NMR. 2004;28:165–172. doi: 10.1023/B:JNMR.0000013824.93994.1f. [DOI] [PubMed] [Google Scholar]
- Tzeng SR, Kalodimos CG. Dynamic Activation of an Allosteric Regulatory Protein. Nature. 2009;462:368–372. doi: 10.1038/nature08560. [DOI] [PubMed] [Google Scholar]
- Vanwart A, Eargle J, Luthey-Schulten ZA, Amaro RE. Exploring Residue Component Contributions to Dynamical Network Models of Allostery. J Chem Theor Comput. 2012;8:2949–2961. doi: 10.1021/ct300377a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velyvis A, Schachman HK, Kay LE. Application of Methyl-TROSY NMR to Test Allosteric Models Describing Effects of Nucleotide Binding to Aspartate Transcarbamoylase. J Mol Biol. 2009;387:540–547. doi: 10.1016/j.jmb.2009.01.066. [DOI] [PubMed] [Google Scholar]
- Wolfenden R, Snider MJ. The depth of chemical time and the power of enzymes as catalysts. Acc Chem Res. 2001;34:938–945. doi: 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.