

Supplemental Digital Content is available in the text.
Keywords: apoptosis, lymphangiogenesis, phosphorylation, proteoglycans, VEGF receptor
Abstract
Rationale:
Lymphatic vessel growth is mediated by major prolymphangiogenic factors, such as vascular endothelial growth factor (VEGF-C) and VEGF-D, among other endothelial effectors. Heparan sulfate is a linear polysaccharide expressed on proteoglycan core proteins on cell membranes and matrix, playing roles in angiogenesis, although little is known about any function(s) in lymphatic remodeling in vivo.
Objective:
To explore the genetic basis and mechanisms, whereby heparan sulfate proteoglycans mediate pathological lymphatic remodeling.
Methods and Results:
Lymphatic endothelial deficiency in the major heparan sulfate biosynthetic enzyme N-deacetylase/N-sulfotransferase-1 (Ndst1; involved in glycan-chain sulfation) was associated with reduced lymphangiogenesis in pathological models, including spontaneous neoplasia. Mouse mutants demonstrated tumor-associated lymphatic vessels with apoptotic nuclei. Mutant lymphatic endothelia demonstrated impaired mitogen (Erk) and survival (Akt) pathway signaling and reduced VEGF-C–mediated protection from starvation-induced apoptosis. Lymphatic endothelial-specific Ndst1 deficiency (in Ndst1f/fProx1+/CreERT2 mice) was sufficient to inhibit VEGF-C–dependent lymphangiogenesis. Lymphatic heparan sulfate deficiency reduced phosphorylation of the major lymphatic growth receptor VEGF receptor-3 in response to multiple VEGF-C species. Syndecan-4 was the dominantly expressed heparan sulfate proteoglycan in mouse lymphatic endothelia, and pathological lymphangiogenesis was impaired in Sdc4(−/−) mice. On the lymphatic cell surface, VEGF-C induced robust association between syndecan-4 and VEGF receptor-3, which was sensitive to glycan disruption. Moreover, VEGF receptor-3 mitogen and survival signaling was reduced in the setting of Ndst1 or Sdc4 deficiency.
Conclusions:
These findings demonstrate the genetic importance of heparan sulfate and the major lymphatic proteoglycan syndecan-4 in pathological lymphatic remodeling. This may introduce novel future strategies to alter pathological lymphatic-vascular remodeling.
Growth and remodeling of lymphatic vasculature in the tumor microenvironment may be supported by a variety of growth factors that stimulate cognate receptors on host lymphatic vessels.1,2 The process has been shown to contribute to lymph node metastasis.3–5 It is also known that a dominant prolymphangiogenic growth factor, vascular endothelial growth factor (VEGF-C), is frequently overexpressed in primary carcinomas. Along with a unique role in driving developmental lymphangiogenesis,2,6 VEGF-C also plays critical roles in tumor lymphangiogenesis along with the stimulatory actions of other tumor vascular growth effectors, such as VEGF-A and fibroblast growth factor-2, as well as cytokines.6,7
Heparan sulfate is a linear glycan polymer expressed on a variety of proteoglycans, which plays important roles in endothelial growth factor binding in unique pathological contexts, including tumor angiogenesis.8–10 Heparan sulfate proteoglycans (HSPGs) secreted into tumor matrix may release growth factors on the action of tumor heparinase, mobilizing banks of proangiogenic factors bound to sulfated domains on heparan sulfate in tumor matrix.11,12 Endothelial-surface proteoglycans may act in cis in a cell-autonomous manner or in trans to promote endothelial proliferation in response to growth factors.10,13 Although less is known with respect to lymphatic biology, preliminary work points to a role for heparan sulfate in VEGF-C–dependent proliferation of lymphatic endothelial cells (LECs) in culture.14 However, the genetic importance, mechanisms, and proteins involved in vivo remain poorly understood.
We generated lymphangiogenesis models in mice bearing a lymphatic deficiency in the heparan sulfate biosynthetic enzyme N-deacetylase/N-sulfotransferase-1 (Ndst1), involved in initiating sulfate modifications of nascent heparan sulfate chains.8 We demonstrate that lymphangiogenesis is inhibited in models of oil granuloma–induced lymphangiogenesis, wound inflammation, and carcinomas on the Ndst1 mutant background, including VEGF-C–dependent lymphangiogenesis on a stringent lymphatic-specific Ndst1-deficient background. The mutation is associated with defects in lymphatic mitogen and survival signaling, and VEGF receptor-3 (VEGFR-3) phosphorylation in response to VEGF-C. Using proteoglycan expression analyses, gene-targeted mice and primary-cell mechanistic analyses, we further highlight syndecan-4 (Sdc4) as a major HSPG coreceptor required for VEGF-C mitogen and survival signaling, which complexes with VEGFR-3 in a glycan-dependent manner on VEGF-C exposure.
Methods
Cells and Cell Lines
Primary LECs were isolated from mouse mesenteric oil granuloma/lymphangiomas, as previously described,15 and tested for LYVE-1/podoplanin expression.14 For some studies, LECs were purified from lungs of Ndst1f/fTekCre+ mutants.16 Primary human lung LEC (hLEC; Lonza; previously shown >99% pure at third passage by Prox1 staining) were also used. For Lewis lung carcinoma (LLC) models, retroviruses expressing pLTR-mVEGFC-GFP were used to transduce LLCs (kindly provided by G. Thurston; Regeneron) with full-length VEGF-C (LLC-VC cells),17 or GFP-expressing empty vector (LLC-ev) cells as controls. The mouse transformed mesenteric LEC line (svLEC)18 was kindly obtained from Dr Alexander (LSU Health Sciences Center).
Mice and Pathological Lymphatic Proliferation Models
Details on mouse models targeting lymphangiogenesis in Ndst1 and Sdc4 mutants, including relevant references, are presented in the expanded Online Data Supplement.
Pathological Tissue Processing and Analysis
Tumor/tissue specimens were formalin-fixed, paraffin-embedded, and hematoxylin and eosin stained, with immunostaining details outlined in the Online Data Supplement.
Flow Cytometry
Lung digests were filtered through 100-μm strainers (Fisher), subjected to red-cell lysis (eBioscience), and stained with PE-labeled antimouse podoplanin (eBioscience) and APC-labeled antimouse LYVE-1 (R&D), with Aqua (Biolegend) viability marker for dead-cell exclusion. Dual PE/APC+ live cells were analyzed by an LSRII (BD) cytometer. The quantity of dual-positive cells as a percentage of total cells was analyzed/plotted and used in statistical analyses comparing lungs from mutant versus control mice.
Quantitative Polymerase Chain Reaction Analyses
RNA was isolated from primary LECs, reverse transcribed (Superscript III, Invitrogen), amplified using gene-specific primers to each core protein, and quantified (triplicate assays) using the 2−ΔΔCt method relative to β-actin. Primers included those for mouse HSPGs (Online Table I). For Ndst, the same method was used with primers for mouse Ndst1–Ndst4 isoenzymes.14
siRNA Transfections
Primary hLEC at near confluence were transfected with siRNA targeting heparan sulfate biosynthetic enzymes xylosyltransferase 2 (XylT2; siXylT2) or Ndst1 (siNdst1), the HSPG core protein Sdc-4 (siSdc4), or receptors VEGFR-2 (siVEGFR-2) or VEGFR-3 (siVEGFR-3); with scrambled-duplex RNA mock transfectants (siDS) as controls. Transfections (20-nmol/L siRNA) were carried out using Lipofectamine (Invitrogen) after manufacturer recommendations. Transfection complex was added in Opti-Mem (Gibco), and incubated for 6 hours, with cell recovery overnight in normal growth medium.
VEGF-C Species
Human recombinant mature VEGF-C was purchased (R&D). Untagged pro–VEGF-C was expressed from full-length cDNA using a chinese hamster ovary dhfr gene-amplification system.19 This was predominantly a mixture of unprocessed and partially processed propeptide forms of VEGF-C. Highly expressed clones were identified by the ability of the culture supernatant to sustain growth of VEGFR-3/EpoR Ba/F3 cells.20 The affinity of pro–VEGF-C for heparin was used in the capture step from serum-free culture supernatant (salt elution from heparin-sepharose column at 0.48 M NaCl, with minor peak at 0.53 M). Cation exchange chromatography (pH 6.6) and gel filtration were used to increase prep homogeneity. Identity was confirmed by Western blotting. A short form of VEGF-C consisting of minimum-binding domain residues A112-L215 was prepared as a strep-II–tagged protein in drosophila S2 cells and purified using streptactin resin. It did not bind to heparin. A mutant form of human VEGF-C (VEGF-CCys156Ser R&D) that binds exclusively to VEGFR-3 was used in some studies.
Immunoblotting
Detailed methods for Western blotting of lysates after VEGF-C stimulation of LECs are reported in the Online Data Supplement.
Receptor Tyrosine Kinase Phospho Arrays
Serum-starved hLEC were stimulated ± pro–VEGF-C (1 μg/mL) for 15 minutes. Lysates from 6-well plates were collected in 500-μL Lysis Buffer (R&D Phospho-RTK Array–assay instructions) and cleared with supernatant used in receptor tyrosine kinase-assay after manufacturer protocol. After blocking, diluted lysates were incubated with slide arrays (4°C overnight) and washed, and antiphosphotyrosine horseradish peroxidase–tagged antibody was added (2 hours at room temperature), followed by wash, chemiluminescent development, and digital-imaging densitometry. Further array details, including incorporation of mitogen-activated protein kinase array after VEGF-C stimulation, are described in the Online Data Supplement.
VEGFR-3 Phosphorylation Assays
Serum-starved cells were treated with VEGF-C species (1 µg/mL) and assayed using a human phospho-VEGFR3 Elisa Kit (R&D). Treated cells were lysed (R&D lysis buffer; 30 minutes, 4°C), spun-down, diluted, added to a precoated anti–VEGFR-3 plate overnight (4°C), followed by antiphosphotyrosine horseradish peroxidase (included in Kit). Biotin anti–VEGFR-3 (Reliatech) labeled with streptavidin-horseradish peroxidase (Vector) was added to detect total VEGFR3. After incubating in substrate solution (R&D), reactions were stopped with 2N sulfuric acid. Plates were assayed at absorbance A450 nm, with values corrected against total VEGFR-3.
Proximity Ligation Assays
Chamber slides (Lab-Tek) coated with 50-μg/mL PurCol (Advanced Biomatrix), layered with serum-starved hLEC were stimulated with human mature VEGF-C (R&D) for 5 minutes ± pre treatment with heparinase in some experiments. Cells were fixed in ice-cold methanol for 10 minutes and blocked (Olink PLA-blocking reagent; 30 minutes at 37°C). Rabbit antihuman VEGFR-3 (Reliatech; or for some studies, anti–VEGFR-2; Cell Signaling) and goat antihuman Sdc-4 (R&D) were then added (2 µg/mL) together (in blocking solution overnight; 4°C). After wash, Rabbit(−) and Goat(+) PLA Probes (Duolink assay; Olink Bioscience) were added (1:10 dilution). In other experiments, rabbit anti–VEGFR-3 was paired with either mouse antihuman Sdc-1 (Abcam) or mouse antihuman Sdc-2 (kind gift from G. David) antibodies. Antibodies were directed against extracellular domains, as per manufacturer protocol (imaging: 40× objective, room temperature).
Statistics
Mean values (±SD) were obtained for lymphatic vessel density (LVD) or apoptotic body index for each genotype. For some analyses, means were compared using Student t test, with normalization to wild-type (or control) baseline values. Paired t tests were applied for comparing means of paired values (eg, for multiple experiments examining western phosphorylation responses pre- versus post–VEGF-C species in siRNA versus control-transfected cells; comparisons of change in caspase signal in response to VEGF-C in siRNA versus control-transfected cells). For some experiments in which binary-type response data were examined (eg, terminal deoxynucleotidyl transferase dUTP nick-end labeling [TUNEL] positivity versus negativity of lymphatic vessels or the presence versus the absence of LYVE-1/podoplanin dual-positive cells in flow cytometry analyses of tumor LECs) the Wald χ2 statistic was used. Two-way ANOVA was used in the analyses for experiments in which the significance of any interaction between genotype and biological response to growth factor (eg, VEGF-C–dependent lymphangiogenesis) was examined.
A 2-way ANOVA was also applied to assess for phospho–VEGFR-3 responses to VEGF-C as they depend on siRNA status and lymphatic endothelial PLA responses to VEGF-C as they depend on treatment ± heparinase. SPSS version 19, general linear model function, was used to compute these ANOVAs. A P value of ≤0.05 was considered significant for all analyses.
Results
Endothelial Mutation Resulting in Lymphatic Ndst1 Deficiency Is Associated With Reduced Pathological Lymphangiogenesis and Altered Lymphatic Signaling
We assessed lymphangiogenesis in models of granulomatous inflammation, wound inflammation, and tumor lymphatic remodeling on the Ndst1f/fTekCre+ mutant background. We first used an established model of oil granuloma/lymphangioma induction in the mouse abdomen,15 wherein plaque-like lesions develop intense proliferation of LYVE-1+ lymphatic endothelium expressing VEGFR-3/Flt-4.15,21 In this model, although the Ndst1 mutation exists in all endothelia, lesion-associated LYVE-1+ vessel density in Ndst1f/fTekCre+ mutants was reduced (Figure 1A), suggesting that lymphatic Ndst1 deficiency affected pathological lymphangiogenesis. Wound lymphangiogenesis associated with early skin-wound remodeling was also reduced on the mutant background (Figure 1B). To examine tumor lymphangiogenesis, we crossed mutants with an MMTV-PyMT spontaneous mammary tumor strain. Tumors demonstrated reduced LVD in Ndst1f/fTekCre+ mutants (Figure 1C, top left). Tumor expression of VEGF-C was confirmed (Figure 1C, top right). TekCre transgene expression is pan-endothelial, and studies quantifying blood-vascular angiogenesis in the mutants (not shown) revealed a 67% reduction in tumor blood-vascular density relative to wild-type by CD105 staining (P<0.001) and 50% reduction by CD31 (P<0.01). The unique effect of mutation on LYVE-1+ vessel-density coupled with marked tumor VEGF-C production prompted us to further explore the effect of altered lymphatic heparan sulfate on VEGF-C–mediated lymphangiogenesis. As a developmental baseline, LVD in the ear bud of newborn mice, which is uniquely VEGF-C dependent,2 was modestly reduced in Ndst1f/fTekCre+ mutants (Online Figure IA). Although the LVD reduction was significant, this did not result in obvious lymphatic developmental defects, such as limb edema or chylous ascites. Knockdown of Ndst1 in primary lung LECs from nonchallenged mutants was confirmed by quantitative polymerase chain reaction (Online Figure IB).
Figure 1.
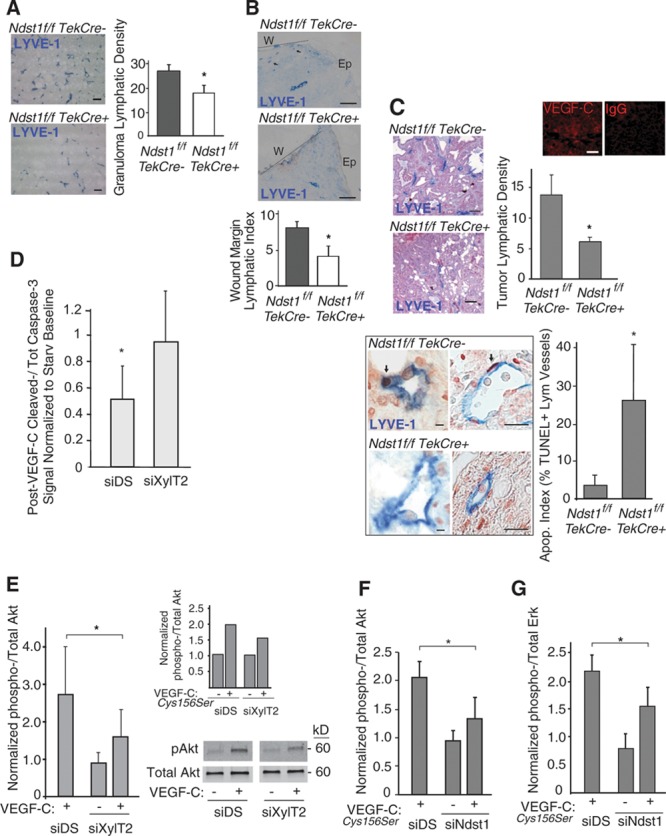
Pan-endothelial mutation in heparan sulfate biosynthesis results in altered pathological lymphangiogenesis, lymphatic-vascular apoptosis, and altered vascular endothelial growth factor-C (VEGF-C) signaling. A, Oil granulomas were generated in Ndst1f/fTekCre+ mutants and Cre− littermates to examine lymphangiogenesis in this model. Sprouting of LYVE-1+ vessels (blue) in lesions was examined by immunohistology (bar=100 μm). Mean lymphatic vessel density graphed to right (n=4 mice/genotype; *P=0.003 for difference). B, Wound lymphangiogenesis after a full-thickness punch-type skin lesion was examined in Ndst1f/fTekCre+ mutants and Cre− controls. LYVE-1+ lymphatic vessels are shown (arrows) with wound edge (dotted line; W) and adjacent epithelial (Ep) surface. Mean lymphatic vessels per wound margin for each genotype graphed below (n=4 mutant and 5 wild-type mice; *P=0.002 for difference; bar=50 μm). C, Lymphangiogenesis was examined in a spontaneous breast carcinoma model. Tumor sections from mutant and control females show LYVE-1+ vessels in blue (bar=100 μm), with vessel density plotted (n=4 mice/genotype; *P=0.004 for difference). Tumor VEGF-C was confirmed (upper-right, immunofluorescence with IgG control; bar=20 μm). Lymphatic apoptotic index (quantity of dual terminal deoxynucleotidyl transferase dUTP nick-end labeling [TUNEL]/LYVE-1+ vessels as a percentage of total LYVE-1+ vessels for each tumor) was examined: Photomicrographs show examples of blue LYVE-1+ vessels with dark TUNEL+ lymphatic nuclei (arrows) in 2 of the Cre+ mutant sections. Apoptotic index is graphed to right (*P<0.001 for difference; n=4 mice/group; left panels bar=20 μm; right panels bar=100 μm). D, Human lung lymphatic endothelial cells (hLEC) were tested for reduction in apoptosis as a result of mature VEGF-C exposure after a 6-h starvation period. In response to VEGF-C exposure, the ratio of cleaved- to total caspase-3 was examined by western, and normalized to densitometry for starved control-transfectant cells (starv baseline); with assays carried out in triplicate wells for each condition. Graph: response of xylosyltransferase 2 (XylT2)–transfected cells (siXylT2; right bar) compared with that of control hLECs transfected with mock/scrambled RNA (siDS; left bar; *P=0.02 for difference; average of 4 experiments). E, VEGF-C–induced phospho-Akt was examined by western in starved siXylT2-transfected vs control hLEC, with phospho/total Akt normalized and plotted relative to value for starved siDS cells (*P=0.05 for indicated difference in poststimulation [+] means; average of 4 experiments; representative blot shown). Inset graph shows response to a human VEGF-C form (VEGF-CCys156Ser), which binds exclusively to VEGFR-3. F, Effect of siNdst1 targeting on Akt phosphorylation in response to VEGF-CCys156Ser (*P<0.01 for difference in means; average of 3 experiments). G, Effect of siNdst1 targeting on Erk1/2 phosphorylation in response to VEGF-CCys156Ser (*P=0.03 for difference in means; average of 3 experiments).
Lymphatic vessels in tumors from MMTV-PyMT Ndst1f/fTekCre+ mutants were characterized by a greater percentage of lymphatic-associated TUNEL+ apoptotic bodies (Figure 1C, bottom). In separate primary human cell–based studies, we questioned whether altered heparan sulfate biosynthesis might impair VEGF-C–mediated protection of primary hLECs from apoptotic stress, as measured by cellular–cleaved caspase levels. In pilot studies, VEGF-C consistently lowered the generation of starvation-induced cleaved caspase by hLEC under several media conditions (Online Figure II). XylT2-deficient LECs (siXylT2; characterized by impaired glycan-chain initiation) were insensitive to VEGF-C during starvation (Figure 1D, right bar), whereas control-transfected hLEC consistently showed reduced apoptosis on starvation in the presence of VEGF-C (Figure 1D, siDS transfection control, left bar). Consistent with this, VEGF-C–dependent Akt phosphorylation in siXylT2-targeted hLEC was reduced relative to control cells (Figure 1E). When stimulated with a VEGF-C ligand that binds exclusively to VEGFR-3 (VEGF-CCys156Ser),22 Akt phosphorylation in siXylT2-transfected cells was also blunted (Figure 1E, inset graph). We next examined how siNdst1 targeting might affect Akt phosphorylation in response to VEGF-CCys156Ser: similar results were found (Figure 1F), implying that VEGFR-3–specific Akt signaling is sensitive to altered glycan sulfation. Moreover, mitogen-activated pathway signaling (phospho-Erk1/2) in response to VEGF-CCys156Ser was also sensitive to hLEC Ndst1 deficiency (Figure 1G; it is noteworthy that in preliminary collagen-attachment studies, hLEC attachment and spreading were somewhat slowed in Ndst1-deficient cells; data not shown).
Lymphatic-Specific Deficiency in the Sulfation of Heparan Sulfate Results in Altered VEGF-C–Driven Tumor Lymphangiogenesis
To examine the effect of a lymphatic-exclusive mutation in heparan sulfate, we used mice bearing a conditional mutation in Ndst1 driven by tamoxifen-inducible Cre under the control of the lymphatic-specific promoter Prox1 (Ndst1f/fProx1+/CreERT2 mutants). In Prox1+/CreERT2Rosa26R reporter studies, inguinal and mediastinal lymph nodes showed a relatively high degree of Cre− LYVE-1 colocalization (Figure 2A, left), noted also in ear dermal lymphatics, albeit in a more patchy distribution (Figure 2A, right). With this in mind, we established subcutaneous VEGF-C overexpressing Lewis lung carcinomas (LLC-VC) in the right flank of tamoxifen-induced Ndst1f/fProx1+/CreERT2 mutant and wild-type (Ndst1f/fProx1−/CreERT2) littermates. LYVE-1 staining showed robust lymphangiogenesis only at the tumor periphery in this model (F4/80 macrophage staining revealed a diffuse-tumor pattern, which decreased toward the tumor periphery, with nearly complete non-overlap of F4/80 with LYVE-1 staining; Online Figure III). The tumors showed no significant difference in size between mutant and wild-type groups (data not shown). Empty-vector control tumors (LLC-ev) were established in the opposite (left) flank of each mouse. In Cre− wild-type mice, LLC-VC tumors showed a significantly higher mean LVD than that of LLC-ev tumors (Figure 2B, graph, left bars), indicating a VEGF-C–dependent boost in LVD caused by tumor-associated VEGF-C expression in wild-type mice (compare representative photomicrographs of LYVE-1 immunofluorescence on left of the panel set, for Cre− animals). However, among Cre+ mutants, LVD was not greater in LLC-VC tumors when compared with that of LLC-ev tumors (Figure 2B right panels and graph, right bars). The findings suggest that the inhibitory effect of lymphatic-targeted Ndst1 mutation on LVD in this model was specifically associated with VEGF-C–mediated lymphatic-vessel growth.
Figure 2.
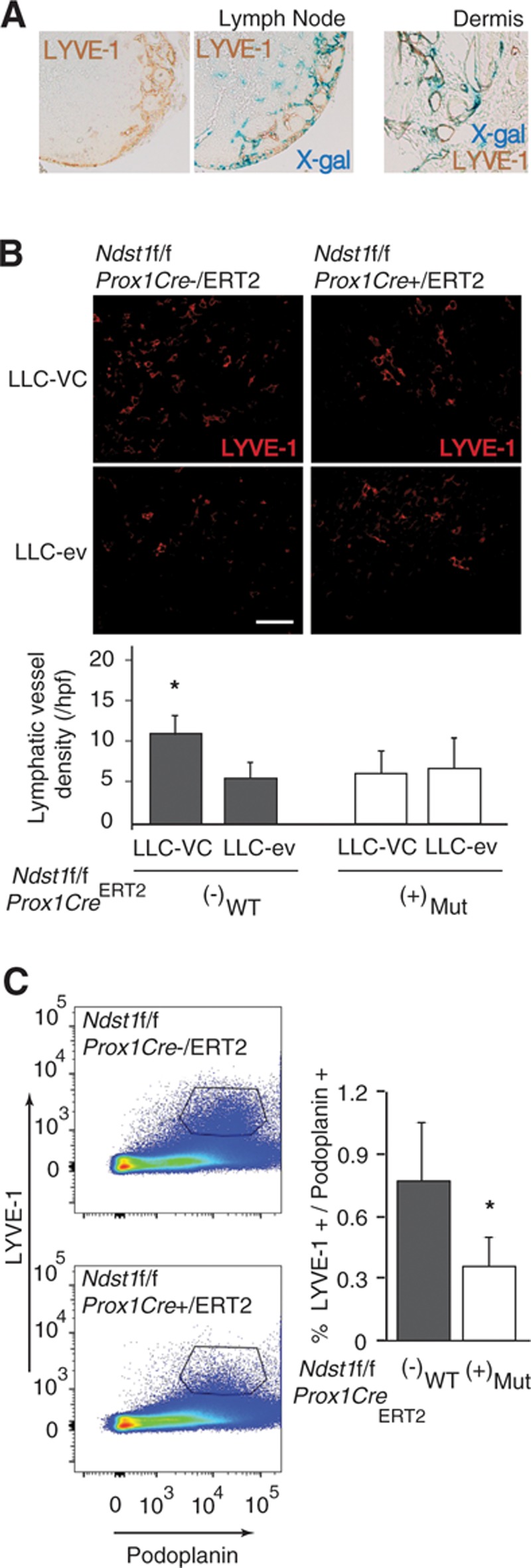
A lymphatic-specific genetic deficiency in the sulfation of heparan sulfate results in altered vascular endothelial growth factor-C (VEGF-C)–driven tumor lymphangiogenesis. Reporter studies in Prox1+/CreERT2 Rosa26R mice examined colocalization of Cre (X-gal positive staining) with LYVE-1+ vessels after tamoxifen induction. A, Lymph nodes (LNs) draining the lung (mediastinum) showed a high degree of colocalization (left), and dermal ear lymphatics (right) demonstrated patches of colocalization. B, Lewis lung carcinoma (LLC) cells that overexpress VEGF-C (LLC-VC) were used to establish subcutaneous tumors in the right flank of Ndst1f/f Prox1Cre+/ERT2 (n=4) mutant mice and Cre− littermate controls (n=4). Simultaneously, control LLC cells (LLC-ev) were injected into the left flank of the same animals to establish VEGF-C–negative control tumors. After 10 d, tumors were resected, and lymphangiogenesis was examined by LYVE-1 immunofluorescence. Representative images show LYVE-1+ lymphatic endothelia (red) in tumor sections from mutants (right) and controls (left). Graph shows mean density of LYVE-1+ lymphatic vessels (±SD) in LLC-VC and LLC-ev tumors from Ndst1f/f Prox1Cre+/ERT2 mutant vs Cre− control littermates (*P=0.05 for the interaction of genotype with VC vs ev tumor status). In the mutant group, the difference in means was not significant. C, To examine tumor, VEGF-C–driven lymphatic proliferation in the lung, LLC-VC cells were intravenously injected into Ndst1f/f Prox1Cre+/ERT2 mutants (n=5) and Cre− (n=5) controls. Mice were euthanized 7 d after injection: Tumor-containing lungs from each mouse were digested into a single-cell suspension, and LYVE-1+/podoplanin+ (double positive) LECs present in the digests were measured by flow cytometry. Representative panels are shown for Cre+ mutant and Cre− control mice; and averages (±SD) for both the groups, expressed as %total cells, are shown in graph to right (*P<0.001 for the difference with wild-type [WT]).
To examine lymphatic proliferation in an orthotopic-tumor setting, LLC-VC cells were intravenously injected into Ndst1f/fProx1+/CreERT2 mutants and Prox1−/CreERT2 controls. The mean quantity of LYVE-1/podoplanin double-positive LECs (as a percentage of total LECs) from lung digests 7 days post injection was reduced in mutants (Figure 2C), indicating an inhibitory effect of mutation on total LECs purified from LLC-VC tumor-harboring lungs.
Phosphorylation of VEGFR-3 Is Sensitive to Altered Biosynthesis of Heparan Sulfate in Cultured Human LECs
Preliminary assessments of VEGF-C produced by cultured LLC-VC cells revealed unprocessed VEGF-C in the supernatants and lysates. The quantity of this species relative to post-translationally processed, including mature, VEGF-C species produced by the tumors in vivo (which we confirmed by Western blotting) is unknown. Because unprocessed VEGF-C contains heparin-binding propeptide extensions,2,23 and because nonmature forms of VEGF-C are variably secreted from tumors,24,25 we first screened the degree to which pro–VEGF-C (a mixture of unprocessed and partially processed VEGF-C propeptides) is able to phosphorylate receptor tyrosine kinase receptors, including VEGFR-3, in heparan sulfate–deficient versus control hLECs (Online Figure IV highlights the composition of pro–VEGF-C separated on a silver-stained gel). Initially, in a highly sensitive receptor tyrosine kinase phospho array, inhibition of hLEC heparan sulfate biosynthesis robustly blocked pro–VEGF-C–mediated VEGFR-3 phosphorylation (Figure 3A), as well as VEGFR-2 phosphorylation. Although phosphorylation of both receptors seemed to be sensitive to the glycan alteration, the baseline phosphorylation of VEGFR-3 on ligand stimulation seemed to be markedly greater in this primary cell line, consistent with its lymphatic endothelial identity. Nevertheless, the marked sensitivity of VEGFR-2 phosphorylation to glycan targeting points to an additional role for heparan sulfate in facilitating VEGFR-2 activation in response to VEGF-C, reminiscent of its importance in VEGF-A signaling.26 The effect of glycan targeting on VEGFR-3 phosphorylation in response to pro–VEGF-C was also tested in a specific (albeit less sensitive) ELISA-based phospho–VEGFR-3 assay (Figure 3B), with phosphorylation blockade that resulted from siXylT2 targeting. VEGFR-3 phosphorylation by mature VEGF-C was also significantly reduced in siXylT2-targeted hLEC (Figure 3C). Interestingly, siXylT2-inhibition of glycan-chain biosynthesis also inhibited receptor phosphorylation by a minimum receptor-binding species of VEGF-C (Figure 3D) that excludes basic amino acids in the C terminus (L216–R227; which may contribute to a weak interaction of mature VEGF-C with heparan sulfate14), implying an important cell-autonomous role for the glycan in receptor activation.
Figure 3.
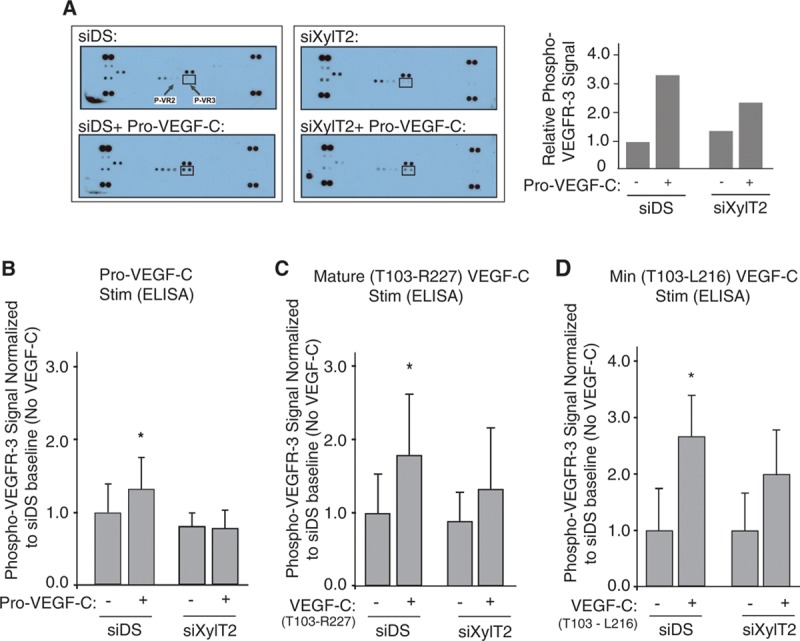
Disruption of lymphatic heparan sulfate biosynthesis in primary human lymphatic endothelial cells (hLECs) results in reduced phosphorylation of vascular endothelial growth factor receptor-3 (VEGFR-3) in response to distinct VEGF-C species. A receptor screening format was used to assess how targeting heparan sulfate biosynthesis might alter VEGFR-3 activation in response to distinct VEGF-C species. A, In a preliminary multiple-receptor screen, cultured serum-starved hLEC transfected with either control/scrambled RNA (siDS) or siXylT2 were stimulated with Pro–VEGF-C, and phosphorylation of growth receptors from poststimulation (vs unstimulated) cell lysates was measured using a receptor tyrosine kinase phospho array, which reports receptor phosphorylation (pair of dots) for all receptors captured from cell-lysate samples. The array for starved control (mock transfected) hLEC shows a weak phospho–VEGFR-3 signal (upper-left slide; dot pair within box, with arrows also pointing to phospho–VEGFR-2 for reference). The array for control cells 15 min poststimulation with Pro–VEGF-C (siDS+ Pro–VEGF-C) is shown in the lower-left slide. Slides to the right show responses for baseline- vs stimulated siXylT2–transfected hLEC (dot pairs on corners of each slide are phosphotyrosine-positive controls.) Autophosphorylation occurred for a few other receptors at baseline, without a major response to Pro–VEGF-C: Those were Flt3 (dot pair immediately above VEGFR-3), VEGFR-1 (to left of VEGFR-2), Tie-2 (lower left), HGFR (hepatocyte growth factor receptor; immediately above/to right of Tie-2), and faintly visible EGFR (upper left). Signal values normalized to that of starved-control unstimulated cells are plotted on graph to right. A separate array repeated under identical conditions showed similar results. B, The ability of Pro–VEGF-C to phosphorylate VEGFR-3 in control- (siDS) or siXylT2-transfected hLEC was then exclusively carried out in a capture-ELISA format. Mean signal values normalized to that of control unstimulated cells (siDS and no VEGF-C) are plotted (*P=0.006 for interaction of siRNA status with VEGF-C stimulation response; average of 3 experiments). C, The same ELISA-based assay was used to examine the effects of hLEC xylosyltransferase 2 (XylT2) silencing on VEGFR-3 phosphorylation in response to stimulation with human mature VEGF-C, with control-normalized signal values plotted on the graph (*P=0.006 for the interaction; average of 6 experiments). D, Responses were examined for stimulation by a short form of VEGF-C (T103-L216) that does not bind heparin, and contains the minimal receptor-binding domain A112-L215 (*P=0.01 for the interaction; average of 5 experiments).
Sdc-4 Is a Dominant Proteoglycan Core Protein on Lymphatic Endothelium With Functional Significance in Pathological Lymphangiogenesis
We explored whether a dominant proteoglycan might present heparan sulfate on the lymphatic cell surface. Although LECs were not easily purified from tumors, pathological primary LECs could be isolated from mesenteric oil granuloma/lymphangioma lesions,14,15 and they were examined for the repertoire of HSPG core proteins quantitative polymerase chain reaction. Sdc-4 was the dominantly expressed lymphatic cell-surface HSPG (Figure 4A, left graph). Cells did not express CD44v3 proteoglycan, known to be expressed by blood-vascular endothelia,27,28 although they did express perlecan, which is secreted into basement membranes, and which has been detected around proliferating and collecting lymphatics.29 The HSPGs expressed by svLEC, an immortalized mouse mesenteric-lymphatic cell line, showed a similar profile (Online Figure V). It should be noted that we were able to measure a moderate increase in Sdc4 expression on Ndst1 silencing in this cell line (Figure 4A, upper-right graph); however, the expression of the other syndecans did not change in that setting, with Sdc4 remaining the dominantly expressed lymphatic HSPG.
Figure 4.
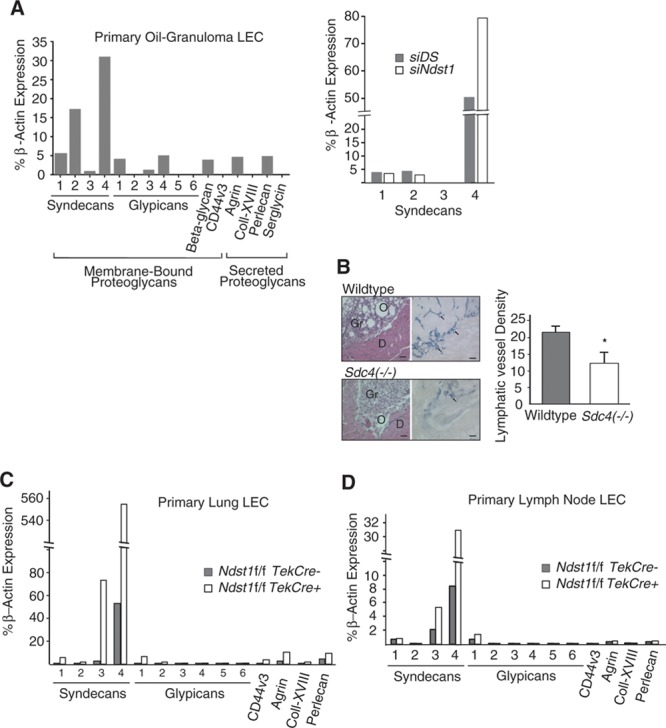
Syndecan 4 is a dominant heparan sulfate proteoglycan (HSPG) in primary lymphatic endothelia, and genetic targeting of syndecan-4 results in altered pathological lymphangiogenesis. The genetic importance of proteoglycan core protein targeting was examined in pathological lymphangiogenesis. A, The repertoire of HSPG core proteins expressed by proliferating lymphatic endothelial cells (LECs) isolated from oil-granuloma lesions in mice was assessed by quantitative polymerase chain reaction (qPCR). RNA was isolated, reverse transcribed, amplified using gene-specific primers to each core protein, and quantified relative to expression of β-actin. Ct values from triplicate assays were used to calculate % expression. Given the unique expression profile of syndecans, with syndecan-4 as a dominantly expressed HSPG, expression of the syndecan members was examined in the svLEC mesenteric LEC cell line in the setting of silencing of Ndst1 (siNdst1), with comparison to expression by control svLECs, transfected with random (scrambled duplex) RNA (siDS); with values in graph to upper right. B, Oil-granuloma lesions were induced in syndecan-4 knockout (Sdc4−/−) mice and wild-type controls. Hematoxylin and eosin–stained sections of the lesions (left) were characterized by dense granuloma cell infiltrates (Gr) surrounding oil droplets (O), with lesions abutting the abdominal diaphragmatic (D) surface. Immunostaining for LYVE-1 revealed marked lesion-associated lymphangiogenesis in sections from wild-type mice, with lymphatic vessels (arrows, right photomicrographs; bar=100 μm) shown in blue. Mean lymphatic vessel densities are graphed to the right (n=4 mice per genotype; *P=0.002 for difference). C, To assess the repertoire of proteoglycan core proteins expressed by other primary nonpathological LECs purified from the mouse as well as the effect of Ndst1 mutation on core protein expression, primary LECs were isolated from the lungs of Ndst1f/fTekCre+ mutants and Cre− littermates. RNA from purified LECs was processed (as in B) for quantitative PCR, and the expression of major HSPGs was quantified relative to that of β-actin (graph). Expression of the dominant core protein, again noted to be syndecan-4, seemed to be markedly upregulated in Ndst1-deficient LECs (light bars in graph). D, Expression was also examined using the same method for primary LECs isolated from the LNs of wild-type and Ndst1f/fTekCre+ mutants, with similar findings.
With this in mind, we generated oil granuloma/lymphangioma lesions in Sdc-4 null (Sdc4−/−) mice, and noted reduced lesion LVD (Figure 4B). Lymphangiogenesis in Sdc4(−/−) Ndst1f/fTekCre+ double mutants examined using this pathological model was not significantly reduced in comparison with that in Sdc4(−/−)Ndst1f/fTekCre− littermates (data not shown), suggesting that Sdc-4 likely serves as a quantitatively dominant functional scaffold for lymphatic cell-surface heparan sulfate, playing a critical role in pathological lymphatic mitogen responses. We also measured HSPG core protein expression in nonpathological LECs purified from the lung (Figure 4C) or lymph nodes (Figure 4D) of wild-type and Ndst1f/fTekCre+ mutants. Sdc-4 was not only the dominant HSPG but was also disproportionately upregulated in the setting of Ndst1 deficiency, indicating that targeting the sulfation of lymphatic heparan sulfate upregulates major HSPG core protein expression.
Sdc-4 Specifically Associates With VEGFR-3 in Response to VEGF-C in Human LECs in a Heparan Sulfate–Dependent Manner and Mediates VEGF-C–Dependent Signaling
We asked whether Sdc-4 might associate with VEGFR-3 (as a possible ternary complex) in hLECs on VEGF-C stimulation. In human LECs, we previously found that expression of Sdc2 was somewhat greater than that of Sdc4.16 (as a reference, for primary human dermal microvascular blood-endothelial cells, Sdc4 seems to be the dominantly expressed transmembrane HSPG; Online Figure VI). Core protein studies also revealed that the dominant cell-surface HSPGs on hLECs were Sdc-2 and Sdc-4 (as assessed by HSPG core protein blotting; Online Figure VII). Nevertheless, proximity ligation analysis (PLA) revealed that resting starved hLEC (ie, pre-VEGF-C stimulation) are characterized by a significant degree of Sdc-4–VEGFR3 association at baseline. Exposure to mature VEGF-C strikingly increased Sdc-4–VEGFR-3 association, whereas Sdc-2–VEGFR-3 association under identical conditions was minimal (Figures 5A and 5B, left side of graph; hLEC). The magnitude of Sdc-1–VEGFR-3 association (not shown) was comparable with that of Sdc-2–VEGFR-3, with signals remaining <10% that of baseline Sdc-4–VEGFR-3 signal. Examination of Sdc-4–VEGFR-3 PLA in the mouse oil granuloma–derived svLEC line also revealed a marked rise in Sdc-4–VEGFR-3 association on VEGF-C exposure (quantified in Figure 5B, right graph; svLEC). In separate experiments examining VEGFR-2, stimulation of hLECs with mature VEGF-C did not lead to engagement of Sdc-4 with VEGFR-2 (Figure 5C; representative panels), suggesting that Sdc-4 serves as a specific coreceptor for VEGFR-3. To further explore mechanism, we found that formation of Sdc-4–VEGFR-3 complexes on VEGF-C treatment was sensitive to hLEC pretreatment with heparanase (Figure 5D), suggesting that lymphatic heparan sulfate is required for stabilizing the proteoglycan-receptor complex on ligand exposure.
Figure 5.
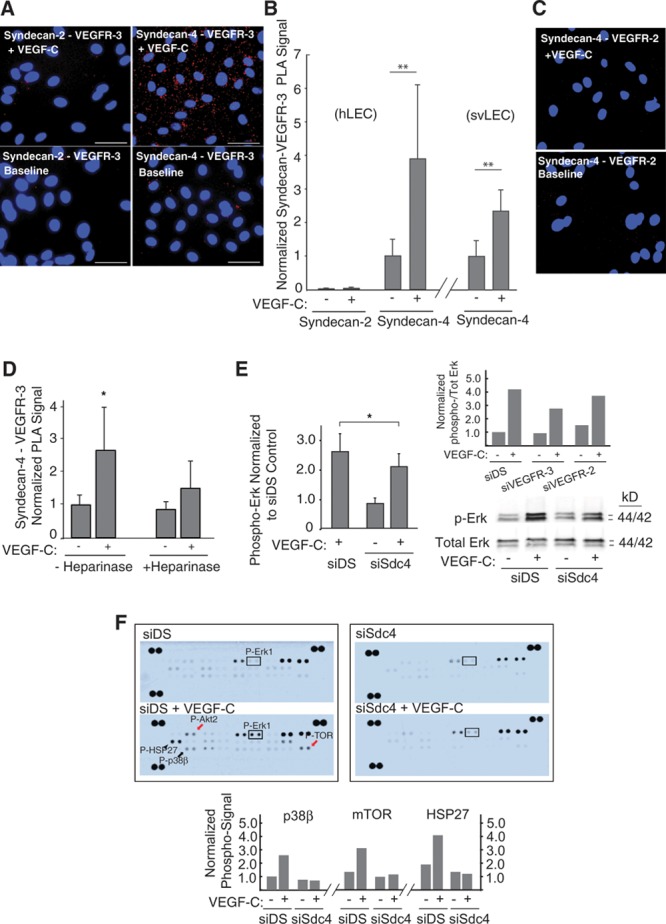
Proteoglycan-dependent signaling and complexing of syndecan-4 (Sdc-4) with vascular endothelial growth factor-3 (VEGFR-3) on VEGF-C stimulation. A, Dynamic association of VEGFR-3 with 2 highly expressed heparan sulfate proteoglycans (Sdc-2 and Sdc-4) on the lymphatic cell surface was tested in response to mature VEGF-C on serum-starved human lung lymphatic endothelial cells (hLECs) via proximity ligation assay (PLA). Proximity of Sdc-4 to VEGFR-3 is shown at baseline (lower right; PLA signal, red dots) and after stimulation with mature VEGF-C (upper right; DAPI [4',6-diamidino-2-phenylindole] nuclei in blue; bar=50 μm). Left, PLA for Sdc-2 and VEGFR-3. B, Split-graph on left shows mean PLA signals for hLECs from multiple experiments (n=3 for Sdc-2 and n=5 for Sdc-4; ±SEM), normalized to mean for Sdc-4/VEGFR-3 association at baseline (**P=0.03 for difference between baseline and +VEGF-C means). Split-graph to right shows mean Sdc-4/VEGFR-3 PLA signals for mouse svLECs (n=4 experiments; ±SEM), normalized to baseline (no VEGF-C; **P=0.03 for difference between baseline and +VEGF-C means). C, PLA to examine Sdc-4/VEGFR-2 association was carried out ± VEGF-C: representative photomicrographs shown. D, To examine the importance of heparan sulfate, PLA signals in hLECs treated ± heparinase (destroys heparan sulfate chains) were quantified, normalized to baseline for Sdc-4/VEGFR-3, and graphed (*P=0.02 for interaction of ± heparinase status with VEGF-C stimulation response; average of 5 experiments). E, The effect of siRNA targeting of Sdc-4 Sdc4 (siSdc4) on Erk phosphorylation in response to mature VEGF-C was examined in cultured hLEC (*P=0.04 for difference; average of 4 experiments; representative blot at lower right). Upper-right inset graph shows representative histogram of effect of siRNA targeting of VEGFR-3 or VEGFR-2 on Erk phosphorylation in the same cells as a mitogen receptor-signaling control. F, Lysates from control (siDS) or siSdc4 hLECs pre-/post–VEGF-C stimulation were applied to a phosphosignaling array reporting phosphorylation of several mitogen-activated protein kinase and survival-signaling intermediates (as dot pairs) on the membranes. Arrays for control cells (siDS) pre-/post–VEGF-C are shown to left. Slides to right show corresponding signals for siSdc4-transfected hLEC. A box is placed around P-Erk1 for reference, showing marked stimulation in control hLEC with blunted response in Sdc-4–deficient cells, resembling western pattern in D. Other notable blunted VEGF-C responses in Sdc4-deficient cells (graphed below) included p38β (black arrow), survival pathway intermediates (Akt2 and TOR; red arrows); and the heat-shock protein HSP27 (arrowhead).
With these findings in mind, we asked whether Sdc-4 deficiency might affect signaling by mature VEGF-C: phosphorylation of the mitogen-pathway intermediate Erk1/2 was sensitive (Figure 5E; representative immunoblot shown to right). As a receptor-signaling control, Erk1/2 phosphorylation in response to VEGF-C was comparatively sensitive to VEGFR-3 deficiency, and somewhat less sensitive to VEGFR-2 deficiency (Figure 5E, upper-right representative histogram). These Erk1/2 signaling findings further prompted us to use a commercial phosphosignaling array to examine patterns in VEGF-C–dependent activation of other lymphatic endothelial mitogen-activated protein kinase–associated intermediates in the setting of Sdc-4 silencing. In addition to replicating the pattern we found in Erk phosphorylation in the array (with predominantly Erk1 showing a blunted response to VEGF-C stimulation in the setting of Sdc-4 deficiency), we also noted inhibition of a second mitogen-activated protein kinase intermediate (p38β) along with associated inhibition of HSP27 phosphorylation in the same setting (Figure 5F, with quantified responses below). The array also demonstrated concomitant reduction in VEGF-C–dependent Akt2 and TOR activation, which corroborates the original findings in Figure 1, showing altered lymphatic endothelial survival signaling as a result of targeting the glycan chain.
Discussion
We examine herein the genetic importance of lymphatic heparan sulfate and that of a key proteoglycan core protein in pathological lymphangiogenesis. Genetic targeting of the glycan impairs pathological lymphangiogenesis in vivo and lymphatic mitogen and survival signaling and phosphorylation of VEGFR-3 in response to VEGF-C. We also demonstrate the genetic importance of Sdc-4 as a key HSPG that scaffolds heparan sulfate on the lymphatic surface, and propose that it functions as a major coreceptor in VEGF-C–mediated pathological lymphangiogenesis.
In Ndst1f/fTekCre+ mutants, Ndst1 inactivation under the Tek-promoter generates a pan-endothelial mutation, and Ndst1 expression in LECs purified from mutants was markedly reduced. Lymphatic signaling via VEGF-C/VEGFR-3 seemed to contribute to lymphatic proliferation in both oil granuloma/lymphangioma models21 (Figure 1A) as well as transgenic carcinoma (Figure 1C) models. Although altering lymphatic Ndst1 inhibits VEGF-C–dependent sprouting and growth signaling in vivo, other heparin-binding growth factors such as VEGF-A, fibroblast growth factor-2, or PDGF may also contribute to lymphatic growth and remodeling. Nevertheless, deficiency in the glycan not only altered VEGF-C–mediated protection of primary LECs from apoptotic stress (Figure 1C, bottom) but consistent with this, Erk- and Akt-mediated signaling in primary hLECs was also inhibited in mutants (Figure 1E through 1G). Pathological blood-vascular angiogenesis seemed to be altered in such pan-endothelial Ndst1 mutants, consistent with previous work.10 Although an indirect effect of the blood-vascular mutation on lymphangiogenesis is possible in the setting of pathological angiogenesis, findings using high-specificity lymphatic gene targeting in vivo (discussed below) together with ex-vivo and cell-based work herein point to an important and direct role for lymphatic-specific heparan sulfate in VEGF-C–mediated VEGFR-3 activation.
To stringently target heparan sulfate in VEGF-C–mediated lymphatic-specific remodeling, we used a VEGF-C expressing lung carcinoma model on a genetic background wherein Ndst1 is specifically inactivated in lymphatic endothelium through the Prox1+/CreERT2 transgene. The findings point to the genetic importance of appropriately sulfated heparan sulfate in mediating the action of VEGF-C on lymphatic endothelium in vivo (Figure 2B). It is possible that nonmature forms of VEGF-C produced by this or other neoplastic cell lines24 may be more sensitive to the effects of tumor-lymphatic Ndst1 mutation on VEGFR-3 activation because such species have a greater affinity for heparan sulfate than shorter (eg, mature) forms of VEGF-C. This may have pathophysiological importance in neoplasia, where nonmature forms of VEGF-C may play important roles in tumor-lymphatic remodeling, with possibly additional regulation through binding to HSPGs secreted into matrix (eg, perlecan). It is noteworthy that the predominant species present in pro–VEGF-C used in Figure 3A and 3B (ie, 29/31-kD propeptide; Online Figure IV) may compete with other species for VEGFR-3 binding,30 and thus contribute to negative regulation. This may explain the relatively weak stimulation by pro–VEGF-C in Figure 3B. Nevertheless, siXylT2 targeting resulted in complete inhibition of VEGFR-3 phosphorylation in this setting, possibly as a result of greater heparan sulfate binding by unprocessed and propeptide VEGF-C species. In tumors, this binding may allow for greater presence of propeptide VEGF-C on the lymphatic cell surface, where proteases (eg, ADAMTS3 tethered to endothelium30) may yield local release of mature VEGF-C. Interestingly, VEGFR-3 activation by a nonheparin-binding short form of VEGF-C remained sensitive to altered heparan sulfate biosynthesis (Figure 3D), pointing to the importance of intact lymphatic cell-surface heparan sulfate in VEGFR-3 activation/function. This is reminiscent of altered VEGFR-2 responses to a key nonheparin-binding form of VEGF-A (ie, VEGF121) when endothelial heparan sulfate is genetically altered.26
A variety of proteoglycans may tether heparan sulfate to the lymphatic cell surface or pericellular matrix.8 Although we found abundant expression of Sdc-4 on lymphatic endothelium (Figure 4), a model limitation is that Sdc4−/− mutation is not tissue specific. However, pairing the genetic importance of appropriate glycan sulfation in lymphangiogenesis with the finding that Sdc-4 forms a specific and robust association with VEGFR-3 in response to VEGF-C (Figure 5A through 5C) suggests that Sdc-4 plays a critical role in VEGFR-3–mediated lymphatic growth. Importantly, the VEGFR-3–specific mitogen response to VEGF-C seems to be glycan dependent (Figure 5D) and reduced in the setting of Sdc-4 deficiency. In light of the altered signaling with this mutation (Figure 5E), the collective findings suggest that lymphatic-endothelial Sdc-4 deficiency (or Ndst1 deficiency, which would affect glycans on all HSPGs, including the dominant membrane-bound pool of Sdc-4) results in both altered mitogen-pathway as well as altered survival/Akt signaling (Figure 1F and 1G as well as Figure 5E). Although other HSPGs could theoretically partially compensate through the collective actions of their glycans to partially support mitogen-pathway responses in Sdc-4–deficient lymphatic endothelium, the silencing of this unique proteoglycan seems to critically alter lymphatic VEGFR-3–dependent growth/survival signaling and possibly pathways that affect cytoskeletal rearrangement during pathological lymphangiogenesis. Interestingly, the cooperative activation of Erk1/2 along with p38 mitogen-activated protein kinase and HSP27 (which we found altered in VEGF-C–treated Sdc-4–deficient primary LECs; Figure 5F) has been reported to play an important role in endothelial actin cytoskeletal reorganization in response to VEGF-A, with known inhibition of HSP27 phosphorylation in response to several angiogenesis-pathway inhibitors.31–33 The findings suggest that VEGF-C–mediated activation of this pathway during lymphatic endothelial cytoskeletal remodeling may also be important, and sensitive to alterations in Sdc-4 as a coreceptor. More generally, the findings point to a critical coreceptor role for the dominant lymphatic HSPG Sdc-4 in pathological lymphangiogenesis.
Figure 6 shows a model to illustrate the functional importance of these molecules in lymphatic receptor functions: in 1 mode, glycans on the lymphatic surface may serve in a cell-autonomous role as a depot for VEGF-C that, in turn, may affect the availability for VEGFR-3 interactions (Figure 6, pathway B to right). The degree of this may depend on the species of VEGF-C available and the heparin-binding affinity of that species; with signaling by the different species variably sensitive to genetic absence of the glycan on the cell surface (Figure 3B through 3D). However, the glycan seems to play an essential role in stabilizing a ternary complex that mediates proximity of the proteoglycan core protein to the growth receptor (Figure 5), with the HSPG thus serving as a coreceptor (Figure 6, bottom). This may occur either simultaneously when ligand becomes available (Figure 6, pathway A), or step-wise via initial concentration and availability of the ligand for receptor-binding events before complex stabilization (Figure 6, 2-step pathway B). Conceptually, in considering distinct vascular beds, this leaves the possibility of proteoglycan core proteins other than Sdc-4 that could depend on the common glycan (heparan sulfate) in mediating receptor activation in response to VEGF-C. However, in the proof-of-concept studies shown here, we have demonstrated important roles for heparan sulfate expressed on lymphatic endothelial Sdc-4.
Figure 6.
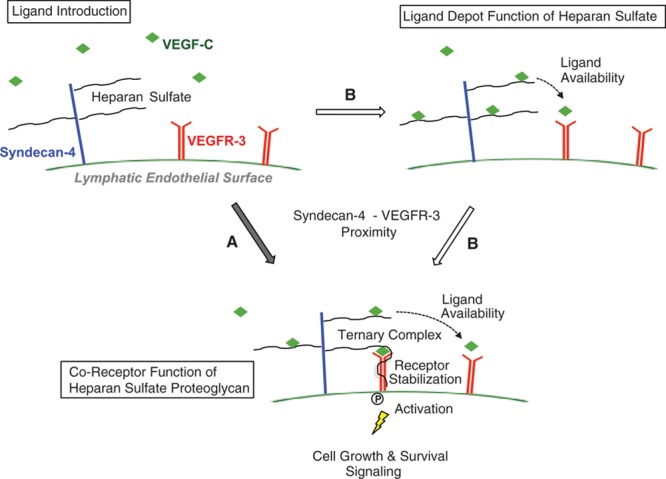
Schematic diagram showing functions of the lymphatic endothelial heparan sulfate proteoglycan syndecan-4 at the cell surface on ligand stimulation by vascular endothelial growth factor-C (VEGF-C). In 1 pathway (A), introduction of VEGF-C induces association between the growth factor (VEGF-C), proteoglycan (syndecan-4), and receptor (VEGFR-3), wherein the proximity of syndecan-4 to the receptor as well as binding of the ligand is stabilized by the glycan chain. This coreceptor function is necessary for efficient receptor phosphorylation and activation, leading to mitogen-activated cell growth and survival signaling. Absence of the proteoglycan, or lack of an appropriately sulfated glycan chain, is associated with impaired lymphatic growth signaling in response to VEGF-C. An alternative pathway (B) highlighting cell-autonomous ligand-depot functions of lymphatic endothelial heparan sulfate illustrates the ability of the appropriately sulfated glycan chain to bind VEGF-C, making it available for receptor binding via VEGFR-3 receptors on the cell surface. This may eventually lead to further#8232;syndecan-4–VEGFR-3 proximity and ternary complex formation (bottom). The depot function of heparan sulfate for species of VEGF-C with greater heparin-binding affinity (ie, preproteolytically processed VEGF-C > mature VEGF-C) may be particularly important in regulating availability of those species for interaction with receptor at the cell surface. Regardless, the glycan chain ultimately is necessary to stabilize a proteoglycan coreceptor complex that optimizes cell signaling (bottom, via pathway B).
These findings may have translational potential. In tumors, although alterations in LVD might not primarily affect primary tumor size, the alteration in lymphatic conduit may reduce the potential for lymphatic metastasis; and in many invasive tumors, upregulation of VEGF-C as well as the expression of other heparin-binding growth factors and cytokines contributes to tumor lymphangiogenesis.2,6,34 Considering also the roles of both Ndst1 (appropriate glycan chain sulfation) and Sdc-4 in the proliferation of lymphatic vessels as well as the mechanistic importance of Sdc-4 as a coreceptor for lymphatic signaling (Figure 6), approaches that target the expression of these molecules in the lymphatic microenvironment may serve as new selective strategies to modulate critical lymphatic remodeling in disease.
Acknowledgments
We appreciate advice and review by Dr Jeffrey Esko, and histology assistance by Dr Nissi Varki as well as members of her core-histology team. We acknowledge Dr Goetinck for Sdc4 homozygous-null mice as well as Dr Guillermo Oliver for original provision of Prox1Cre/ERT2 transgenic mice. We thank Dr Steven Alexander for providing the SV-LEC cell line; and Dr Guido David for 3G10 and Syndecan-2 antibodies. We thank Dr Gavin Thurston (of Regeneron) for providing LLC-VC and LLC-ev cell lines. Finally, we acknowledge Dr Paul Clopton of the VA San Diego Healthcare System and James Proudfoot of the UCSD Clinical and Translational Research Institute for assistance with statistical analyses. We thank Dr Philip Gordts for valuable insights as well.
Sources of Funding
We acknowledge funding from National Institutes of Health (NIH)/National Heart, Lung, and Blood Institute (NHLBI; R01-HL107652 to M.M. Fuster and P01-HL57345), the VA Career Development and Merit Review Programs (VA-CDTA Award and VA-Merit I01BX000987-01 to M.M. Fuster), the American Cancer Society (RSG-08-153-01-CSM to M.M. Fuster). We also acknowledge NIH/NHLBI (T32 HL098062 to X. Yin and R. El Ghazal), and appreciate support from the Veterans Medical Research Foundation (M.M. Fuster) as well as the Academy of Finland (decisions 265982 and 273612 to M. Jeltsch) and the K. Albin Johansson Foundation (to M. Jeltsch).
Disclosures
None.
Supplementary Material
Nonstandard Abbreviations and Acronyms
- HSPG
- heparan sulfate proteoglycan
- hLEC
- human lung LEC
- LEC
- lymphatic endothelial cells
- LLC
- Lewis lung carcinoma
- LVD
- lymphatic vessel density
- Ndst
- N-deacetylase/N-sulfotransferase
- PLA
- proximity ligation assay
- Sdc4
- syndecan-4
- siDS
- siRNA duplex scrambled control
- VEGF
- vascular endothelial growth factor
- VEGFR-2
- VEGF receptor-2
- VEGFR-3
- VEGF receptor-3
- XylT2
- xylosyltransferase 2
In April 2016, the average time from submission to first decision for all original research papers submitted to Circulation Research was 15.28 days.
S.C. Johns and X.Yin are joint first authors.
The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA.116.308504/-/DC1.
Novelty and Significance
What Is Known?
Lymphatic-vascular remodeling in disease states such as neoplasia or inflammation may facilitate important downstream pathophysiological events, such as lymphatic metastasis or organ fibrosis, among other consequences depending on the tissue and disease process.
Although the major lymphatic vascular mitogen vascular endothelial growth factor-C (VEGF-C) is critical in driving pathological lymphangiogenesis primarily through the activation of the major lymphatic receptor VEGFR-3, coreceptors that critically regulate ligand and receptor activation are poorly understood.
Cell-surface proteoglycans displaying sulfated carbohydrate chains known as heparan sulfate are known to play key roles in endothelial growth factor binding and receptor signaling, although the genetic importance and function(s) of these molecules in lymphatic remodeling in vivo remain unknown.
What New Information Does This Article Contribute?
The sulfation of heparan sulfate on lymphatic endothelium is important in mediating the actions of VEGF-C on lymphatic endothelial growth in pathological models in vivo.
Syndecan-4 is a major heparan sulfate proteoglycan expressed on lymphatic endothelium, and it serves as a novel coreceptor for lymphatic VEGFR3 signaling, forming a glycan-dependent complex with ligand and receptor on VEGF-C stimulation.
Syndecan-4 deficiency results in reduced pathological lymphangiogenesis and reduced VEGFR-3 mitogen signaling in primary lymphatic endothelial cells, introducing a novel mode of biological modulation and possibly therapeutic targeting.
The process of lymphangiogenesis plays critical roles in the pathological progression of several important diseases. These include metastasis-promoting lymphatic remodeling in cancer, lymphatic proliferation associated with lymphagioleiomyomatosis, or fibrotic progression in idiopathic fibrosis and renal tubulointerstitial disease, among others. Overexpression of VEGF-C in the lymphatic microenvironment of these disorders is a central requirement, and growth signaling primarily through the cognate lymphatic VEGFR-3 receptor plays a critical molecular role. We report the genetic importance of a novel glycan coreceptor for pathological VEGF-C–dependent lymphangiogenesis in vivo. Mechanistic work points to important roles for appropriately sulfated lymphatic heparan sulfate in mediating Akt- and Erk-dependent lymphatic signaling as well as activation of lymphatic endothelial VEGFR-3 by multiple VEGF-C species. We also discovered that syndecan-4 is the major lymphatic heparan sulfate proteoglycan involved in mediating lymphatic VEGF-C–VEGFR-3 complex formation and signaling in a manner that critically depends on its glycan chains, and we propose a novel role for heparan sulfate proteoglycans in mediating this biological process. The findings may guide development of novel glycan-biosynthesis inhibitors or proteoglycan-targeting strategies to inhibit carcinoma spread, fibrosis in a variety of inflammatory states, or conditions where lymphangiogenesis contributes to pathological progression.
References
- 1.Lohela M, Bry M, Tammela T, Alitalo K. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr Opin Cell Biol. 2009;21:154–165. doi: 10.1016/j.ceb.2008.12.012. doi: 10.1016/j.ceb.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 2.Alitalo K, Tammela T, Petrova TV. Lymphangiogenesis in development and human disease. Nature. 2005;438:946–953. doi: 10.1038/nature04480. doi: 10.1038/nature04480. [DOI] [PubMed] [Google Scholar]
- 3.Achen MG, McColl BK, Stacker SA. Focus on lymphangiogenesis in tumor metastasis. Cancer Cell. 2005;7:121–127. doi: 10.1016/j.ccr.2005.01.017. doi: 10.1016/j.ccr.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 4.Kyzas PA, Geleff S, Batistatou A, Agnantis NJ, Stefanou D. Evidence for lymphangiogenesis and its prognostic implications in head and neck squamous cell carcinoma. J Pathol. 2005;206:170–177. doi: 10.1002/path.1776. doi: 10.1002/path.1776. [DOI] [PubMed] [Google Scholar]
- 5.Tammela T, Alitalo K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell. 2010;140:460–476. doi: 10.1016/j.cell.2010.01.045. doi: 10.1016/j.cell.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 6.Hirakawa S. Regulation of pathological lymphangiogenesis requires factors distinct from those governing physiological lymphangiogenesis. J Dermatol Sci. 2011;61:85–93. doi: 10.1016/j.jdermsci.2010.11.020. doi: 10.1016/j.jdermsci.2010.11.020. [DOI] [PubMed] [Google Scholar]
- 7.Karpanen T, Egeblad M, Karkkainen MJ, Kubo H, Ylä-Herttuala S, Jäättelä M, Alitalo K. Vascular endothelial growth factor C promotes tumor lymphangiogenesis and intralymphatic tumor growth. Cancer Res. 2001;61:1786–1790. [PubMed] [Google Scholar]
- 8.Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- 9.Iozzo RV, Sanderson RD. Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J Cell Mol Med. 2011;15:1013–1031. doi: 10.1111/j.1582-4934.2010.01236.x. doi: 10.1111/j.1582-4934.2010.01236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuster MM, Wang L, Castagnola J, Sikora L, Reddi K, Lee PH, Radek KA, Schuksz M, Bishop JR, Gallo RL, Sriramarao P, Esko JD. Genetic alteration of endothelial heparan sulfate selectively inhibits tumor angiogenesis. J Cell Biol. 2007;177:539–549. doi: 10.1083/jcb.200610086. doi: 10.1083/jcb.200610086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Y, Macleod V, Miao HQ, Theus A, Zhan F, Shaughnessy JD, Jr, Sawyer J, Li JP, Zcharia E, Vlodavsky I, Sanderson RD. Heparanase enhances syndecan-1 shedding: a novel mechanism for stimulation of tumor growth and metastasis. J Biol Chem. 2007;282:13326–13333. doi: 10.1074/jbc.M611259200. doi: 10.1074/jbc.M611259200. [DOI] [PubMed] [Google Scholar]
- 12.Vlodavsky I, Friedmann Y. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. J Clin Invest. 2001;108:341–347. doi: 10.1172/JCI13662. doi: 10.1172/JCI13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jakobsson L, Kreuger J, Holmborn K, Lundin L, Eriksson I, Kjellén L, Claesson-Welsh L. Heparan sulfate in trans potentiates VEGFR-mediated angiogenesis. Dev Cell. 2006;10:625–634. doi: 10.1016/j.devcel.2006.03.009. doi: 10.1016/j.devcel.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 14.Yin X, Johns SC, Lawrence R, Xu D, Reddi K, Bishop JR, Varner JA, Fuster MM. Lymphatic endothelial heparan sulfate deficiency results in altered growth responses to vascular endothelial growth factor-C (VEGF-C). J Biol Chem. 2011;286:14952–14962. doi: 10.1074/jbc.M110.206664. doi: 10.1074/jbc.M110.206664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mancardi S, Stanta G, Dusetti N, Bestagno M, Jussila L, Zweyer M, Lunazzi G, Dumont D, Alitalo K, Burrone OR. Lymphatic endothelial tumors induced by intraperitoneal injection of incomplete Freund’s adjuvant. Exp Cell Res. 1999;246:368–375. doi: 10.1006/excr.1998.4270. doi: 10.1006/excr.1998.4270. [DOI] [PubMed] [Google Scholar]
- 16.Yin X, Truty J, Lawrence R, Johns SC, Srinivasan RS, Handel TM, Fuster MM. A critical role for lymphatic endothelial heparan sulfate in lymph node metastasis. Mol Cancer. 2010;9:316. doi: 10.1186/1476-4598-9-316. doi: 10.1186/1476-4598-9-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gale NW, Prevo R, Espinosa J, Ferguson DJ, Dominguez MG, Yancopoulos GD, Thurston G, Jackson DG. Normal lymphatic development and function in mice deficient for the lymphatic hyaluronan receptor LYVE-1. Mol Cell Biol. 2007;27:595–604. doi: 10.1128/MCB.01503-06. doi: 10.1128/MCB.01503-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ando T, Jordan P, Joh T, Wang Y, Jennings MH, Houghton J, Alexander JS. Isolation and characterization of a novel mouse lymphatic endothelial cell line: SV-LEC. Lymphat Res Biol. 2005;3:105–115. doi: 10.1089/lrb.2005.3.105. doi: 10.1089/lrb.2005.3.105. [DOI] [PubMed] [Google Scholar]
- 19.Kaufman RJ. Selection and coamplification of heterologous genes in mammalian cells. Methods Enzymol. 1990;185:537–566. doi: 10.1016/0076-6879(90)85044-o. [DOI] [PubMed] [Google Scholar]
- 20.Achen MG, Roufail S, Domagala T, Catimel B, Nice EC, Geleick DM, Murphy R, Scott AM, Caesar C, Makinen T, Alitalo K, Stacker SA. Monoclonal antibodies to vascular endothelial growth factor-D block its interactions with both VEGF receptor-2 and VEGF receptor-3. Eur J Biochem. 2000;267:2505–2515. doi: 10.1046/j.1432-1327.2000.01257.x. [DOI] [PubMed] [Google Scholar]
- 21.Kasten P, Schnöink G, Bergmann A, Papoutsi M, Buttler K, Rössler J, Weich HA, Wilting J. Similarities and differences of human and experimental mouse lymphangiomas. Dev Dyn. 2007;236:2952–2961. doi: 10.1002/dvdy.21298. doi: 10.1002/dvdy.21298. [DOI] [PubMed] [Google Scholar]
- 22.Joukov V, Kumar V, Sorsa T, Arighi E, Weich H, Saksela O, Alitalo K. A recombinant mutant vascular endothelial growth factor-C that has lost vascular endothelial growth factor receptor-2 binding, activation, and vascular permeability activities. J Biol Chem. 1998;273:6599–6602. doi: 10.1074/jbc.273.12.6599. [DOI] [PubMed] [Google Scholar]
- 23.McColl BK, Baldwin ME, Roufail S, Freeman C, Moritz RL, Simpson RJ, Alitalo K, Stacker SA, Achen MG. Plasmin activates the lymphangiogenic growth factors VEGF-C and VEGF-D. J Exp Med. 2003;198:863–868. doi: 10.1084/jem.20030361. doi: 10.1084/jem.20030361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lopez de Cicco R, Watson JC, Bassi DE, Litwin S, Klein-Szanto AJ. Simultaneous expression of furin and vascular endothelial growth factor in human oral tongue squamous cell carcinoma progression. Clin Cancer Res. 2004;10:4480–4488. doi: 10.1158/1078-0432.CCR-03-0670. [DOI] [PubMed] [Google Scholar]
- 25.Weich HA, Bando H, Brokelmann M, Baumann P, Toi M, Barleon B, Alitalo K, Sipos B, Sleeman J. Quantification of vascular endothelial growth factor-C (VEGF-C) by a novel ELISA. J Immunol Methods. 2004;285:145–155. doi: 10.1016/j.jim.2003.10.015. doi: 10.1016/j.jim.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 26.Xu D, Fuster MM, Lawrence R, Esko JD. Heparan sulfate regulates VEGF165- and VEGF121-mediated vascular hyperpermeability. J Biol Chem. 2011;286:737–745. doi: 10.1074/jbc.M110.177006. doi: 10.1074/jbc.M110.177006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forster-Horváth C, Mészáros L, Rásó E, Döme B, Ladányi A, Morini M, Albini A, Tímár J. Expression of CD44v3 protein in human endothelial cells in vitro and in tumoral microvessels in vivo. Microvasc Res. 2004;68:110–118. doi: 10.1016/j.mvr.2004.05.001. doi: 10.1016/j.mvr.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 28.Seiter S, Engel P, Föhr N, Zöller M. Mitigation of delayed-type hypersensitivity reactions by a CD44 variant isoform v3-specific antibody: blockade of leukocyte egress. J Invest Dermatol. 1999;113:11–21. doi: 10.1046/j.1523-1747.1999.00635.x. doi: 10.1046/j.1523-1747.1999.00635.x. [DOI] [PubMed] [Google Scholar]
- 29.Rutkowski JM, Boardman KC, Swartz MA. Characterization of lymphangiogenesis in a model of adult skin regeneration. Am J Physiol Heart Circ Physiol. 2006;291:H1402–H1410. doi: 10.1152/ajpheart.00038.2006. doi: 10.1152/ajpheart.00038.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeltsch M, Jha SK, Tvorogov D, Anisimov A, Leppänen VM, Holopainen T, Kivelä R, Ortega S, Kärpanen T, Alitalo K. CCBE1 enhances lymphangiogenesis via A disintegrin and metalloprotease with thrombospondin motifs-3-mediated vascular endothelial growth factor-C activation. Circulation. 2014;129:1962–1971. doi: 10.1161/CIRCULATIONAHA.113.002779. doi: 10.1161/CIRCULATIONAHA.113.002779. [DOI] [PubMed] [Google Scholar]
- 31.Rousseau S, Houle F, Landry J, Huot J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene. 1997;15:2169–2177. doi: 10.1038/sj.onc.1201380. doi: 10.1038/sj.onc.1201380. [DOI] [PubMed] [Google Scholar]
- 32.Keezer SM, Ivie SE, Krutzsch HC, Tandle A, Libutti SK, Roberts DD. Angiogenesis inhibitors target the endothelial cell cytoskeleton through altered regulation of heat shock protein 27 and cofilin. Cancer Res. 2003;63:6405–6412. [PubMed] [Google Scholar]
- 33.Nguyen A, Chen P, Cai H. Role of CaMKII in hydrogen peroxide activation of ERK1/2, p38 MAPK, HSP27 and actin reorganization in endothelial cells. FEBS Lett. 2004;572:307–313. doi: 10.1016/j.febslet.2004.06.061. doi: 10.1016/j.febslet.2004.06.061. [DOI] [PubMed] [Google Scholar]
- 34.Liersch R, Detmar M. Lymphangiogenesis in development and disease. Thromb Haemost. 2007;98:304–310. [PubMed] [Google Scholar]