

Abstract
Arsenic exposure has been implicated as a risk factor for cardiovascular diseases, metabolic disorders, and cancer, yet the role mitochondrial dysfunction plays in the cellular mechanisms of pathology is largely unknown. To investigate arsenic-induced mitochondrial dysfunction in vascular smooth muscle cells (VSMCs), we exposed rat aortic smooth muscle cells (A7r5) to inorganic arsenic (iAs(III)) and its metabolite monomethylarsonous acid (MMA(III)) and compared their effects on mitochondrial function and oxidative stress. Our results indicate that MMA(III) is significantly more toxic to mitochondria than iAs(III). Exposure of VSMCs to MMA(III), but not iAs(III), significantly decreased basal and maximal oxygen consumption rates and concomitantly increased compensatory extracellular acidification rates, a proxy of glycolysis. Treatment with MMA(III) significantly increased hydrogen peroxide and superoxide levels compared to iAs(III). Exposure to MMA(III) resulted in significant decreases in mitochondrial ATP, aberrant perinuclear clustering of mitochondria, and decreased mitochondrial content. Mechanistically, we observed that mitochondrial superoxide and hydrogen peroxide contribute to mitochondrial toxicity, as treatment of cells with MnTBAP (a mitochondrial superoxide dismutase mimetic) and catalase significantly reduced mitochondrial respiration deficits and cell death induced by both arsenic compounds. Overall, our data demonstrates that MMA(III) is a mitochondria-specific toxicant that elevates mitochondrial and non-mitochondrial sources of ROS.
Keywords: Arsenic, Vascular Smooth Muscle Cells, Methyliiodoarsine, Mitochondria, Superoxide, Antioxidant
Graphical abstract
Introduction
Exposure to arsenic has been associated with cancer, diabetes, cardiovascular disease, and metabolic syndrome (Singh et al. 2011, Maull et al. 2012, Kim et al. 2015, Mohammed Abdul et al. 2015). It has been suggested that arsenic alters common pathways such as those involved in oxidative stress and inflammatory signaling, which underlie a variety of arsenic-associated diseases (States et al. 2009, Straif et al. 2009, Mo et al. 2011, Kim et al. 2015). The generation of ROS in particular is a major pathological mechanism induced by arsenic and is involved in the pathogenesis of cancer (Shi et al. 2004), insulin resistance (Padmaja Divya et al. 2015), metabolic syndrome (Ando et al. 2009), and cardiovascular disease (States et al. 2009).
Arsenic has been shown to induce oxidative stress in numerous cell lines, including human vascular smooth muscle cells (VSMCs) (Lynn et al. 2000, Lantz et al. 2006, Naranmandura et al. 2011, Calatayud et al. 2014, Pan et al. 2014), by causing the release of iron from ferritin (Ahmad et al. 2000), or by altering antioxidant activity and increasing inflammation (Flora 2011). Additional tissue pathology in the vascular endothelium caused by exposure to arsenic includes an increase in oxidative stress caused by increased NADPH oxidase activity in VSMCs and endothelial cells, leading to an increase in superoxide and hydrogen peroxide levels (Lynn et al. 2000), and a vicious cycle of dysregulated calcium and nitric oxide signaling. Arsenic-induced ROS couples with nitric oxide (NO) producing peroxynitrite (Bunderson et al. 2002) and decreases the bioavailability of NO to vascular endothelium and smooth muscle, which likely contributes to cardiovascular complications. ROS have also been shown to result in cell damage and cell death in numerous cell lines and models of arsenic-induced disease pathology (Li et al. 2010, Shi et al. 2010, Gu et al. 2016, Aposhian et al. 2003, Abhyankar et al. 2012).
Inorganic arsenic (iAs(III)) undergoes biomethylation in the liver to form the toxic intermediate metabolites monomethyl-arsonous acid (MMA(III)) and dimethylarsonous acid (DMA(III)). These trivalent organic arsenic species have demonstrated elevated toxicity relative to iAs(III) in several studies (Styblo et al. 2000, Cohen et al. 2007, Calatayud et al. 2013) and may be responsible for many of arsenic’s pathological effects. Indeed, cells with higher rates of arsenic metabolism show increased susceptibility to oxidative damage to DNA and induction of tumorigenesis (Styblo et al. 2000, Sakurai et al. 2006). These organic arsenic species can also promote cytotoxicity and tissue pathology by decreasing the levels of several antioxidants. For instance, in comparison to iAs(III), much lower concentrations of MMA(III) over shorter periods of time can alter the activity of glutathione peroxidase and catalase, induce expression of stress proteins and metallothioneins, and increase ROS levels (Calatayud et al. 2013). Other proposed mechanisms for the enhanced toxicity of MMA(III) include greater rates of cell permeation and accumulation (Wang et al. 2015), differences in subcellular distribution, and selective toxicity towards subcellular compartments such as the mitochondria (Naranmandura et al. 2011).
Several studies have investigated the effects of arsenic on VSMCs, which comprise the medial cell layer of vascular arteries and are known to be exposed to arsenic through contact with blood plasma. In vivo, it has been reported that iAs(III) increases blood pressure through calcium sensitization in VSMCs (Lee et al. 2005), and that exposure to MMA(III) at high doses induces VSMC dysfunction by impairing voltage-sensitive Ca2+ channels and reducing vascular reactivity to contractile agonists. In addition to inducing apoptosis of VSMCs (Lee et al. 2005, Bae et al. 2008, Martin-Pardillos et al. 2013), arsenic trioxide has been shown to induce apoptosis in human coronary smooth muscle cells (Luan et al. 2009), microvascular cells (Suriyo et al. 2012), human umbilical vein endothelial cells (Shi et al. 2010), and intestinal endothelial cells (Calatayud et al. 2013). However, some studies have demonstrated a proliferative effect of arsenic on VSMCs (Soucy et al. 2004) and endothelial cells (Barchowsky et al. 1999), presumably by activating mitogenic signals and inducing the release of vascular endothelium growth factor. Bimodal effects of MMA(III) have also been demonstrated. Lim et al reported that MMA(III) elicited higher vasopressor responses at low doses while suppressing vasoconstriction at high doses in vivo (Lim et al. 2011). Collectively these data suggest that the variable effects of these arsenical species may be caused by differences in concentrations and exposure duration, differences in cell culture conditions, tissue preparations of ex vivo models of arsenic toxicity, and cell type analyzed in each study.
As previously mentioned, mitochondria may be a primary target of arsenic in several tissues. Indeed, exposure of arsenic is associated with loss of mitochondrial membrane potential in human pulmonary cell lines (Han et al. 2008), and reduced ATP content in rat liver mitochondria (Hosseini et al. 2013). In addition, MMA(III), but not iAs(III) can selectively target and inhibit mitochondrial complexes II and IV in isolated mitochondria from rat liver (Naranmandura et al. 2011), and induce ROS levels in intact mitochondria in human epithelial cells (Calatayud et al. 2013). The effects of arsenic species on mitochondrial structure/function and mechanisms of toxicity in VSMCs however, remain to be elucidated.
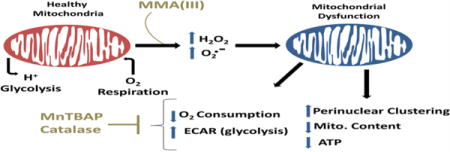
Our study is the first to comparatively analyze the effects MMA(III) and iAs(III) on mitochondrial structure and function in immortalized rat aortic smooth muscle A7r5 cells, a tissue culture model of VSMCs. The doses of iAs(III) and MMA(III) used in this study are in accordance with environmental exposures of at-risk populations reported in several studies (Lerda 1994, Warner et al. 1994, Gebel 2001, Wang et al. 2002) and have been used in previously published studies (Styblo et al. 2000, Naranmandura et al. 2011, Calatayud et al. 2013). In this study, we report that MMA(III), but not iAs(III), promotes mitochondrial dysfunction, metabolic and morphological alterations, and oxidative stress compared to untreated VSMCs. We also found that MMA(III)-mediated induction of ROS preceded loss of mitochondrial content and cell death. Overall, our data supports a conceptual model that suggests that MMA(III) impairs mitochondrial function by eliciting mitochondrial and non-mitochondrial sources of ROS that contribute to cytopathology of VSMCs.
Materials
Dulbecco’s Modified eagle’s media (DMEM), trypsin-EDTA solution (0.25% Trypsin, 0.02% EDTA), fetal bovine serum (FBS), antibiotic-antimycotic (ABAM) (100×), and Amplex® Red Hydrogen Peroxide/Peroxidase Assay kit were purchased from Invitrogen (Carlsbad, CA). Trypan Blue solution (0.4%), RIPA buffer, inorganic arsenic (As2O3) (iAs(III)), 4′,6-diamidino-2-phenylindole (DAPI) nuclear stain, sodium pyruvate solution, sodium hydroxide pellets, methyl iodide, hydrochloric acid, sulfur dioxide, protease inhibitor cocktail, adenosine 5′ -triphosphate (ATP) disodium salt hydrate, dithiothreitol (DTT), catalase from bovine liver, and Bradford Assay Kit were obtained from Sigma-Aldrich (St. Louis, MO). GlutaMax (GIBCO) and MitoSOX (Molecular Probes) were purchased from Thermo Fisher Scientific (Waltham, MA). MnTBAP chloride hydrate was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The CellTiter-Glo® Luminescent Cell Viability Assay kit and the CellTiter 96® AQueous One Solution Cell Proliferation Assay kit were obtained from Promega (Madison, WI). The XF Cell Mito Stress Test Kit was purchased from Seahorse Biosciences (Billerica, MA). The broad-spectrum caspase inhibitor Z-VAD-FMK was purchased from Enzo Life Sciences, Inc. (Farmingdale, NY).
The following antibodies were used: mouse anti-OXPHOS (Abcam), rabbit anti-β-tubulin (Abcam), rabbit anti-Tom20 (Santa Cruz Biotech), rabbit anti-cleaved caspase-3 (Cell Signaling Technology), rabbit IgG and mouse IgG secondary antibodies conjugated to horse radish peroxidase (GE Healthcare Bio-Sciences, Pittsburgh, PA), and Alexa 568-conjugated donkey anti-rabbit IgG secondary antibody (Molecular Probes, Eugene, CA).
Diiodomethylarsine (CH3AsI2), a precursor of monomethylarsonous acid (MMA(III)), was synthesized in the Angermann lab as described by Millar et al (1960). In brief, arsenic(III) oxide (As2O3) and sodium hydroxide were combined in a solution, to which ethyl alcohol and 1.6M methyl iodide were added. The resulting solution was cooled in an ice bath under continuous stirring for 20 hours, and evaporated to dryness under vacuum. A fraction of the solid generated was diverted, reconstituted in ethanol, and recrystallized 2× from ethanol as disodium methanearsonate (Na2(CH3AsO3); ‘MMA(V)’), which presented as white crystals melting at 161°C. The remainder of the residue was dissolved in water, to which concentrated hydrochloric acid, sulfur dioxide, and potassium iodide (KI) were added, generating a precipitate which was then filtered out, washed twice, and discarded; the lower layer of diiodomethylarsine (the diiodo- salt of MMA(III)) was subsequently separated from the filtrate and recrystallized 2× from methanol. The final product was washed with ice water, vacuum dried, and the resultant yellow crystals were verified as diiodomethylarsine by capillary melting point determination at 30°C (Millar et al. 1960). Purity of MMA(III) was further confirmed via 400 MHz 1H NMR spectroscopy on a Varian 400-MR instrument (Figure S1). Diiodomethylarsine crystals were stored at −80°C. MMA(III) working solutions were made fresh as needed by dissolving diiodomethylarsine in ultrapure water, storing at 4°C, and using within 24 hours. Arsenic compounds (iAs(III) and MMA(III)) are carcinogens and skin irritants.
Health Precautions
Proper precautions should be taken to avoid inhalation or direct skin contact by wearing proper laboratory attire including a laboratory coat, safety goggles, and nitrile gloves when handling these compounds.
Methods
Vascular Smooth Muscle Cell Cultures
Immortalized rat aortic smooth muscle cells (A7r5) were generously provided by Dr. Dean Burkin (Department of Pharmacology, University of Nevada School of Medicine). Cells were seeded in polystyrene tissue culture treated T-75 flasks (Sigma-Aldrich) in DMEM containing 4500 mg/L glucose and 100 mg/mL sodium pyruvate supplemented with 10% FBS and 1% ABAM. Cells were maintained at 37°C/5% CO2 and passaged at 80% confluency. All experiments were conducted on cells derived from passages between 5 and 25.
Bright Field Microscopy
A7r5 cells were seeded at 30,000 cells per well in tissue culture treated 24-well plates and adhered for 24 hours prior to treatment with increasing concentrations of iAs(III) (0.1μM, 1μM, 10μM) or MMA(III) (0.1μM, 1μM, 10μM). Live cells were imaged under bright field phase for morphological changes 24 hours post exposure to arsenic species at a magnification of 10× or 20× on an AxioVert 200M microscope (Zeiss) or with an EVOS-FL microscope at the same magnification (Life Technologies).
Trypan Blue Exclusion Assay for Cellular Mortality
Cells were plated at 30,000 cells per well in a 24-well plate for 24 hours, after which cells were treated with increasing concentrations of iAs(III) or MMA(III) for 24 hours. Cells were then trypsinized with 0.25% trypsin to obtain aliquots of cell suspension. Active measurements of cell death induced by arsenic toxicity were conducted by manually counting aliquots of cell suspension by using a 1:1 dilution of Trypan Blue Exclusion dye. The lethal concentrations at 50% (LC50) for MMA(III) and iAs(III) at 24 hours were determined using Origin software (version 6.0). To complement our Trypan Blue Exclusion assays, cell viability was determined by performing cell counts of cell suspensions of A7r5 cells treated with increasing concentrations of arsenicals by using a Z™ series-Coulter Counter (Beckman Coulter) (Figure S3).
Cell Survival Assays
To measure the effects of iAs(III) and MMA(III) on cell viability of VSMCs, cells were seeded in opaque-walled, clear bottom 96 well plates at 10,000 cells per well. Twenty-four hours after seeding, cells were treated with either 0.5μM MMA(III) or 10μM iAs(III) for 24 hours, with or without the broad spectrum caspase inhibitor Z-VAD-FMK at 40μM, followed by incubation with the CellTiter 96® AQueous One Solution MTS reagent for 1 hour per manufacturer’s instructions.
Absorbance was measured at 490 nm with a Multi-mode SpectraMax M4 96 well plate reader (Molecular Devices, Sunnyvale, CA). It is important to note that the amount of iodide present in MMA(III) stocks does not significantly affect cell viability compared to water-treated cells or untreated cells as determined by performing the MTS assay (Figure S2A) or DAPI counts (data not shown), nor does it impact mitochondrial content (data not shown). Hence, all ensuing survival assays were done in vehicle-treated cells compared to MMA(III)-treated cells.
Mitochondrial respiration assay
Cellular respiration was analyzed in cells seeded in a XF24 V7 Cell Culture Microplate by using an XFe24 Extracellular Flux Analyzer (Seahorse Biosciences, North Billerica, MA). In brief, A7r5 cells were plated at a density of 10,000 cells per well for 24 hours prior to analysis of mitochondrial respiration. The oxygen consumption rates (OCRs) and extracellular acidification rates (ECARs) were measured in quadruplicate wells for basal respiration by performing four measuring cycles (~30 minutes) followed by injecting the wells with vehicle or arsenic compounds to analyze for toxic effects of arsenic species on mitochondrial respiration. Cells were then sequentially treated with 1μM oligomycin to measure ATP-linked mitochondrial respiration, 1μM carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP) to measure the maximal respiratory capacity, and with 1μM rotenone/antimycin-A to measure mitochondria-specific respiration as previously described (Dagda et al. 2009, Dagda et al. 2011). We also performed concentration-dependent studies of MMA(III) to measure the lowest dose at which MMA(III) induces a deficit in mitochondrial respiration in A7r5 cells. Following each assay, OCRs and ECARs were normalized to protein concentration by using the Bradford Assay. The line trace of the oxygraph for each experiment was plotted and the mean values of OCRs and ECARs with standard errors of the mean (SEM) were calculated by using the WAVE 2.0 software (Seahorse Biosciences). All cells not exposed to MMA(III) were treated with water. However, it is worth noting that the iodide contained in stocks of MMA(III) did not significantly contribute to a decrease in mitochondrial respiration, suggesting negligible potential for MMA(III) solution-derived iodide toxicity in this model (Figure S2B).
Intracellular detection of Hydrogen peroxide levels
A7r5 cells were seeded at 30,000 cells per well in 6-well plates for 24 hours prior to analysis of extracellular levels of hydrogen peroxide by using the Amplex Red kit. On the day of the assay, the media was replaced with fresh media containing a dilution of 1:1000 Amplex Red, 1:100 horseradish peroxidase (HRP), and iAs(III) (1μM) or MMA(III) (100nM, 1μM, 10μM) and incubated at 37 °C for 2 hours. A Tecan Ultra microplate reader (Männedorf, Switzerland) was used to measure absorption and fluorescence emission at 530 nm and 590 nm, respectively. The data was plotted as the mean fluorescence derived from at least 5 wells per condition.
Mitochondrial Superoxide Assays
To measure the effect of arsenic species on the levels of mitochondria-derived superoxide at different time points, cells were stained with MitoSOX (Molecular Probes) as described previously (Kulich et al. 2007, Dagda et al. 2009). As a control for induction of mitochondrial superoxide, cells were treated with 1μM rotenone for 2 or 4 hours. We used Image J (version 1.44, Bethesda, MD) to calculate the mean fluorescence intensity of MitoSOX per cell for at least 30 cells per condition as previously described (Dagda et al. 2009, Dagda et al. 2011).
Western blot analysis
Cell lysates were prepared by harvesting cells in lysis buffer (1.0% Triton X-100 with phosphatase inhibitors sodium orthovanadate and sodium pyrophosphate, and protease inhibitors E64 and PMSF). Twenty-five micrograms of cell lysates were resolved on 10% SDS-PAGE Tris-buffered polyacrylamide gels by using the mini-gel BioRad system as previously described (Dagda et al. 2014). At the end of the SDS-PAGE run, the proteins were transferred onto methanol pre-activated PVDF membranes by using the Transblot SD Semi-Dry Transfer electrophoresis system (BioRad, Hercules, CA). The PVDF membrane was then blocked in either 2% BSA or 5% non-fat dry milk for 1 hour, washed in phosphate buffered saline with 0.1% Triton X-100 (PBST), and incubated overnight at 4°C with primary antibodies specific for the outer mitochondrial membrane (OMM) -localized protein TOM 20 (1:2000), for subunits of various mitochondrial-encoded mitochondrial complexes (OXPHOS) (1:2000), and for β-tubulin (1:10,000) as a loading control. PVDF membranes were incubated with the appropriate secondary antibodies conjugated to horseradish peroxidase (1:5,000) in PBST and immunoreactive bands were imaged by chemiluminescence by using a BioRad Imager available at the Nevada Proteomics Center.
Immunofluorescence
To image mitochondria, VSMCs were fixed in 4% paraformaldehyde, permeabilized in PBST containing 0.1% Triton X-100 and stained for mitochondria by exposing cells to rabbit-anti-human TOM 20 antibody overnight at 4°C. The cells were then washed extensively in PBST and incubated with Alexa 568-conjugated donkey anti-rabbit IgG (1:1,000), and counterstained with 1.25 μg/ml DAPI to stain for nuclei. Immunolabeled cells were imaged at 25°C by using an EVOS-FL Cell Imaging System equipped with EVOS Light cubes specific for GFP (Ex/Em of 470/510), RFP (Ex/Em of 531/593) and Cy5 (Ex/Em of 628/692) and at a magnification of 20× (0.45NA) or 40× (0.60NA). NA: numerical aperture.
Fluorescence-based image analyses of cell density and active cell death
To determine whether MMA(III) induces caspase-3 mediated apoptosis, we assessed cell density and cell death in VSMCs that were treated with LD50 concentrations of arsenicals by performing image analyses of caspase-3 and DAPI-stained cells. To assess cell density, the number of nuclei per epifluorescence micrograph was manually counted blind to the observer for at least 300 cells per condition. By using the semi-automated Apoptosis Cell Density Image J macro, (http://imagejdocu.tudor.lu/doku.php?id=plugin:morphology:apoptosis_and_cell_count_macro:start), we also analyzed for cell death induced by arsenicals by counting the percentage of cells containing fragmented, pyknotic, or irregular-shaped nuclei per epifluorescence micrograph, morphological hallmarks of cell death (Figure S5). Arsenical-treated cells were also stained for cleaved caspase-3 to determine the extent by which MMA(III) or iAs(III) promote late stage apoptosis in A7r5 cells. Immunolabeled cells were imaged at 25°C by using the EVOS-FL Cell Imaging System as described above. The percentage of cells that stained for caspase-3 per epifluorescence micrograph was determined to assess the ability of MMA(III) or iAs(III) to induce apoptosis.
Mitochondrial morphology
To analyze the effects of arsenic species on mitochondrial morphology 5 hours post treatment, the average area/perimeter ratio per cell was measured as an index of mitochondrial interconnectivity in arsenic-treated VSMCs by using the “Mitochondrial Morphology Macro” (available for free on the Wiki site http://imagejdocu.tudor.lu/) on Image J software (v1.39) as previously described (Dagda et al. 2008). We chose 5 hours as this is the longest duration of treatment with iAs(III) or MMA(III) for functional and non-cytotoxic assays done in this study.
ATP Assay
Mitochondrial ATP concentrations were measured by using the CellTiter-GloR Luminescent Cell Viability Assay (Promega, USA) and a SpectraMax Microplate Reader (Molecular Devices, Sunnyvale, CA) as previously described (Dagda et al. 2009, Dagda et al. 2011). In brief, cells were plated at 10,000 cells per well in opaque-walled 96-well microplates and cultured in DMEM containing 10mM galactose, 2mM glutamine, 2mM sodium pyruvate and 10% FBS for 24 hours prior to treatment with arsenic. Under these media conditions, the cells predominantly rely on mitochondrial oxidative phosphorylation for ATP production (Rossignol et al. 2004). Cells were exposed to iAs(III) (1μM, 10μM) or MMA(III) (0.5μM, 1μM) for 6 hours. To assess for mitochondrial ATP levels, one half of the wells containing cells were treated with the ATP synthase inhibitor oligomycin for 1 hour at 1μM. The luminescence readings were converted to ATP concentrations (μM) based on a 0–10μM standard curve and normalized to protein concentration by using the Bradford assay. Mitochondrial and cytosolic (non-mitochondrial) ATP levels were determined based on the following equation: Mitochondrial steady-state ATP levels= (Total cellular steady−state ATP levels) – (oligomycin-resistant steady state ATP levels, cytosolic ATP). For some experiments, cells were cultured in high glucose media to induce glycolysis to assess the effects of iAs(III) or MMA(III) on cytosolic ATP levels in glycolytic conditions.
Toxicity reversal studies with antioxidants
To determine whether mitochondrial superoxide and/or hydrogen peroxide play a role in arsenic-mediated mitochondrial dysfunction, A7r5 cells were plated at 10,000 cells per well in a XF24 V7 Microplate and either pre-treated with manganese (III) tetrakis (4-benzoic acid) porphyrin (MnTBAP) -a superoxide dismutase mimetic- at 100μM for 24 hours prior to the addition of MMA(III) (0.5μM, 2hours), or co-treated with 10 units/ml of catalase for the duration of MMA(III) treatment (0.5μM, 2hours) to scavenge hydrogen peroxide. This specific dose of MMA(III) consistently decreases mitochondrial respiration (Figure S6). Following treatments with anti-oxidants, the baseline, maximal, and ATP-linked mitochondrial respiration were measured as described above (Mitochondrial respiration assay section). It is worth noting that the brown color in the media formed in cells co-treated with MnTBAP impaired the ability of the XF24e Analyzer to read OCRs, which precluded an analysis of the effects of co-treating cells with MnTBAP and arsenic compounds on mitochondrial function. Hence, for this reason, it was necessary to pretreat (not co-treat) VSMCs with MnTBAP for 24 hours prior to reading OCRs in arsenic-treated cells.
To study the effects of antioxidants on arsenic-induced cell death, cells were plated in opaque-walled 96-well plates at 10,000 cells per well. Following an adherence time of 24 hours, cells were co-treated with 0.5μM MMA(III) or 10μM iAs(III) in the presence of either 100μM of MnTBAP or 10 units/ml of catalase for 24 hours. Following a 24 hour exposure, cells were incubated with One Solution MTS reagent for 1 hour and absorbance was measured at 490 nm by using a Multi-mode SpectraMax M4 96 well plate reader as described above. The absorbance of wells containing cells and treatments were subtracted from wells lacking cells but containing the respective backgrounds. It is worth noting that cells were treated with the above concentrations of arsenicals for 24 hours in order to elicit a consistent, measurable and significantly consistent decrease in cell viability (~50% cell death) prior to assessing for potential significant reversal of cytotoxicity with antioxidants.
Statistical Analysis
All statistical analyses were performed by using the SPSS software (IBM) (v 23). Unless indicated otherwise, most experiments were performed at least three times and the results are presented as means ± standard errors (SE). By applying an α of 0.05, One-Way ANOVAs followed by the post hoc Tukey’s HSD tests were conducted for all experiments to account for multiple comparisons in data interpretation.
Results
Viability of A7r5 Cells following exposure to iAs(III) and MMA(III)
We conducted several cell survival assays to compare the cytotoxic potentials of iAs(III) and MMA(III) in VSMCs at 24 hours. Qualitative analyses of cell morphology revealed that exposure of cells to iAs(III) induced morphological changes associated with necrosis while MMA(III) induced morphological changes consistent with apoptosis (pyknosis and cell blebbing) (Figure 1A). Consistent with the observation that MMA(III) promotes apoptosis, we observed that treating cells with MMA(III), but not iAs(III), robustly elicited the activation of caspase-3 and nuclear fragmentation, pathological hallmarks of apoptosis (Figure S4A–B). Furthermore, co-treating cells with the pan-caspase inhibitor Z-VAD-FMK conferred partial protection against MMA(III)-mediated cell death suggesting that factors other than caspases also contribute to cell death (Figure S4C–D). This data is in agreement with other reported studies that show that MMA(III) and iAs(III)-mediated cell toxicity is associated with the activation of apoptosis and necrosis respectively (Scholz et al. 2005, Lim et al. 2011, Calatayud et al. 2013). The LC50s at 24 hours for MMA(III) and iAs(III) were determined to be 0.50μM and 10.0μM respectively as assessed by three independent methods: the trypan blue exclusion assays (Figure 1B,C), and further corroborated by the MTS assay, a more quantitative and non-biased method for assessing cell viability (Figure 1D), and by performing cell counts using a Coulter counter apparatus (Figure S3). Collectively, these data show that MMA(III) is 20× more toxic than iAs(III) to VSMCs at 24 hours. Based on these cell death studies, all subsequent mitochondrial function and morphological analyses were done using sublethal exposures of arsenical that do not result in significant cell death (Figure S5).
Figure 1. Differential morphological and toxicological effects of MMA(III) and iAs(III) in A7r5 cells.
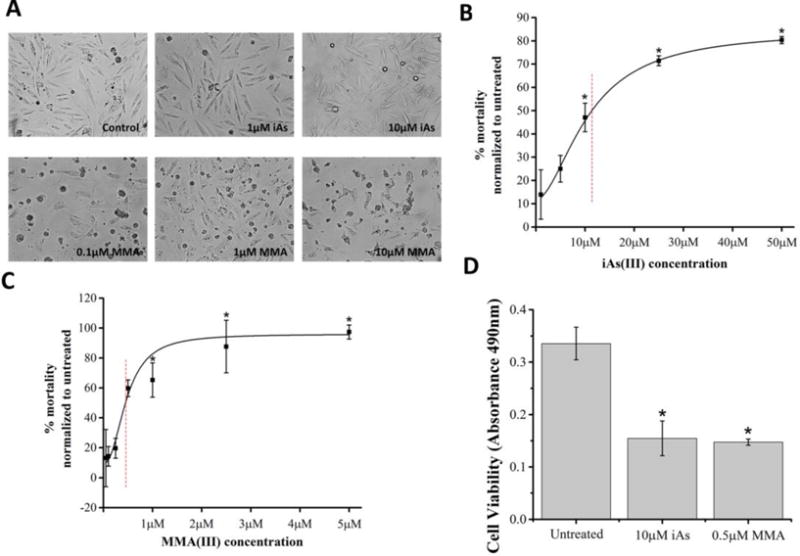
A. Representative images of live A7r5 cells treated with the indicated concentrations of toxicants or vehicle control (Control) for 24 hours. Cells were imaged with an AxioVision (Zeiss) microscope at 10× magnification. Representative micrographs of cells with MMA(III) show pyknosis and blebbing, consistent with apoptosis.
B, C. Cell mortality curves were assessed by performing the Trypan Blue Exclusion Dye assay for cells treated with iAs (panel B) or with MMA(III) (panel C). The percent mortality was calculated as follows: [Cell mortality= Trypan Blue stained cells/total cells * 100]. The 24 hour lethal concentrations at 50% (LC50s) were determined to be 0.5μM for MMA(III) and 11.3μM for iAs(III) as indicated by red-hatched lines. Dose dependent graphs are representative of three experiments (*:p<0.05 vs. untreated, Means ±SE, n=3 samples per condition). One-way ANOVA, Tukey’s HSD.
D. Viability of A7r5 cells was assessed via the MTS assay following 24 hour exposure. Cells treated with 0.5μM MMA(III) or with 10μM iAs(III) showed a significant decrease in cell viability (*:p<0.05 vs. untreated, Means±SE, n=18 wells/condition). One-way ANOVA, Tukey’s HSD. Data was compiled from 3 independent experiments.
Effects of iAs(III) and MMA(III) on mitochondrial respiration
We measured the effects of both inorganic and organic arsenic species on mitochondrial function by using an Extracellular Flux XF24e Analyzer, a well characterized system for measuring real-time oxygen consumption in live cells. In brief, we found that prelethal exposure of cells to 0.5μM of MMA(III), but not iAs(III) at 10μM, progressively induced a significant decrease in the basal respiration of VSMCs within 3 hours of treatment (Figure 2A,B). For comparative purposes, other OCR traces show consistency and little variability in the temporal dynamics of MMA(III)-mediated decrease in OCRs across experiments (Figure S6). It is worth noting that treating cells with MMA(III), but not with iAs(III), led to a progressive reduction in baseline OCR that plateaued by 100 min. of treatment (Figure 2A, Figure S6). In addition, we observed a significant decrease in maximal oxygen consumption rates (OCRs) in cells exposed to 0.5μM MMA(III), but not with 10μM iAs(III) (Figure 2 A,C). MMA(III) is a mitochondria-specific toxicant since treating cells with MMA(V) at similar concentrations (1μM) did not decrease OCRs in A7r5 cells (Figure S7).
Figure 2. Effect of arsenic species on mitochondrial respiration.
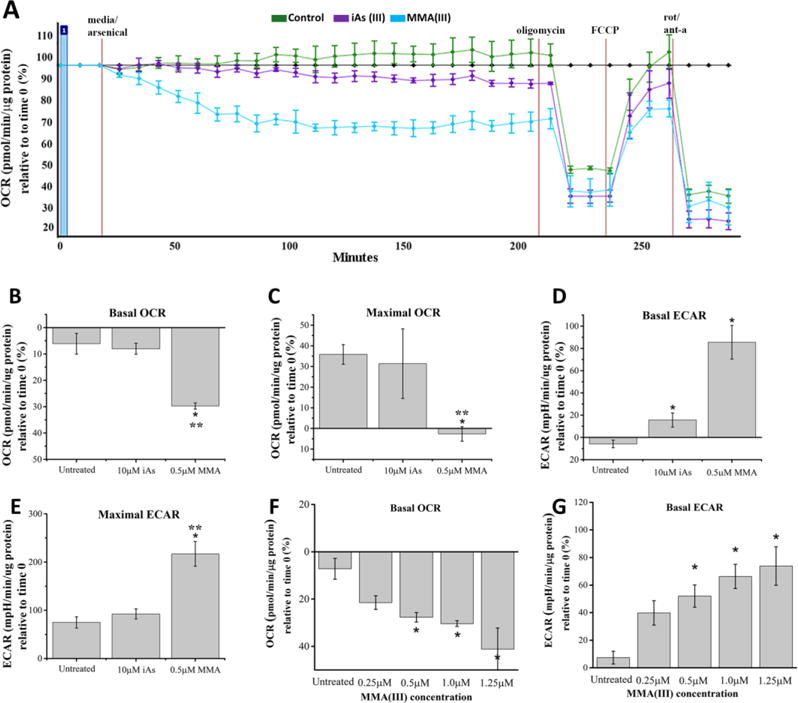
A. Representative mitochondrial respiration profile (oxygraph) of three experiments demonstrates that VSMCs treated with 0.5μM MMA(III), but not with 10μM iAs(III) show a progressive decrease in baseline OCRs followed by a reduction in maximal OCRs compared to control (untreated). The oxygraph is representative of three experiments with similar results and all other traces have been uploaded as supplemental information (Figure S6).
B. Comparative analysis of basal respiration, measured as a percentage relative to time 0, demonstrates that VSMCs treated with 0.5μM MMA(III) for 3 hours showed a significant decrease in baseline OCRs relative to untreated cells or cells treated with 10μM iAs(III) (*:p<0.05 vs. untreated cells, **:p<0.05 vs. 10μM iAs(III) treated cells, Means, ±SE, n=10–16 wells/condition). One-way ANOVA, Tukey’s HSD. Data was compiled from three independent experiments.
C. Comparative analysis of maximal mitochondrial respiration, measured as a percentage relative to time 0, demonstrates that VSMCs exposed for 3 hours to 0.5μM MMA(III) show significantly reduced maximal OCRs compared to untreated cells or cells treated with 10μM iAs(III). To measure maximal mitochondrial respiration, cells were treated with 1μM FCCP and OCRs were measured for three consecutive time points (*:p<0.05 vs. untreated cells,**:p<0.05 vs. 10μM iAs(III) treated cells, graph shows means, ±SE, n=10–16 wells/condition). One-way ANOVA, Tukey’s HSD. Data was compiled from three independent experiments.
D. Comparative analyses of extracellular acidification rate (ECAR) show that treating VSMCs with 0.5μM MMA(III) or with 10μM iAs(III) for 3 hours leads to a significant increase in ECARs (*:p<0.05 vs. untreated cells, Means, ±SE, n=10–16 wells/condition). Treatment with 0.5μM MMA(III) resulted in a non-significant increase in ECAR relative to treatment with 10μM iAs(III). One-way ANOVA, Tukey’s HSD. Data was compiled from three independent experiments.
E. Comparative analyses of maximal ECARs show that treating VSMCs with 0.5μM MMA(III) leads to a significant increase in maximal ECARs compared to untreated cells or cells treated with 10μM iAs(III). (*:p<0.05 vs. untreated cells, **:p<0.05 vs. 10μM iAs(III) treated cells, Means, ±SE, n=10–16 wells/condition). One-way ANOVA, Tukey’s HSD. Data was compiled from three independent experiments.
F. Dose-dependent analyses show that baseline OCRs were significantly reduced in VSMCs treated with MMA(III) in a dose dependent manner. (*:p<0.05 vs. untreated, Means, ±SE n=8–14 wells/condition). One-way ANOVA, Tukey’s HSD. Data was compiled from three independent experiments.
G. Dose-dependent analyses show that baseline ECARs were significantly reduced in VSMCs treated with MMA(III) in a dose dependent manner (*:p<0.05 vs. untreated, Means, ±SE n=8–14 wells/condition). One-way ANOVA, Bonferroni adjusted Tukey’s HSD. Data was compiled from three experiments.
Interestingly, treating VSMCs with MMA(III) led to a significant increase in basal and maximal extracellular acidification rates (ECARs), a proxy of glycolysis. This data suggests that MMA(III) toxicity leads to a compensatory increase in glycolysis when mitochondrial function is impaired, also known as the Warburg effect (Suchorolski et al. 2013) (Figure 2D,E). Treatment with iAs(III) led to a robust increase in basal ECARs (Figure 2D) but maximal ECARs were not significantly affected (Figure 2E), suggesting that treatment of VSMCs with iAs(III) leads to a partial upregulation of ECAR. Next, we wanted to assess the minimal concentration of MMA(III) that can induce alterations in mitochondrial respiration and glycolysis in VSMCs. Dose-dependent analysis of OCRs and ECARs revealed that MMA(III) can induce a significant decrease in basal OCRs and ECARs at concentrations as low as 0.5μM. A non-significant decrease in OCRs was observed in cells exposed to 0.25μM of MMA(III). (Figure 2F, G). Overall, these data suggest that treating A7r5 cells with sublethal doses of MMA(III), but not iAs(III), selectively impair mitochondrial respiration while concomitantly upregulating glycolysis, possibly as a protective mechanism to compensate for mitochondrial dysfunction.
Effects of Arsenic compounds on mitochondrial ATP levels
Given that oxidative phosphorylation drives the synthesis of mitochondrial ATP, we then investigated whether MMA(III)-mediated deficits on mitochondrial respiration are associated with a decline in mitochondrial ATP levels. We observed that exposing VSMCs to 0.5μM MMA(III) for 6 hours led to a significant decrease in mitochondrial ATP levels, while 1μM MMA(III) significantly decreased cytosolic ATP levels (Figure 3). To further corroborate the effects of MMA(III) on cytosolic ATP steady-state levels, we observed that prelethal exposure to MMA(III), but not iAs(III), contributed to a modest but significant decrease in cytosolic ATP levels in cells grown in high glucose-containing media (Control: 2.98μM ±0.12; MMA(III): 2.21 μM ±0.06, p<0.00001). On the other hand, treatment with 1μM or 10μM iAs(III) had no significant effect on either mitochondrial, cytosolic, or total ATP levels compared to control cells in no-glucose media (Figure 3) or under glycolytic conditions (Control: 2.98μM ±0.12; iAs(III): 2.43 μM ±0.18, p=0.25). It is worth noting that treating cells with these doses of arsenicals for 6 hours does not cause significant cell death which rules out the possibility that any observed decreases in ATP levels is due to cytotoxicity (Figure S5). These results suggest that MMA(III) significantly decreases mitochondrial respiration and ATP levels while iAs(III) does not.
Figure 3. Effect of arsenic species on mitochondrial ATP levels.
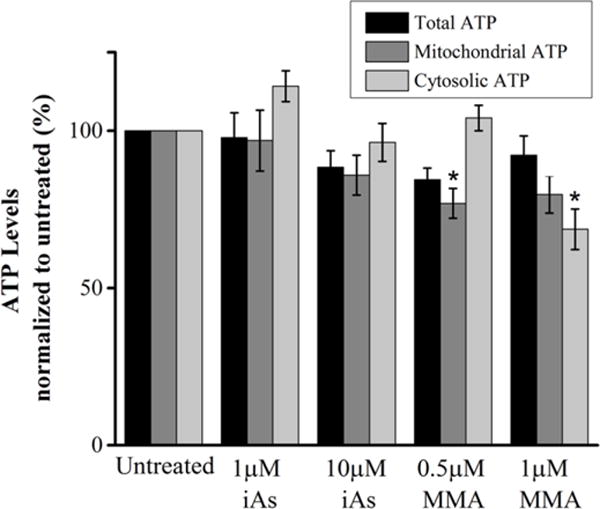
Compiled quantification of ATP levels in VSMCs treated with the indicated concentrations of MMA(III) and iAs(III) for 6 hours. Mitochondrial ATP levels were determined by subtracting the levels of ATP not affected by electron chain complex inhibitors rotenone and antimycin-A (cytosolic ATP) from total ATP levels. Total, mitochondrial or cytosolic ATP levels shown in the compiled quantification was normalized to its respective untreated control within each group (total, mitochondrial or cytosolic).Treatment with 0.5μM MMA(III) resulted in a significant decrease in mitochondrial ATP levels relative to untreated cells, while treatment with 1μM MMA(III) resulted in a significant decrease in cytosolic ATP relative to untreated cells. (*:p<0.05 vs. untreated cells, Means, ±SE, n=18 wells/condition) One-way ANOVA, Tukey’s HDS. Data was compiled from three independent experiments.
Please note that in media conditions that do not have glucose but is supplemented with pyruvate and galactose, we observe that cells produce 60–70% of ATP from mitochondria whereas cells grown in high glucose media, which stimulate glycolysis, predominantly produce non-mitochondrial (cytosolic) ATP (~80–90% of total ATP).
Effects of arsenic compounds on ROS levels
Given that MMA(III) can significantly suppress mitochondrial respiration, we then wanted to identify mechanisms of MMA(III)-mediated mitochondrial dysfunction in VSMCs. Arsenic compounds are known to stimulate the production of hydrogen peroxide in myriad cell types (Li et al. 2001, Jomova et al. 2011). Hence, we surmised that an elevation in the levels of hydrogen peroxide may be correlated with mitochondrial dysfunction induced by MMA(III). In brief, we indirectly measured the levels of hydrogen peroxide in cells exposed to arsenic species by using the Amplex Red assay. We observed that increasing concentrations of MMA(III) induced a significant elevation of hydrogen peroxide levels compared to control cells or iAs(III) at 2 hours post exposure. This data suggests that elevated hydrogen peroxide levels induced by MMA(III) may contribute to mitochondrial and cellular pathology (Figure 4A).
Figure 4. Effects of arsenic species on hydrogen peroxide and mitochondrial superoxide levels.
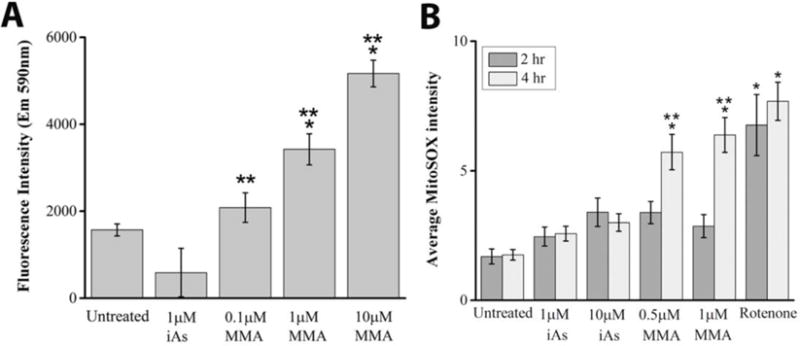
A. Mean hydrogen peroxide levels, as assessed by the Amplex red assay, were significantly elevated in cells exposed to 1μM or 1μM MMA(III) for 2 hours compared to untreated cells or cells treated with 1μM iAs(III) (*:p<0.05 vs. untreated cells, **:p<0.05 vs. 1μM iAs(III) treated cells, graph shows means, ±SE, n=4 wells/condition). One-way ANOVA, Tukey’s HSD. Data is representative of one of three experiments.
B. The average intensity of MitoSOX fluorescence was quantified in untreated cells, and in cells exposed for 2 or 4 hours to the indicated concentrations of MMA(III), iAs(III) or with the complex I inhibitor rotenone as a positive control for mitochondrial superoxide induction. Mitochondrial superoxide was significantly elevated among cells treated for 4 hours with 0.5μM or 1μM MMA(III) compared to untreated cells, or cells treated with iAs(III) at a concentration of 1μM or 10μM (*:p<0.05 vs. untreated, **:p<0.05 vs. iAs(III), Means, ±SE, n=25–46 cells/condition). One way ANOVA, Tukey’s HSD. Data was compiled from three independent experiments.
Generation of Superoxide
Arsenic toxicity has been associated with increased levels of mitochondrial superoxide in other cell lines (Naranmandura et al. 2011). Given that hydrogen peroxide is derived from mitochondrial superoxide, we investigated the extent to which arsenic species can induce the production of mitochondrial superoxide by staining live cells with the red-fluorescent, cell permeable superoxide indicator MitoSOX. Image analyses of mean MitoSOX fluorescence suggests that treating cells with 0.5μM or 1μM MMA(III) can significantly elevate mitochondrial superoxide levels at 4 hours compared to control cells or cells treated with 1μM or 10μM iAs(III) (Figure 4B). MMA(III)-mediated elevation in superoxide is commensurate with oxidative stress induced by exposure to the complex I inhibitor rotenone. Neither arsenic species significantly elevated mitochondrial superoxide levels at 2 hours, while only a non-significant increase in superoxide was induced by iAs(III) at 4 hours (Figure 4B). However, at longer exposures of 6 hours, we found that iAs(III) can significantly increase mitochondrial superoxide levels compared to untreated cells (Figure S8). Our data is consistent with another study that showed a stronger induction of ROS in MMA(III) compared to iAs(III) in liver cells (Naranmandura et al. 2011). Collectively, this data suggests that MMA(III) is a more efficient inducer of mitochondrial superoxide compared iAs(III) in VSMCs.
Effects of Arsenic compounds on mitochondrial morphology and content
Next, we surmised that the decreased mitochondrial respiration and ATP levels induced by MMA(III) leads to a decrease in mitochondrial content in VSMCs. To address this hypothesis, we quantified mitochondrial content in VSMCs by using image-based analysis of mitochondrial content in VSMCs immunostained for mitochondria and by immunoblotting for the total cellular levels of various mitochondrial markers. Western blot analyses of mitochondrial markers revealed that treating VSMCs with 1μM MMA(III) for 5 hours, a concentration that is sufficient to induce mitochondrial damage and oxidative stress but not cell death (Figure 2, 4 and S4), significantly decreased the levels of mitochondrial markers localized either to the outer mitochondrial membrane (TOM20) or the inner mitochondrial membrane (ATP synthase V) (Figure 5A–C). On the other hand, treating cells with 1μM iAs(III) for 5 hours caused a non-significant decrease in mitochondrial protein levels (Figure 5A–C). This data is consistent with the effects of MMA(III) on mitochondria-derived ATP levels (Figure 3). In further agreement with our Western blot data, image analyses of VSMCs immunostained for mitochondria revealed that cells treated with 1μM MMA(III) or 1μM iAs(III) for 5 hours led to a significant decrease in mitochondrial content (% of cytosol occupied by mitochondria) (Figure 5D, S7). Given that mitochondrial content is regulated by mitochondrial turnover through the autophagosomal-lysosomal pathway (mitophagy) and the fact that mitochondrial fragmentation precedes mitophagy (Klionsky et al. 2000, Bursch 2001, Twig et al. 2008), we then surmised that treating cells with both arsenic species causes mitochondrial fragmentation. Unexpectedly, we observed that 1μM MMA(III), but not 1μM iAs(III), promoted perinuclear clustering of mitochondria and a significant increase in mitochondrial interconnectivity at 5 hours post exposure (Figure 5 E–F). However, treatment of cells with longer exposure of 10μM iAs(III) can decrease mitochondrial content eventually and fragment mitochondria (Figure S9, S10). Overall, our immunofluorescence data highlights significant differences in the ability of MMA(III) and iAs(III) for altering mitochondrial morphology and mitochondrial distribution.
Figure 5. Effects of arsenic species on mitochondrial content and morphology.
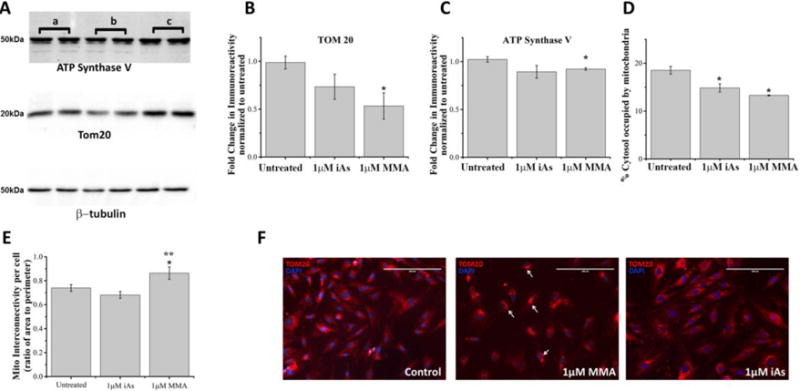
A. Representative Western blot analyses of the total cellular levels of mitochondrial markers (TOM20 and ATP synthase V) and of β-tubulin in cell lysates from untreated cells (lanes indicated by a), and cells exposed for 5 hours to 1μM MMA(III) (lanes indicated by b) or exposed to 1μM iAs(III) (lanes indicated by c).
B. Densitometric analyses of the immunoreactivity of the outer membrane localized protein TOM20 as assessed by Western blot of cell lysates from cells treated with indicated concentrations of MMA(III) and iAs(III) and normalized to untreated cells. (*:p<0.05 vs. untreated, Means, ±SE, n=5 lanes/condition). One-way ANOVA, Tukey’s HSD. Data was compiled from three independent Western blot experiments.
C. Densitometric analyses of the immunoreactivity of the inner membrane localized protein ATP Synthase V as assessed by Western blot of cell lysates derived from cells treated with indicated concentrations of MMA(III) and iAs(III) and normalized to untreated cells. (*:p<0.05 vs. untreated, Means, ±SE, n=5 lanes/condition). One-way ANOVA, Tukey’s HSD. Data was compiled from three independent Western blot experiments.
D. Image-based quantification of mitochondrial content in paraformaldehyde-fixed cells immunolabeled for TOM20 as assessed by measuring the percentage of the cytosolic area occupied by fluorescently-labeled mitochondria-specific pixels and normalized to untreated cells. (*:p<0.05 vs. untreated cells, Means, ±SE, n=44–57 cells/condition). One-way ANOVA, Fishers LSD Tukey’s HSD. Data was compiled from three independent experiments.
E. Mitochondrial interconnectivity, defined as the ratio of the average area to the perimeter of mitochondria per cell (Dagda et al. 2009), was measured in untreated cells and in cells treated with the indicated concentrations of iAs(III) and MMA(III). Cells treated with MMA(III) had significantly increased interconnectivity compared to untreated cells or cells treated with iAs(III) (*:p<0.05 vs. untreated, **:p<.05 vs. 1μM iAs(III), Means, ±SE, n=44–57 cells/condition). One-way ANOVA, Fishers LSD Tukey’s HSD. Data was compiled from three independent experiments.
F. Representative epifluorescence micrographs of paraformaldehyde-fixed cells immunolabeled for TOM20 (red) and counterstained with DAPI to visualize mitochondria and nuclei respectively. Epifluorescence micrographs were acquired at a magnification of 20×. While untreated VSMCs showed a normal mitochondrial morphology (tubular to interconnected), cells treated with MMA(III), but not iAs(III), showed the presence of aberrant mitochondrial clusters (indicated by white arrows). Data is representative of one of three experiments with similar results.
Mitochondrial and non-mitochondrial sources of ROS contribute to arsenic toxicity in VSMCs
Given that MMA(III), but not iAs(III), selectively promotes mitochondrial dysfunction and is a more efficient inducer of ROS than iAs(III), we then surmised that mitochondrial superoxide and hydrogen peroxide induced by MMA(III) contributes to mitochondrial dysfunction in VSMCs. To address this hypothesis, we co-treated VSMCs with catalase or pretreated them with the superoxide dismutase mimetic MnTBAP- two anti-oxidants that can scavenge hydrogen peroxide and mitochondrial superoxide, respectively. Following exposure to antioxidants, VSMCs were treated with 0.5μM MMA(III) and assayed for mitochondrial respiration by employing a XF24e Extracellular Flux Analyzer. We observed that MnTBAP treatment partially but significantly ameliorated MMA(III)-mediated deficits in basal OCRs but was unable to reverse the decrease in maximal OCRs (Figure 6B–D). Interestingly, MnTBAP treatment was able to reverse the increase in maximal ECARs induced by MMA(III) (Figure 6F). Treating VSMCs with catalase, on the other hand, completely reversed MMA(III)-mediated decreases in basal and maximal OCRs and was able to block the increase in ECARs (Figure 6A, C–F). Next, we generated “phenograms” -visual representations of the cell’s energy profile derived by plotting OCRs vs. ECARs- to further understand the metabolic rescue mediated by catalase and MnTBAP treatment in cells exposed to MMA(III). We observed that treating cells with catalase not only reversed mitochondrial dysfunction induced by MMA(III), but it also significantly reversed the energy profile of VSMCs back to a more aerobically active and less glycolytic phenotype compared to MMA(III)-treated cells (Figure 7A) whereas treating VSMCs with MnTBAP had a partial effect (Figure 7B).
Figure 6. Antioxidant rescue of MMA(III)-induced deficits in mitochondrial respiration.
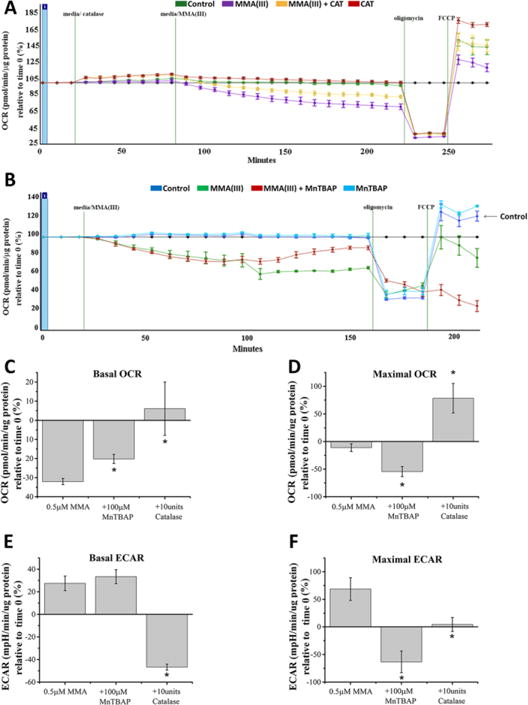
A. Representative mitochondrial respiratory profile (oxygraph) demonstrates that co-treating cells with catalase rescued cells against 0.5μM MMA(III)-mediated decreases in baseline OCRs and maximal respiratory capacity. The oxygraph is representative of one of three experiments showing similar results.
B. Representative mitochondrial respiratory profile (oxygraph) demonstrates that pre-treating cells with MnTBAP rescued cells against 0.5μM MMA(III)-mediated decreases in baseline OCRs but not the maximal respiratory capacity. The oxygraph is representative of one of three experiments showing similar results.
C. Graph showing average baseline OCRs compiled from three experiments in cells treated with MMA(III) and co-treated with catalase or pretreated with MnTBAP for 24 hours at the indicated concentrations for 3 hours. (*:p<0.05 vs. 0.5μM MMA(III), Means, ±SE, n=25–29 wells/condition). One-way ANOVA, Tukey’s HSD.
D. Graph showing average maximal OCRs compiled from three experiments in cells treated with MMA(III) and co-treated with catalase or pretreated with MnTBAP for 24 hours at the indicated concentrations for 3hours. (*:p<0.05 vs. 0.5μM MMA(III), Means, ±SE, n=25–29 wells/condition). One-way ANOVA, Tukey’s HSD.
E. Graph showing average basal ECARs compiled from three experiments in cells treated with MMA(III) and co-treated with catalase or pretreated with MnTBAP for 24 hours at the indicated concentrations for 3hours. (*:p<0.05 vs. 0.5μM MMA(III), Means, ±SE, n=25–29 wells/condition). One-way ANOVA, Tukey’s HSD.
F. Graph showing average maximal ECARs compiled from three experiments in cells treated with MMA(III) and co-treated with catalase or pretreated with MnTBAP for 24 hours at the indicated concentrations for 3 hours. (*:p<0.05 vs. 0.5μM MMA(III), Means, ±SE, n=25–29 wells/condition). One-way ANOVA, Tukey’s HSD.
Figure 7. Antioxidants reverse the metabolic alterations induced by MMA(III).
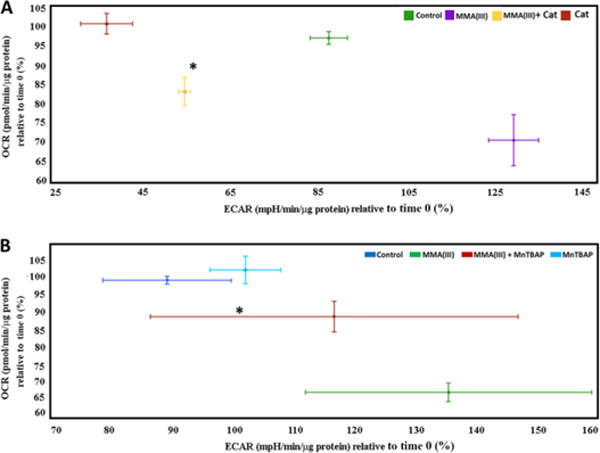
A. Representative phenogram from one of three experiments (baseline OCRs vs. baseline ECARs) showing a robust glycolytic shift in cells treated with 0.5μM MMA(III) for 200 min. compared to control (untreated) but reversed by co-treatment with catalase (*:p<0.05 vs. 0.5μM MMA(III), Means, ±SE, n= 4–6 wells per condition). One-way ANOVA, Tukey’s HSD. The decreased OCR observed in control group relative to its first time point likely reflects a modest induction of oxidative stress in cells incubating for 3 hours required to do the oxygen consumption assay.
B. Representative phenogram from three experiments showing (baseline OCRs vs. baseline ECARs) a robust glycolytic shift in cells treated with 0.5μM MMA(III) compared to control (untreated). MnTBAP pre-treatment was only able to partially reverse the glycolytic shift (*:p<0.05 vs. 0.5μM MMA(III), Means, ±SE, n= 4–6 wells per condition). One-way ANOVA, Fishers LSD. The decreased OCR observed in control group relative to its first time point likely reflects a modest effect oxidative stress in cells incubating for 3 hours required to do the oxygen consumption assay.
Finally, we investigated whether the reversal in mitochondrial dysfunction mediated by anti-oxidants was associated with increased cell survival against MMA(III) and iAs(III)-mediated toxicity. As proof of principle of the ROS quenching effects of antioxidants in cells are cytoprotective, we observed that treating cells with catalase or MnTBAP had a significant effect for enhancing cell survival against both MMA(III) and iAs(III) whereas MnTBAP had a partial protective effect against MMA(III) highlighting the differential contribution of different sources of ROS on cytopathology and the ability of antioxidants to confer protection (Figure 8).
Figure 8. Antioxidants reverse cell death induced by MMA(III) and iAs(III).
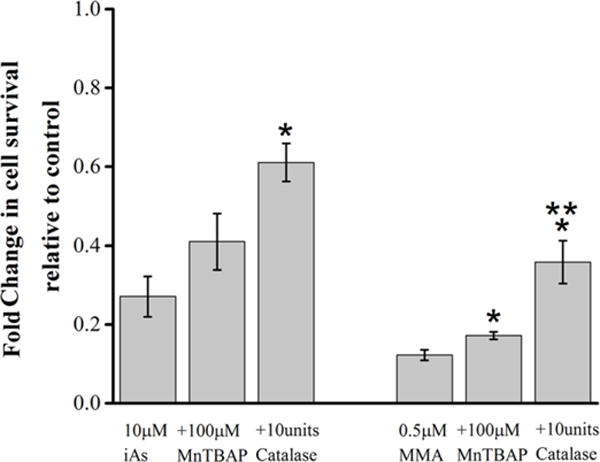
Compiled quantification of cell viability as measured by the MTS assay in cells treated with the indicated concentrations of MMA or iAs(III), and either pretreated with MnTBAP or co-treated with catalase. (*:p<0.05 vs. 10μM iAs(III) or 0.5μM MMA(III) of their respective groups, **:p<.05 comparing MnTBAP to catalase treatment in respective groups, Means, ±SE, n=6–21 wells/condition). One-way ANOVA, Tukey’s HSD. Data was compiled from three experiments.
Discussion
Although it is widely accepted that arsenic species can produce various degrees of mitochondrial dysfunction in myriad cell types, the effects of inorganic and organic species of arsenic on mitochondrial function and structure in VSMCs have not been elucidated. In this study, we demonstrate for the first time that MMA(III) promotes mitochondrial dysfunction and oxidative stress in VSMCs, and that these deficits stem from the generation of mitochondrial and non-mitochondrial ROS following exposure to MMA(III).
MMA(III) induces cytotoxicity in VSMCs
Our findings on the cytotoxicity of arsenic to VSMCs are in agreement with a majority of studies demonstrating arsenic’s capacity to induce apoptosis in various cell lines including aortic, coronary, and mesenteric vascular smooth muscle (Lee et al. 2005, Martin-Pardillos et al. 2013) (Bae et al. 2008, Luan et al. 2009, Shi et al. 2010, Suriyo et al. 2012, Calatayud et al. 2013). Our data shows that MMA(III), but not iAs(III), promotes caspase-dependent apoptosis in A7r5 cells consistent with other studies (Lim et al. 2011, Calatayud et al. 2013), whereas iAs(III) predominantly elicits necrosis (Scholz et al. 2005) (Figure 1, S4). Furthermore, several studies suggest that apoptosis of smooth muscle cells may play a pathological role in cardiovascular disease progression (Blaschke et al. 2004, Su et al. 2004, Li et al. 2010) (Blaschke et al. 2004, Su et al. 2004, Li et al. 2010), suggesting that cell death of vascular smooth muscle cells may underlie pathology in CVDs associated with arsenic intoxication.
Arsenic targets mitochondria
Organic species of arsenic demonstrate higher toxicity compared to iAs in a variety of cell types. For instance, MMA(III) can induce apoptosis in the human intestinal epithelial cell line Caco-2 by elevating ROS, inducing a reduction in intracellular GSH, and increasing peroxidation of lipids. MMA(III) also alters the antioxidant activities of glutathione peroxidase and catalase and concomitantly induces expression of heat shock proteins and metallothioneins (Calatayud et al. 2013). There is a wealth of evidence that supports selective toxicity of arsenic species towards mitochondria. Previous studies have shown that arsenic induces widespread mitochondrial dysfunction via loss of mitochondrial membrane potential, generation of ROS, diminution of cytochrome c oxidase function, and suppression of oxygen consumption via depletion of mtDNA copy number (Partridge et al. 2007, Han et al. 2008). Consistent with the toxic effects of MMA(III) on mitochondria in many cell types, our data also show that MMA(III) is a mitochondria-specific toxicant in VSMCs. Our study is the first to show an interplay and temporal dynamics of MMA(III) exposure, generation of mitochondrial superoxide and non-mitochondrial ROS, and alterations in mitochondrial function and metabolism in VSMCs (Figure 2A–C, Figure 4).
Our mitochondrial functional assays suggest that MMA(III) is a stronger inducer of mitochondrial dysfunction upstream of cell death compared to iAs(III) or to MMA(V) at equimolar concentrations that are sufficient to induce significant mitochondrial superoxide levels but not high enough to induce cytotoxicity (Figure 2, Figure S5). Although some doses of arsenicals used in this study may be considered high, humans have been acutely and chronically exposed to similar concentrations of arsenicals environmentally (Gebel 2001) and in the clinical treatment of acute promyelocytic leukemia (Wang et al. 2002). Furthermore many epidemiological studies have demonstrated the pathological effects of long-term accumulation of arsenicals in human bodies at high doses (Chen et al. 1996, Rahman et al. 1999). Nevertheless, our study raises the possibility that mitochondrial dysfunction/metabolic alterations induced by MMA(III), at the prelethal doses used, may contribute to pathology and metabolic alterations in humans exposed to arsenic. Indeed, MMA(III)-induced perturbations of glycolytic flux, as discussed in the following section, are particularly relevant to vascular smooth muscle function due to the observation that ATP generated from glycolysis may have a separate ‘metabolic purpose’ (e.g., provision of energy for the Na+/K+ antiporter) than ATP derived from oxidative phosphorylation, which has been demonstrated to provide energy to primarily support contractile functions (Paul 1983). MMA(III)-induced shifts in ATP generation may therefore have profound consequences for VSMC function, especially since these cells are reliant on phosphocreatine (PCr) for short-term, instantaneous metabolic demands and PCr is resynthesized mainly via oxidative phosphorylation (Butler et al. 1985). Overall, our data strongly suggest that MMA(III) is a highly selective arsenic metabolite that targets mitochondrial function at sublethal concentrations and time points. However, we do not rule out the possibility that iAs(III) may also contribute to mitochondrial pathology, albeit less efficiently than MMA(III), in VSMCs in instances of prolonged exposure to iAs(III).
Selective toxicity of MMA(III) to mitochondria activates metabolic compensation in VSMCs
Our results show that treatment of VSMCs with MMA(III) led to significant decreases in basal and maximal OCRs while concomitantly enhancing basal and maximal ECARs compared to cells treated with iAs(III) (Figure 2). The ECAR values detected by the XF24e analyzer are a reliable measure of glycolytic rate. Hence, a decrease in the ratio of the mean OCR to ECAR associated with MMA(III) exposure (Figure 7) is an indicator of a compensatory upregulation of glycolysis as a result of mitochondrial dysfunction. Given that MMA(III) is known to adversely affect the functioning of the electron transport chain (ETC) (Naranmandura et al. 2011), an increase in glycolysis may be a compensatory mechanism to ensure cellular survival, somewhat akin to the Warburg effect (Suchorolski et al. 2013) due to mitochondrial impairment. However, it is worth noting that this increase in ECARs caused by MMA(III) exposure does not lead to an increase in cytosolic ATP levels under conditions that favor mitochondrial ATP production (Figure 3), or in glycolytic conditions (data not shown), suggesting that either MMA(III) elicits a futile upregulation of glycolysis or that non-mitochondrial sources of ROS (e.g. hydrogen peroxide from ER) shut down compensatory glycolysis as suggested by our ATP assays that show a significant decrease in cytosolic ATP at 1μM MMA(III). Toxic stimuli that elicit classical apoptosis (e.g. staurosporine) can significantly elevate cytosolic ATP levels to promote cell death (Zamaraeva et al. 2005). Although MMA(III) elicits apoptosis as evident by increased active caspase-3 levels and nuclear fragmentation (Figure S4A–B), our luminescence-based ATP assays show that MMA(III) does not elicit an increase, but modestly decreases, cytosolic steady-state ATP levels at 2 or 6 hour of exposure (data not shown). This observation suggests that MMA(III) may promote apoptosis through distinct mechanisms that do not involve a transient rise in cytosolic ATP levels compared to “classical” apoptosis induced by staurosporine, or that non-mitochondrial ROS may prevent an upregulation of cytosolic ATP levels by inactivating glycolytic enzymes.
Our data also suggests that the decline in mitochondrial respiration resulting from MMA(III) is an early event that occurs in response to elevated mitochondrial superoxide levels and precedes the loss of mitochondrial ATP and content (Figures 2–5). Although iAs(III) toxicity does not lead to early mitochondrial dysfunction, prolonged exposure of VSMCs with iAs(III) can lead to a reduction in mitochondrial content, fragment mitochondria, and elevate mitochondrial superoxide levels suggesting that iAs(III) induces delayed mitochondrial toxicity without inducing significant cell death (Figure, S4, S7–9).
What are the mechanisms by which arsenic decreases mitochondrial content in VSMCs?
We observed that treating VSMCs with MMA(III) was associated with robust aberrant perinuclear aggregation (clustering) of mitochondria in a subpopulation of cells (Figure 5F) and significant loss of mitochondria (Figure 5B–E). The effects of MMA(III) on mitochondrial aggregation (clumping or perinuclear clustering) is consistent with another study which reported mitochondrial aggregation in the arsenic trioxide -sensitive A172 human glioblastoma cells (Haga et al. 2005). In yet another study, chronic treatment of AL human-hamster hybrid CHOK1 cells with arsenic resulted in an abnormal distribution of mitochondria that varied considerably between cells including increased mitochondrial fusion or filamentous morphology. This effect was associated with decreased mitochondrial function (Partridge et al. 2007).
In mammalian cells, the E3 ubiquitin ligase Parkin is known to promote perinuclear clustering of damaged mitochondria to facilitate mitophagy (Youle et al. 2011). Indeed, Watanabe et al. recently reported that ubiquitin proteasome system activity is increased by arsenic trioxide and the autophagy lysosomal system is increased in mouse atrial cardiomyocytes suggesting that the elimination of dysfunctional mitochondria is enhanced by the E3 ubiquitin ligase Parkin in arsenic-treated cells (Watanabe et al. 2014). Based on our morphological analyses of perinuclear clustering and mitochondrial loss, it is conceivable that the process of elimination of mitochondria induced by both MMA(III) and iAs(III) may involve Parkin-mediated mitophagy. Hence, our study warrants future investigations to elucidate mechanisms of mitochondrial elimination by MMA(III) in VSMCs.
Contribution of different sources of ROS induced by arsenic
There is significant evidence in the scientific literature indicating that arsenic exposure results in increased production of ROS in different cell types including Neuro-2a cells, β- pancreatic cells, human hepatocellular carcinoma HepG2 cells, and human breast and lung carcinoma cell lines (Jiang et al. 2015, Pozo-Molina et al. 2015). However, little is known regarding the subcellular sources of ROS induced by arsenic compounds. In our study, MMA(III) induced a significant elevation of mitochondrial superoxide and hydrogen peroxide levels (Figure 4A–B). At 4 hours post exposure to MMA(III), the levels of superoxide generated are similar to levels achieved in cells treated with the complex I inhibitor rotenone. iAs(III) treatment also induces a modest increase in mitochondrial superoxide production at 4 and 6 hours post treatment suggesting that MMA(III) is a more potent inducer of mitochondrial ROS in VSMCs.
Although it is clear that mitochondrial superoxide induced by MMA(III) contributes to mitochondrial dysfunction in VSMCs (Figure 2 and 3), our data suggest that non-mitochondrial sources of hydrogen peroxide also contribute to mitochondrial dysfunction (Figure 8). While pretreating VSMCs with catalase was sufficient to reverse MMA(III)-mediated decreases in basal and maximal OCRs and block the increase in ECARs (Figure 6C–D), MnTBAP was incapable of rescuing all aspects of mitochondrial function (e.g. maximal respiration) (Figure 6B and D). Unexpectedly, although MnTBAP pretreatment partially rescued basal OCRs, it worsened the reduction in maximal OCRs induced by MMA(III) treatment (Figure 6D). Thus, treating VSMCs with catalase, but not MnTBAP, reversed the energy profile from a predominantly glycolytic to normal phenotype as untreated cells (Figure 6A).
It is worth noting that mitochondria may be the primary target of different arsenic compounds, but are not the sole contributors to ROS production. Indeed, the endoplasmic reticulum and peroxisomes have a greater capacity to produce ROS than mitochondria in liver tissue (Brown et al. 2012). Additionally, arsenic has been shown to induce ROS by deregulating NADH/NADPH oxidase activity in VSMCs and endothelial cells, leading to an increase in superoxide and hydrogen peroxide levels (Lynn et al. 2000), and arsenic-induced ROS couples with nitric oxide (NO) to produce peroxynitrite (Bunderson et al. 2002). Thus, in addition to mitochondrially derived ROS, non-mitochondrial ROS induced by iAs(III) and MMA(III) likely elicit oxidative damage in other organelles in our experimental model. Hence, our work warrants future studies that identify all mitochondrial sources of ROS, and verify non-mitochondrial sources of ROS induced by MMA(III) that contribute to cellular pathology in VSMCs through previously elucidated pathways (Lynn et al. 2000, Bunderson et al. 2002).
A major limitation of the present study is the use of an immortal cell line for linking pathological effects of arsenic toxicity with chronic effects of arsenic toxicity as related to toxicity in humans. However, the benefits of using the immortalized A7r5 cell line as an in vitro model of arsenic toxicity in smooth muscle cells are to unveil mechanisms of arsenic toxicity in VSMCs using an acute dosing paradigm include robust consistency, reproducibility of results, a high availability of biomass to do in vitro studies in lieu of primary cells which precludes the need to sacrifice animals, and a lack of phenotypic change due to sub-selection in passaging. Thus, given the mitochondrial-toxic effects of MMA(III) as shown in this report, our studies warrant future experiments that analyze the toxic effects of MMA(III) vs. iAs(III) in a chronic cell culture model of arsenic exposure to elucidate the role that MMA(III) plays in VSMC toxicity.
Conclusion
In summary, our study is the first to show that MMA(III) can induce concomitant mitochondrial dysfunction, mitochondrial ROS and metabolic alterations in VSMCs. Our study suggests that MMA(III) elicits mitochondrial and non-mitochondrial ROS to promote early mitochondrial dysfunction by affecting oxidative phosphorylation, ATP content, and mitochondrial morphology resulting in a pathology that precedes the loss of mitochondrial content. Our work warrants future studies that probe for molecular mechanisms by which MMA(III) selectively targets mitochondria and elicits the release of superoxide in VSMCs.
Supplementary Material
Research Highlights.
MMA(III) is significantly more toxic to mitochondria than iAs(III) in VSMCs
MMA(III) decreases mitochondrial content, respiration and upregulates glycolysis
MMA(III) elevates mitochondrial ROS and mitochondrial interconnectivity
Antioxidants can reverse MMA(III)-induced mitochondrial dysfunction
Acknowledgments
We would like to acknowledge Dr. Carlos Martino for his technical support and assistance in optimizing the Amplex Red assays. We would like to also acknowledge Dr. Stephen Spain and Casey Philbin of the UNR Chemistry Departments’ Shared Instrumentation Laboratory (SIL) for their assistance with assessment of the purity and synthesis of MMA(III) stocks by 1H-NMR. This research was funded in part by NIH/NIGMS #GM103554 (a COBRE grant in “Cell Signaling Across Membranes”), and by start-up funds from the University of Nevada School of Community Health Sciences and the Department of Pharmacology of the University of Nevada School of Medicine.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
The authors have no conflict of interests to declare for this study.
References
- Abhyankar LN, Jones MR, Guallar E, Navas-Acien A. Arsenic exposure and hypertension: a systematic review. Environ Health Perspect. 2012;120(4):494–500. doi: 10.1289/ehp.1103988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad S, Kitchin KT, Cullen WR. Arsenic species that cause release of iron from ferritin and generation of activated oxygen. Arch Biochem Biophys. 2000;382(2):195–202. doi: 10.1006/abbi.2000.2023. [DOI] [PubMed] [Google Scholar]
- Ando K, Fujita T. Metabolic syndrome and oxidative stress. Free Radic Biol Med. 2009;47(3):213–218. doi: 10.1016/j.freeradbiomed.2009.04.030. [DOI] [PubMed] [Google Scholar]
- Aposhian HV, Zakharyan RA, Avram MD, Kopplin MJ, Wollenberg ML. Oxidation and detoxification of trivalent arsenic species. Toxicol Appl Pharmacol. 2003;193(1):1–8. doi: 10.1016/s0041-008x(03)00324-7. [DOI] [PubMed] [Google Scholar]
- Bae ON, Lim EK, Lim KM, Noh JY, Chung SM, Lee MY, Yun YP, Kwon SC, Lee JH, Nah SY, Chung JH. Vascular smooth muscle dysfunction induced by monomethylarsonous acid (MMA III): a contributing factor to arsenic-associated cardiovascular diseases. Environ Res. 2008;108(3):300–308. doi: 10.1016/j.envres.2008.06.012. [DOI] [PubMed] [Google Scholar]
- Barchowsky A, Roussel RR, Klei LR, James PE, Ganju N, Smith KR, Dudek EJ. Low levels of arsenic trioxide stimulate proliferative signals in primary vascular cells without activating stress effector pathways. Toxicol Appl Pharmacol. 1999;159(1):65–75. doi: 10.1006/taap.1999.8723. [DOI] [PubMed] [Google Scholar]
- Blaschke F, Bruemmer D, Yin F, Takata Y, Wang W, Fishbein MC, Okura T, Higaki J, Graf K, Fleck E, Hsueh WA, Law RE. C-reactive protein induces apoptosis in human coronary vascular smooth muscle cells. Circulation. 2004;110(5):579–587. doi: 10.1161/01.CIR.0000136999.77584.A2. [DOI] [PubMed] [Google Scholar]
- Brown CG, Borutaite V. There is no evidence that mitochondria are the main source of reactive oxygen species in mammalian cells. Mitochondrion. 2012;12(1):1–4. doi: 10.1016/j.mito.2011.02.001. [DOI] [PubMed] [Google Scholar]
- Bunderson M, Coffin JD, Beall HD. Arsenic induces peroxynitrite generation and cyclooxygenase-2 protein expression in aortic endothelial cells: possible role in atherosclerosis. Toxicol Appl Pharmacol. 2002;184(1):11–18. [PubMed] [Google Scholar]
- Bursch W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 2001;8(6):569–581. doi: 10.1038/sj.cdd.4400852. [DOI] [PubMed] [Google Scholar]
- Butler MT, Siegman MJ. High-energy phosphate metabolism in vascular smooth muscle. Annu Rev Physiol. 1985;47:629–643. doi: 10.1146/annurev.ph.47.030185.003213. [DOI] [PubMed] [Google Scholar]
- Calatayud M, Devesa V, Vélez D. Differential toxicity and gene expression in Caco-2 cells exposed to arsenic species. Toxicol Lett. 2013;218(1):70–80. doi: 10.1016/j.toxlet.2013.01.013. [DOI] [PubMed] [Google Scholar]
- Calatayud M, Gimeno-Alcañiz JV, Vélez D, Devesa V. Trivalent arsenic species induce changes in expression and levels of proinflammatory cytokines in intestinal epithelial cells. Toxicol Lett. 2014;224(1):40–46. doi: 10.1016/j.toxlet.2013.09.016. [DOI] [PubMed] [Google Scholar]
- Chen CJ, Chiou HY, Chiang MH, Lin LJ, Tai TY. Dose-response relationship between ischemic heart disease mortality and long-term arsenic exposure. Arterioscler Thromb Vasc Biol. 1996;16(4):504–510. doi: 10.1161/01.atv.16.4.504. [DOI] [PubMed] [Google Scholar]
- Cohen SM, Ohnishi T, Arnold LL, Le XC. Arsenic-induced bladder cancer in an animal model. Toxicol Appl Pharmacol. 2007;222(3):258–263. doi: 10.1016/j.taap.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Dagda RK, Cherra SJ, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem. 2009;284(20):13843–13855. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda RK, Gusdon AM, Pien I, Strack S, Green S, Li C, Van Houten B, Cherra SJ, 3rd, Chu CT. Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinson’s disease. Cell Death Differ. 2011;18(12):1914–1923. doi: 10.1038/cdd.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda RK, Pien I, Wang R, Zhu J, Wang KZ, Callio J, Banerjee TD, Dagda RY, Chu CT. Beyond the mitochondrion: cytosolic PINK1 remodels dendrites through protein kinase A. J Neurochem. 2014;128(6):864–877. doi: 10.1111/jnc.12494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda RK, Zhu J, Kulich SM, Chu CT. Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: implications for Parkinson’s disease. Autophagy. 2008;4(6):770–782. doi: 10.4161/auto.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flora JS. Arsenic-induced oxidative stress and its reversibility. Free Radic Biol Med. 2011;51(2):257–281. doi: 10.1016/j.freeradbiomed.2011.04.008. [DOI] [PubMed] [Google Scholar]
- Gebel WT. Genotoxicity of arsenical compounds. Int J Hyg Environ Health. 2001;203(3):249–262. doi: 10.1078/S1438-4639(04)70036-X. [DOI] [PubMed] [Google Scholar]
- Gu S, Chen C, Jiang X, Zhang Z. ROS-mediated endoplasmic reticulum stress and mitochondrial dysfunction underlie apoptosis induced by resveratrol and arsenic trioxide in A549 cells. Chem Biol Interact. 2016;245:100–109. doi: 10.1016/j.cbi.2016.01.005. [DOI] [PubMed] [Google Scholar]
- Haga N, Fujita N, Tsuruo T. Involvement of mitochondrial aggregation in arsenic trioxide (As2O3)-induced apoptosis in human glioblastoma cells. Cancer Sci. 2005;96(11):825–833. doi: 10.1111/j.1349-7006.2005.00114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YH, Kim SZ, Kim SH, Park WH. Arsenic trioxide inhibits the growth of Calu-6 cells via inducing a G2 arrest of the cell cycle and apoptosis accompanied with the depletion of GSH. Cancer Lett. 2008;270(1):40–55. doi: 10.1016/j.canlet.2008.04.041. [DOI] [PubMed] [Google Scholar]
- Hosseini MJ, Shaki F, Ghazi-Khansari M, Pourahmad J. Toxicity of Arsenic (III) on Isolated Liver Mitochondria: A New Mechanistic Approach. Iran J Pharm Res. 2013;12(Suppl):121–138. [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Wang L, Chen L, Cai GH, Ren QY, Chen JZ, Shi HJ, Xie YH. As2O3 induces apoptosis in human hepatocellular carcinoma HepG2 cells through a ROS-mediated mitochondrial pathway and activation of caspases. Int J Clin Exp Med. 2015;8(2):2190–2196. [PMC free article] [PubMed] [Google Scholar]
- Jomova K, Jenisova Z, Feszterova M, Baros S, Liska J, Hudecova D, Rhodes CJ, Valko M. Arsenic: toxicity, oxidative stress and human disease. J Appl Toxicol. 2011;31(2):95–107. doi: 10.1002/jat.1649. [DOI] [PubMed] [Google Scholar]
- Kim HS, Kim YJ, Seo YR. An Overview of Carcinogenic Heavy Metal: Molecular Toxicity Mechanism and Prevention. J Cancer Prev. 2015;20(4):232–240. doi: 10.15430/JCP.2015.20.4.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky JD, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulich SM, Horbinski C, Patel M, Chu CT. 6-Hydroxydopamine induces mitochondrial ERK activation. Free Radic Biol Med. 2007;43(3):372–383. doi: 10.1016/j.freeradbiomed.2007.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lantz CR, Hays AM. Role of oxidative stress in arsenic-induced toxicity. Drug Metab Rev. 2006;38(4):791–804. doi: 10.1080/03602530600980108. [DOI] [PubMed] [Google Scholar]
- Lee MY, Lee YH, Lim KM, Chung SM, Bae ON, Kim H, Lee CR, Park JD, Chung JH. Inorganic arsenite potentiates vasoconstriction through calcium sensitization in vascular smooth muscle. Environ Health Perspect. 2005;113(10):1330–1335. doi: 10.1289/ehp.8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PC, Ho IC, Lee TC. Oxidative stress mediates sodium arsenite-induced expression of heme oxygenase-1, monocyte chemoattractant protein-1, and interleukin-6 in vascular smooth muscle cells. Toxicol Sci. 2005;85(1):541–550. doi: 10.1093/toxsci/kfi101. [DOI] [PubMed] [Google Scholar]
- Lerda D. Sister-chromatid exchange (SCE) among individuals chronically exposed to arsenic in drinking water. Mutat Res. 1994;312(2):111–120. doi: 10.1016/0165-1161(94)90015-9. [DOI] [PubMed] [Google Scholar]
- Li D, Morimoto K, Takeshita T, Lu Y. Arsenic induces DNA damage via reactive oxygen species in human cells. Environ Health Prev Med. 2001;6(1):27–32. doi: 10.1007/BF02897306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JX, Shen YQ, Cai BZ, Zhao J, Bai X, Lu YJ, Li XQ. Arsenic trioxide induces the apoptosis in vascular smooth muscle cells via increasing intracellular calcium and ROS formation. Mol Biol Rep. 2010;37(3):1569–1576. doi: 10.1007/s11033-009-9561-z. [DOI] [PubMed] [Google Scholar]
- Lim KM, Shin YS, Kang S, Noh JY, Kim K, Chung SM, Yun YP, Chung JH. Potentiation of vasoconstriction and pressor response by low concentration of monomethylarsonous acid (MMA(III)) Toxicol Lett. 2011;205(3):250–256. doi: 10.1016/j.toxlet.2011.06.008. [DOI] [PubMed] [Google Scholar]
- Luan TZ, Fu SB, Zhou LJ, Li WM, Huang YL. [Effects of arsenic trioxide on human coronary smooth muscle cells: experiment in vitro] Zhonghua Yi Xue Za Zhi. 2009;89(2):133–137. [PubMed] [Google Scholar]
- Lynn S, Gurr JR, Lai HT, Jan KY. NADH oxidase activation is involved in arsenite-induced oxidative DNA damage in human vascular smooth muscle cells. Circ Res. 2000;86(5):514–519. doi: 10.1161/01.res.86.5.514. [DOI] [PubMed] [Google Scholar]
- Martin-Pardillos A, Sosa C, Sorribas V. Arsenic increases Pi-mediated vascular calcification and induces premature senescence in vascular smooth muscle cells. Toxicol Sci. 2013;131(2):641–653. doi: 10.1093/toxsci/kfs313. [DOI] [PubMed] [Google Scholar]
- Maull EA, Ahsan H, Edwards J, Longnecker MP, Navas-Acien A, Pi J, Silbergeld EK, Styblo M, Tseng CH, Thayer KA, Loomis D. Evaluation of the association between arsenic and diabetes: a National Toxicology Program workshop review. Environ Health Perspect. 2012;120(12):1658–1670. doi: 10.1289/ehp.1104579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar IT, Heany H, Heinekey DM, C FW. Methyliiodoarsine. Inorg Synth. 1960;6:113–115. [Google Scholar]
- Mo J, Xia Y, Wade TJ, DeMarini DM, Davidson M, Mumford J. Altered gene expression by low-dose arsenic exposure in humans and cultured cardiomyocytes: assessment by real-time PCR arrays. Int J Environ Res Public Health. 2011;8(6):2090–2108. doi: 10.3390/ijerph8062090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed Abdul KS, Jayasinghe SS, Chandana EP, Jayasumana C, De Silva PM. Arsenic and human health effects: A review. Environ Toxicol Pharmacol. 2015;40(3):828–846. doi: 10.1016/j.etap.2015.09.016. [DOI] [PubMed] [Google Scholar]
- Naranmandura H, Xu S, Sawata T, Hao WH, Liu H, Bu N, Ogra Y, Lou YJ, Suzuki N. Mitochondria are the main target organelle for trivalent monomethylarsonous acid (MMA(III))-induced cytotoxicity. Chem Res Toxicol. 2011;24(7):1094–1103. doi: 10.1021/tx200156k. [DOI] [PubMed] [Google Scholar]
- Padmaja Divya S, Pratheeshkumar P, Son YO, Vinod Roy R, Andrew Hitron J, Kim D, Dai J, Wang L, Asha P, Huang B, Xu M, Luo J, Zhang Z. Arsenic Induces Insulin Resistance in Mouse Adipocytes and Myotubes Via Oxidative Stress-Regulated Mitochondrial Sirt3-FOXO3a Signaling Pathway. Toxicol Sci. 2015;146(2):290–300. doi: 10.1093/toxsci/kfv089. [DOI] [PubMed] [Google Scholar]
- Pan X, Jiang L, Zhong L, Geng C, Jia L, Liu S, Guan H, Yang G, Yao X, Piao F, Sun X. Arsenic induces apoptosis by the lysosomal-mitochondrial pathway in INS-1 cells. Environ Toxicol. 2014 doi: 10.1002/tox.22027. [DOI] [PubMed] [Google Scholar]
- Partridge MA, Huang SX, Hernandez-Rosa E, Davidson MM, Hei TK. Arsenic induced mitochondrial DNA damage and altered mitochondrial oxidative function: implications for genotoxic mechanisms in mammalian cells. Cancer Res. 2007;67(11):5239–5247. doi: 10.1158/0008-5472.CAN-07-0074. [DOI] [PubMed] [Google Scholar]
- Paul JR. Functional compartmentalization of oxidative and glycolytic metabolism in vascular smooth muscle. Am J Physiol. 1983;244(5):C399–409. doi: 10.1152/ajpcell.1983.244.5.C399. [DOI] [PubMed] [Google Scholar]
- Pozo-Molina G, Ponciano-Gómez A, Rivera-González GC, Hernández-Zavala A, Garrido E. Arsenic-induced S phase cell cycle lengthening is associated with ROS generation, p53 signaling and CDC25A expression. Chem Biol Interact. 2015;238:170–179. doi: 10.1016/j.cbi.2015.06.040. [DOI] [PubMed] [Google Scholar]
- Rahman M, Tondel M, Ahmad SA, Chowdhury IA, Faruquee MH, Axelson O. Hypertension and arsenic exposure in Bangladesh. Hypertension. 1999;33(1):74–78. doi: 10.1161/01.hyp.33.1.74. [DOI] [PubMed] [Google Scholar]
- Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, Capaldi RA. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004;64(3):985–993. doi: 10.1158/0008-5472.can-03-1101. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Kojima C, Kobayashi Y, Hirano S, Sakurai MH, Waalkes MP, Himeno S. Toxicity of a trivalent organic arsenic compound, dimethylarsinous glutathione in a rat liver cell line (TRL 1215) Br J Pharmacol. 2006;149(7):888–897. doi: 10.1038/sj.bjp.0706899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz C, Wieder T, Stärck L, Essmann F, Schulze-Osthoff K, Dörken B, Daniel PT. Arsenic trioxide triggers a regulated form of caspase-independent necrotic cell death via the mitochondrial death pathway. Oncogene. 2005;24(11):1904–1913. doi: 10.1038/sj.onc.1208233. [DOI] [PubMed] [Google Scholar]
- Shi H, Shi X, Liu KJ. Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol Cell Biochem. 2004;255(1–2):67–78. doi: 10.1023/b:mcbi.0000007262.26044.e8. [DOI] [PubMed] [Google Scholar]
- Shi Y, Wei Y, Qu S, Wang Y, Li Y, Li R. Arsenic induces apoptosis of human umbilical vein endothelial cells through mitochondrial pathways. Cardiovasc Toxicol. 2010;10(3):153–160. doi: 10.1007/s12012-010-9073-z. [DOI] [PubMed] [Google Scholar]
- Singh AP, Goel RK, Kaur T. Mechanisms pertaining to arsenic toxicity. Toxicol Int. 2011;18(2):87–93. doi: 10.4103/0971-6580.84258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucy NV, Klei LR, Mayka DD, Barchowsky A. Signaling pathways for arsenic-stimulated vascular endothelial growth factor-a expression in primary vascular smooth muscle cells. Chem Res Toxicol. 2004;17(4):555–563. doi: 10.1021/tx034193q. [DOI] [PubMed] [Google Scholar]
- States JC, Srivastava S, Chen Y, Barchowsky A. Arsenic and cardiovascular disease. Toxicol Sci. 2009;107(2):312–323. doi: 10.1093/toxsci/kfn236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straif K, Benbrahim-Tallaa L, Baan R, Grosse Y, Secretan B, El Ghissassi F, Bouvard V, Guha N, Freeman C, Galichet L, Cogliano V, W. H. O. I. A. f. R. o. C. M. W. Group A review of human carcinogens–Part C: metals, arsenic, dusts, and fibres. Lancet Oncol. 2009;10(5):453–454. doi: 10.1016/s1470-2045(09)70134-2. [DOI] [PubMed] [Google Scholar]
- Styblo M, Del Razo LM, Vega L, Germolec DR, LeCluyse EL, Hamilton GA, Reed W, Wang C, Cullen WR, Thomas DJ. Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Arch Toxicol. 2000;74(6):289–299. doi: 10.1007/s002040000134. [DOI] [PubMed] [Google Scholar]
- Su J, Li J, Li W, Altura B, Altura B. Cocaine induces apoptosis in primary cultured rat aortic vascular smooth muscle cells: possible relationship to aortic dissection, atherosclerosis, and hypertension. Int J Toxicol. 2004;23(4):233–237. doi: 10.1080/10915810490471361. [DOI] [PubMed] [Google Scholar]
- Suchorolski MT, Paulson TG, Sanchez CA, Hockenbery D, Reid BJ. Warburg and Crabtree effects in premalignant Barrett’s esophagus cell lines with active mitochondria. PLoS One. 2013;8(2):e56884. doi: 10.1371/journal.pone.0056884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suriyo T, Watcharasit P, Thiantanawat A, Satayavivad J. Arsenite promotes apoptosis and dysfunction in microvascular endothelial cells via an alteration of intracellular calcium homeostasis. Toxicol In Vitro. 2012;26(3):386–395. doi: 10.1016/j.tiv.2011.12.017. [DOI] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. Embo J. 2008;27(2):433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CH, Jeng JS, Yip PK, Chen CL, Hsu LI, Hsueh YM, Chiou HY, Wu MM, Chen CJ. Biological gradient between long-term arsenic exposure and carotid atherosclerosis. Circulation. 2002;105(15):1804–1809. doi: 10.1161/01.cir.0000015862.64816.b2. [DOI] [PubMed] [Google Scholar]
- Wang QQ, Zhou XY, Zhang YF, Bu N, Zhou J, Cao FL, Naranmandura H. Methylated arsenic metabolites bind to PML protein but do not induce cellular differentiation and PML-RARα protein degradation. Oncotarget. 2015;6(28):25646–25659. doi: 10.18632/oncotarget.4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner ML, Moore LE, Smith MT, Kalman DA, Fanning E, Smith AH. Increased micronuclei in exfoliated bladder cells of individuals who chronically ingest arsenic-contaminated water in Nevada. Cancer Epidemiol Biomarkers Prev. 1994;3(7):583–590. [PubMed] [Google Scholar]
- Watanabe M, Funakoshi T, Unuma K, Aki T, Uemura K. Activation of the ubiquitin-proteasome system against arsenic trioxide cardiotoxicity involves ubiquitin ligase Parkin for mitochondrial homeostasis. Toxicology. 2014;322:43–50. doi: 10.1016/j.tox.2014.04.008. [DOI] [PubMed] [Google Scholar]
- Youle JR, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12(1):9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamaraeva MV, Sabirov RZ, Maeno E, Ando-Akatsuka Y, Bessonova SV, Okada Y. Cells die with increased cytosolic ATP during apoptosis: a bioluminescence study with intracellular luciferase. Cell Death Differ. 2005;12(11):1390–1397. doi: 10.1038/sj.cdd.4401661. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.