

SUMMARY
Despite its eponymous association with the heat shock response, yeast heat shock factor 1 (Hsf1) is essential even at low temperatures. Here we show that engineered nuclear export of Hsf1 results in cytotoxicity associated with massive protein aggregation. Genome-wide analysis revealed that Hsf1 nuclear export immediately decreased basal transcription and mRNA expression of 18 genes, which predominately encode chaperones. Strikingly, rescuing basal expression of Hsp70 and Hsp90 chaperones enabled robust cell growth in the complete absence of Hsf1. With the exception of chaperone gene induction, the vast majority of the heat shock response was Hsf1-independent. By comparative analysis of mammalian cell lines, we found that only heat shock-induced but not basal expression of chaperones is dependent on the mammalian Hsf1 homolog (HSF1). Our work reveals that yeast chaperone gene expression is an essential housekeeping mechanism and provides a roadmap for defining the function of HSF1 as a driver of oncogenesis.
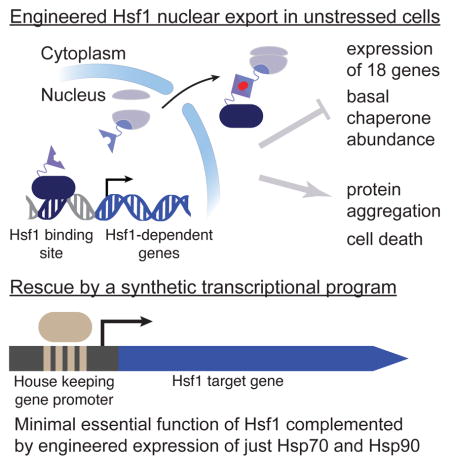
Graphical Abstarct
INTRODUCTION
Cells maintain protein homeostasis (proteostasis) in the face of proteotoxic stress, such as heat shock, by inducing expression of genes encoding factors for protein folding and degradation (Hartl et al., 2011; Richter et al., 2010). Failure to maintain proteostasis by regulating gene expression has been linked to aging and neurodegeneration (Balch et al., 2008), while many cancers are associated with elevated expression of proteostasis factors (Tang et al., 2015). Yeast heat shock factor 1 (Hsf1) was the first eukaryotic proteostasis transcription factor to be discovered (Sorger and Pelham, 1987), which enabled identification of homologous transcription factors across the eukaryotic lineage (Clos et al., 1990; Jakobsen and Pelham, 1991; Rabindran et al., 1991; Scharf et al., 1990). Hsf1 is constitutively nuclear and essential for yeast growth under all conditions (Jakobsen and Pelham, 1988; Sorger and Pelham, 1988). By contrast, mammalian HSF1 is normally dispensable for cell growth absent stress and resides in a repressed state under physiological conditions (Sarge et al., 1993). Following heat shock, however, HSF1 enables thermotolerance by inducing gene expression of proteostasis factors (McMillan et al., 1998; Zhang et al., 2002). Despite these species differences, the core function of mammalian HSF1 is conserved such that a constitutively active version can complement the yeast deletion (Liu et al., 1997). Moreover, as in yeast, HSF1 is constitutively active and essential for proliferation of many cancer cell types (Dai et al., 2007).
Efforts to systematically define genes whose stress-induced expression is dependent on either yeast Hsf1 or mammalian HSF1 have been challenging (Akerfelt et al., 2010). Part of the difficulty comes from gene co-regulation by other transcription factors (Zhang et al., 2002). For example, many heat-activated yeast genes, including several that encode heat shock proteins (HSPs), can be induced by the general stress response factors Msn2 and Msn4 (Boy-Marcotte et al., 1999; Gasch et al., 2000; Treger et al., 1998). There is also the additional challenge in yeast of defining Hsf1’s basal transcriptional program, which is presumed to be essential for cell viability because mutations that disrupt either Hsf1 DNA binding or transcriptional activation are lethal (Jakobsen and Pelham, 1991; Torres and Bonner, 1995). Viable partial loss-of-function mutants of Hsf1 that disrupt heat shock-induced gene expression (Eastmond and Nelson, 2006; Morano et al., 1999; Zarzov et al., 1997) cannot resolve whether the basal and heat-induced transcriptional programs are qualitatively distinct or if heat primarily tunes the magnitude of Hsf1’s basal transcriptional program. Chromatin immunoprecipitation combined with microarray (ChIP-chip) studies have favored the former possibility by showing that Hsf1 associates with additional promoters under heat stress (Hahn et al., 2004; Lee et al., 2002). By intersecting these data with heat-induced changes in mRNA abundance detected using DNA microarrays, a list of >160 Hsf1-dependent genes has emerged that is partly devoted to proteostasis and partly to disparate cellular functions such as energy generation, carbon metabolism and vesicle transport (Eastmond and Nelson, 2006; Hahn et al., 2004).
Hsf1’s essential function in the absence of stress is presumably to drive basal gene expression of essential proteostasis factors. However, closer inspection of the Hsf1 target genes challenges this simple conclusion in several ways. First, among Hsf1 gene targets not involved in protein folding, some are individually essential, while pairwise deletions of many non-essential ones result in synthetic lethality (Hahn et al., 2004). Second, it is not known to what extent other basal transcription factors would maintain expression of Hsf1 gene targets involved in protein folding were Hsf1 to be acutely inhibited. Finally, deletion of many non-essential Hsf1 targets results in elevated basal Hsf1 activity (Brandman et al., 2012), suggesting that overexpression of some targets can compensate for the loss of others. One unbiased strategy for defining essential Hsf1 targets would be to systematically place all targets under the control of Hsf1-independent promoters and find which among them are minimally required for life without Hsf1. This synthetic biology approach is conceptually simple but the lengthiness of the target list renders the prospect of its execution fanciful.
Our starting point was to develop a chemical genetics tool for inducing rapid Hsf1 nuclear export. This enabled us to measure immediate changes in genome-wide basal transcription. From this analysis emerged a list of 18 genes—all but one of which encodes a chaperone—that are strongly dependent on Hsf1 for their basal expression. In addition, we find that Hsf1’s repertoire of gene targets is not significantly expanded by heat shock; instead, Hsf1 drives heat-induced chaperone overexpression. With a greatly reduced list of Hsf1-dependent genes, we systematically placed all targets under the control of Hsf1-independent promoters to find which among them are minimally required for life without Hsf1. This analysis defined Hsp70 and Hsp90 as the two critical chaperones. Cells engineered to live without Hsf1 were thermosensitive, arguing that survival under severe proteotoxic stress necessitates chaperone overexpression by Hsf1. Lastly, we used CRISPR/Cas9 to create two distinct hsf1−/− mouse cell lines, which enabled us to define HSF1’s core transcriptional program: a set of 9 genes—8 of which encode chaperones—that are functionally akin to Hsf1-dependent genes in yeast. However, in mammalian cells basal chaperone expression is independent of HSF1; rather, HSF1 induced chaperone overexpression following heat shock.
RESULTS
A chemical genetics approach enables acute Hsf1 nuclear depletion at the physiological temperature
To develop a tool that acutely inactivates Hsf1 in the absence of stress we used the “Anchor Away” (AA) approach (Haruki et al., 2008). Briefly, we created a yeast strain (Hsf1-AA) that is resistant to Tor1 inhibition by rapamycin and co-expresses an Hsf1-FRB (FKBP rapamycin-binding domain) fusion and a ribosomal protein L13a-FKBP12 fusion (Rpl13A-FKBP12). In this strain, rapamycin should induce FRB-FKBP12 heterodimerization, thus tethering Hsf1 to nascent ribosome subunits prior to their nuclear export (Figure 1A). Real-time imaging of cells by fluorescence microscopy revealed that rapamycin treatment induced a rapid change in in Hsf1-FRB-GFP localization within ~10 minutes (Figure S1A). Co-expression of a nuclear marker (NLS-mKate2) with Hsf1-FRB-GFP showed that Hsf1-FRB-GFP co-localized with NLS-mKate2 in the nucleus of untreated cells, but after rapamycin treatment Hsf1-FRB-GFP became cytosolic while NLS-mKate2 remained nuclear (Figure S1B, C). Chromatin immunoprecipitation analysis of Hsf1-AA cells demonstrated that rapamycin treatment induced rapid loss of Hsf1 promoter binding for two major chaperone genes (HSP82 and SSA1) (Figure S1D). To functionally test if Hsf1 was removed from chromatin, we employed a transcription factor competition assay (Jakobsen and Pelham, 1988) in which GFP is driven by a synthetic promoter with overlapping binding sites for both Hsf1 and a chimeric, β-estradiol-responsive transcription factor (Figure S1E, F) (McIsaac et al., 2011). Rapamycin treatment facilitated GFP reporter induction as a function of β-estradiol concentration, suggesting that chromatin-associated Hsf1 was removed as an obstacle to promoter access (Figure S1G). Consistent with this interpretation, rapamycin pretreatment was sufficient to render a distinct GFP reporter of Hsf1-dependent transcription (Brandman et al., 2012) unresponsive to heat shock (Figure S1H). Together, these results demonstrate that Hsf1-AA is a useful tool for acute chromatin and nuclear depletion of Hsf1.
Figure 1. Acute inactivation of Hsf1 induces proteotoxicity even in the absence of stress.

(A) Schematic of Hsf1 Anchor Away (Hsf1-AA). (B) Hsf1-AA cells with indicated plasmids were spotted at two cell densities onto rapamycin or mock plates. Shown are images of plates incubated for 3 days at 30°C. (C) Representative confocal micrographs of Hsf1-AA cells expressing endogenous Hsp104-GFP and plasmid-borne mCherry-Ubc9wt or -ubc9ts taken after logarithmic growth at 30°C, after treatment with heat shock (20′ at 37°C), and after treatment with rapamycin (360′ at 30°C). (D) Hsf1-AA cells described in part (C) were treated with rapamycin for indicated times at 30°C and imaged by epifluorescence microscopy. Blinded images containing at least 100 cells were scored for cells containing mCherry foci and the fraction of scored cells plotted. See also Figure S1.
Hsf1 prevents protein aggregation at the physiological temperature
To measure the phenotypic consequences of Hsf1 nuclear export, we incubated Hsf1-AA cells on plates containing rapamycin and observed no colony formation (Figure 1B). This phenotype recapitulates the lethal phenotype of hsf1Δ cells and was dependent on Hsf1-Rpl13a heterodimerization (Figure S1I), while a second copy of Hsf1 not fused to FRB was able to rescue growth (Figures 1B and S1I). Even though rapamycin treatment of Hsf1-AA cells induced Hsf1 export in a matter of minutes, it took ~2.5 hours before cells became arrested at various stages of the cell cycle (measured by budding index). After extended treatment (>13 hours) some cells lysed asynchronously (Figure S1J), and removal of rapamycin failed to restore cell growth, arguing for irreversible cytotoxicity. Since basal expression of several chaperone genes is dependent on Hsf1 binding sites in their promoters (Erkine et al., 1996; Gross et al., 1993; Nicholls et al., 2009; Sakurai and Ota, 2011), we reasoned that rapid Hsf1 nuclear export is followed by a slower decrease in chaperone concentration leading to protein aggregation and cytotoxicity. To test this, we measured Hsp70 (Ssa1) and Hsp90 (Hsc82) protein levels following rapamycin treatment by quantitative Western blotting. Indeed, two hours after Hsf1 inactivation at physiological temperature Ssa1 and Hsc82 proteins dropped below 30% and 50% of their initial levels, respectively (Figure S1K). Next, we monitored protein aggregation using mCherry fused to a metastable, temperature-sensitive (ts) allele of Ubc9, an established reporter of protein misfolding in yeast that forms cytosolic aggregates visible as fluorescent puncta by microscopy (Kaganovich et al., 2008). In a control experiment, we confirmed that heat shock at 37°C induced mCherry-ubc9ts—but not stable mCherry-Ubc9wt—to form puncta that co-localized with Hsp104-GFP, a protein disaggregase that localizes to protein aggregates (Glover and Lindquist, 1998) (Figure 1C). Consistent with the timing of the reduction in Hsp70 and Hsp90 levels, we observed mCherry-ubc9ts aggregation at the physiological temperature in the majority of cells within 2.5 hours of rapamycin addition (Figure 1D). Importantly, mCherry-Ubc9wt remained apparently soluble even after extended rapamycin treatment (Figure 1C, D) despite the appearance of Hsp104-GFP puncta in the same cells, which likely mark aggregates of endogenous metastable proteins with similar folding requirements to ubc9ts (Figure 1C). Thus, depletion of Hsf1 leads first to decreased expression of chaperones followed by proteostasis collapse and cell death.
Yeast Hsf1 drives basal expression of 18 genes
To define the immediate transcriptional effects of Hsf1 nuclear export, we used native elongating transcript sequencing (NET-seq) (Churchman and Weissman, 2011) to globally track transcription of individual genes in Hsf1-AA cells during a rapamycin treatment time course (15, 30, and 60 minutes) (Table S1). Statistical analysis (see Experimental Procedures) defined 25 genes that were transcriptionally repressed and 5 that were induced by 15 minutes of drug treatment (p-value < 10−4) (Figure S2A, left panel), and these changes persisted in the later time points (Figure S2A, middle and right panels). To substantiate that the transcriptional changes identified by the NET-seq analysis resulted in bona fide changes in mRNA abundance, we analyzed Hsf1-AA cells treated for 60 minutes with rapamycin by RNA-seq (Table S2). We observed statistically significant changes in mRNA abundance for 18/25 transcriptionally repressed genes and none of the induced genes (Figures 2A and S2B, C; see Experimental Procedures). Importantly, the genes defined by our combined NET-seq/RNA-seq analysis had a strong correlation between the fold-decrease in their transcription and their mRNA abundance (Figure S2D).
Figure 2. Hsf1 drives basal expression of a diverse set of protein folding factors.

(A) Hsf1-AA cells were grown logarithmically at 30°C and harvested for analysis by NET-seq or RNA-seq immediately prior to and after 15′, 30′ and 60′ of rapamycin treatment. Shown is a gene scatter plot of transcription versus mRNA changes induced by treatment with rapamycin for 15′ and 60′, respectively. (B) Venn diagram comparing Hsf1 target genes defined by ChIP-, NET- and RNA-seq, with the names of the 18 Hsf1-dependent genes (HDGs) detected by all 3 techniques indicated. (C) Gene scatter plot of change in HDG transcription resulting from 15′ of rapamycin treatment versus Hsf1 occupancy at HDG promoters. (D) Bioinformatic analysis of the 18 HDGs defined by ChIP-, NET- and RNA-seq. Solid bars show the number of HDGs with the given annotation (GO term or promoter motif) and dashed bars show the remaining number of HDGs. The fill color indicates the significance level for the enrichment of the annotated HDGs versus other genes. See Supplementary Figure 2J for a similar bioinformatic analysis of Hsf1 targets defined by each individual genome-wide approach or by combining any two approaches. See also Figure S2 and Tables S1–3.
Given that we defined ~10-fold fewer Hsf1 targets by NET-seq analysis than anticipated (Eastmond and Nelson, 2006; Hahn et al., 2004; Lee et al., 2002), we considered the possibility that technical or biological variability limited our statistical power to detect additional biologically significant transcriptional changes. However, there was no significant difference in the width of confidence intervals—which inversely relate to statistical power—between genes defined by our analysis and other genes (Figure S2E). Another possible source of error was to miss slower transcriptional effects of Hsf1 nuclear export. However, we defined only 3 significant changes in transcription after 30 or 60 minutes of rapamycin treatment (Table S1), and none of these were corroborated by mRNA analysis (Table S2). Rather, transcription of most genes was consistent and largely unperturbed by Hsf1 inactivation, both globally—R2 = 0.98 for all adjacent time points (Figure S2A)—and at the level of individual loci (Figure S2F) across a wide range of expression levels (Figure S2G). Thus, detection of additional Hsf1 targets was not limited by the sensitivity or reproducibility of our analysis.
To independently validate these targets, we monitored Hsf1 DNA binding by chromatin immunoprecipitation followed by deep sequencing (ChIP-seq). We isolated chromatin using tandem affinity purification of dual epitope-tagged Hsf1-FLAG-V5 expressed as the only copy of Hsf1 in an otherwise wild type genetic background. Analysis using a stringent peak-calling algorithm (see Experimental Procedures) and bioinformatics identified 40 gene targets (Table S3) with a strong promoter enrichment for Hsf1-binding sites (p-value < 10−9). The ChIP-seq analysis identified none of the 12 NET-seq-only gene targets while it identified all 18 gene targets corroborated by NET-seq and RNA-seq (p-value < 10−42) (Figure 2B and S2H). Importantly, the genes defined by combined NET-seq/ChIP-seq analysis showed a strong correlation between fold decrease in transcription and ChIP enrichment (Figure 2C and S2I), arguing that Hsf1 binding to their promoters drives their basal expression.
In summary, we combined three genome-wide approaches (NET-seq, RNA-seq, and ChIP-seq) to define 18 Hsf1-dependent genes (HDGs) (Figure 2B), which collectively encode two Hsp70 paralogs (SSA1 and SSA2), both Hsp90 paralogs (HSC82 and HSP82), nucleotide exchange factors and co-chaperones for Hsp70 and Hsp90 (YDJ1, SIS1, FES1, AHA1, HCH1, STI1, CPR6), nuclear and cytoplasmic aggregases (BTN2, CUR1, and HSP42), mitochondrial protein folding factors (HSP78, MDJ1), a disaggregase (HSP104), and a cell cycle transcriptional regulator (MBF1). Bioinformatic analysis of HDG promoters revealed a strong enrichment for consensus Hsf1 binding sites (Sorger and Pelham, 1987) (p-value < 10−10), and gene ontology analysis revealed a strong enrichment for protein folding function (p-value < 10−21) (Figure 2D and S2J). We note that our analysis excludes genes that are regulated by Hsf1 under non-basal conditions, genes that Hsf1 controls redundantly with other transcription factors and genes that utilize Hsf1 as a pioneer factor (Fujimoto et al., 2012). We conclude that Hsf1 drives a compact transcriptional program in basal conditions.
The majority of the yeast heat shock response is Hsf1-independent
We considered the possibility that Hsf1-AA cells attempt to counteract proteotoxicity induced by prolonged rapamycin treatment by secondary changes in gene expression. Indeed, RNA-seq revealed that prolonged rapamycin treatment, which we defined as a comparison between 4 hours and 1 hour of treatment, caused a >4-fold induction of ~200 genes (Figure S3A), including 4 HDGs (HSP42, HSP72, HSP104 and HSP82). Bioinformatic analysis suggested these changes were associated with alternative metabolism and various stress responses, including heat shock (Figure S3B). To identify the regulators of this response, we analyzed the promoters of induced genes to identify enriched sequence motifs (Carlson et al., 2007), which were cross-referenced with the known sequence specificities of yeast transcription factors (de Boer and Hughes, 2011). This analysis defined a highly enriched (p-value < 10−34), ubiquitous motif that was found in 78% of induced genes, including the 4 strongly up-regulated HDGs (Figure S3C). This motif corresponded to the consensus binding site for Msn2 and Msn4 (jointly Msn2/4), two redundant transcription factors activated by a variety of environmental stresses, including heat shock (Causton et al., 2001; Schmitt and McEntee, 1996). Msn2/4 activity is regulated by Protein Kinase A (PKA), which phosphorylates Msn2/4 under non-stress conditions preventing their nuclear entry (Görner et al., 1998). To test if Msn2/4 activation mimics gene activation induced by prolonged rapamycin treatment, we treated cells expressing analog-sensitive PKA (PKAas) with the ATP-analog 1-NM-PP1, an established chemical genetics approach for inducing Msn2/4 nuclear localization (Hao and O’Shea, 2012). RNA-seq analysis revealed that 1-NM-PP1 treatment of PKAas cells was well correlated with prolonged rapamycin treatment (R2 = 0.70, Figure S3D). Comparative analysis of PKAas msn2Δ msn4Δ cells established that Msn2/4 targets (p-value < 10−103) were significantly attenuated (Figure S3E and F). These data show that PKA inhibition mimics gene expression changes induced by prolonged rapamycin treatment.
Despite its eponymous association with the heat shock response, we observed a remarkable resemblance between the heat shock response and the response to prolonged Hsf1 inactivation (Figure 3A). The similarity between these two responses suggested that gene induction by heat was largely Hsf1-independent and Msn2/4-dependent. To test this, we used NET-seq and RNA-seq to measure the effect of a short rapamycin pre-treatment on the heat shock response of Hsf1-AA cells. Consistent with our previous observations, HDGs with multiple Msn2/4 binding sites were still induced by heat shock while the remaining HDGs were repressed (Figure 3B). Genome wide, the heat shock response remained generally intact in the absence of nuclear Hsf1 (R2 = 0.81), including activation of Msn2/4 targets (Figure 3C). This was supported by Hsf1 ChIP-seq analysis during heat shock, which revealed a list of target genes that significantly overlapped the list of basal targets (p-value < 10−59) (Figure S3G). Unfortunately, our efforts to exploit Hsf1-AA to rigorously define the proximal transcriptional effects of Hsf1 inactivation on the heat shock response were hampered by their convolution with other indirect, secondary changes. When we bioinformatically analyzed gene expression that was significantly reduced by rapamycin pretreatment (Figure S3H), we could not find compelling evidence that Hsf1 directly regulated all but 8 of these these genes (Figure S3I,J and Supplemental Experimental Procedures). Rather, we found that well-established Hsf1-independent components of the heat shock response were exaggerated in its absence (Figure S3H–J), which we speculate is due to enhanced heat-induced proteotoxicity absent Hsf1-dependent chaperones (see Supplementary Experimental Procedures).
Figure 3. Induction of most genes by heat shock is Hsf1-independent and Msn2/4-dependent.

(A) Hsf1-AA cells were grown at 30°C, heat shocked (39°C for 30′) or treated with rapamycin for either 60′ or 240′ at 30°C prior to harvesting for analysis by RNA-seq. Shown is a gene scatter plot of mRNA changes induced by prolonged rapamycin treatment (240′ vs. 60′) (y-axis) versus changes induced by heat shock (x-axis). Msn2/4 targets were defined as genes with at least one Msn2/4 promoter binding site (AGGGG) that were in the top 10% of genes induced by PKA inhibition (see Figure 3D). (B) Left: Locations of predicted bindings sites for Hsf1 (TTCnnGAA and TTC-n7-TTC-n7-TTC) and Msn2/4 (AGGGG) in HDG promoters. Right: Hsf1-AA cells were grown logarithmically at 30°C (control) or treated with rapamycin (30°C for 30′) followed by heat shock (39°C for 30′) prior to harvesting for RNA-seq analysis. Shown are HDG mRNA changes induced by sequential treatment relative to control. (C) Hsf1-AA cells were grown at 30°C (control) or treated with either rapamycin (30′ at 30°C) and then heat shock (39°C for 30′) or carrier-only and then heat shock prior to harvesting for RNA-seq analysis. Shown is a gene scatter plot of mRNA changes induced by the two treatments relative to the control. (D) Hsf1-AA PKAas cells were grown at 30°C (control) or treated with heat shock (39°C for 30′) or the PKA inhibitor 1-NM-PP1 (30°C for 30′) prior to harvesting for RNA-seq analysis. Shown is a gene scatter plot comparing mRNA changes induced by these two treatments relative to control. (E) Hsf1-AA PKAas cells were grown at 30°C or after treatment with either rapamycin (30′ at 30°C) and then 1-NM-PP1 (30′ at 30°C) or carrier-only and then 1-NM-PP1 prior to harvesting for RNA-seq analysis. Shown is a gene scatter plot comparing mRNA changes induced by these two treatments relative to control. (F) Left: HDG promoter locations of Hsf1 and Msn2/4 binding sites as in part (B). Right: Hsf1-AA PKAas cells were grown at 30°C (control) or after treatment with rapamycin (30′ at 30°C) and then 1-NM-PP1 (30′ at 30°C) prior to harvesting for RNA-seq analysis. Shown are HDG mRNA changes induced by sequential treatment relative to control. See also Figure S3.
As an alternative to heat stress, we took advantage of the known observation that heat shock and PKA inhibition have similar effects on cell growth, protein synthesis and the activity of multiple transcription factors, including Msn2/4 (Causton et al., 2001; Smith et al., 1998; Thevelein and de Winde, 1999). Indeed, 1-NM-PP1 treatment of PKAas Hsf1-AA cells induced a change in the transcriptome that resembled the heat-shock response (R2 = 0.76) (Figure 3D), as well as prolonged rapamycin treatment (Figure S3D). Yet, unlike these responses, PKA inhibition did not trigger mCherry-ubc9ts aggregation (Figure S3K), affording us the opportunity to probe Hsf1’s role in co-activating the bulk of the heat shock response. Strikingly, rapamycin pre-treatment of PKAas Hsf1-AA cells had a minor effect on genome-wide expression changes induced by 1-NM-PP1 treatment (R2 = 0.95) (Figure 3E), including the induction of Msn2/4 targets (Figure S3L). Rapamycin only led to the reduced expression of HDGs without multiple Msn2/4 binding sites in their promoters (Figure 3F). These data support the notion that Msn2/4 drive expression of the majority of heat shock-induced genes independent of Hsf1.
Mammalian HSF1 drives a core transcriptional program of 9 genes during heat shock
Mammalian cells have multiple heat shock factors (e.g., HSF1, 2, 4, 5, X and Y in the human genome) homologous to yeast Hsf1 (Akerfelt et al., 2010). None are normally essential for viability in the absence of stress, but loss of HSF1 uniquely renders cells heat sensitive and unable to acquire thermotolerance (McMillan et al., 1998; Zhang et al., 2002). Comparison of heat-induced changes in mRNA abundance measured by DNA microarray analysis in wild-type mouse embryonic fibroblasts (MEFs) versus those derived from a hsf1−/− mouse has led to a long list of HSF1-dependent genes (Trinklein et al., 2004). To define what fraction of these changes represent the core HSF1 transcriptional program that is shared across many cell types and differentiation states, we used CRISPR/Cas9-mediated genome editing to generate hsf1−/− mouse embryonic stem cells (mESCs) and hsf1−/− MEFs (Figure S4A, B). Next, we measured mRNA abundance by RNA-seq in hsf1−/− mESCs and MEFs in reference to their wild type counterparts both in unstressed cells and following heat shock (Table S4). In unstressed cells, the transcriptomes of the wild type and hsf1−/− cells were remarkably similar (Figure S4C). Only 2 genes were significantly upregulated and 2 genes were significantly downregulated in both cell types (both p-values = 0.33) (see Experimental Procedures), approximately the number of significant observations you would expect by random chance. There was no functional enrichment among these genes, and none encode chaperones. We conclude that HSF1 has little or no effect on basal transcription that is common to both mESCs and MEFs.
To define HSF1’s core transcriptional program during heat shock, we first asked which genes are heat-induced in both wild type mESCs and MEFs and found 20 genes (p-value < 10−18), many of which encode chaperones (Figure 4A). Next we asked which genes showed reduced expression in hsf1−/− cells compared to wild type cells following heat shock in both mESCs and MEFs and found 15 genes (p-value < 10−11), again including many chaperone genes (Figure 4B). By intersecting these two gene lists, we found only 9 genes that were induced by heat shock in both wild type mESCs and MEFs and repressed in both heat shocked hsf1−/− cell types (Figure 4C). We define these 9 genes as the core mammalian HSF1-dependent genes (HDGs) and note that 8/9 form a densely linked interaction network with the cytosolic Hsp40, Hsp70 and Hsp90 chaperones at its center (Figure 4D). The structure of this network is remarkably similar to the one formed by yeast HDGs (Figure 4E).
Figure 4. Mammalian HSF1 enables heat induction of a chaperone network similar to the yeast HDG network.

(A) Wild-type mESCs and MEFs were cultured at 37°C or treated with heat shock (42°C for 60′) prior to harvesting for RNA-seq analysis. Shown is a gene scatter plot of mRNA abundances in treated versus control samples for each cell type. Dark lines indicate statistical thresholds used to define genes with significant changes in expression (see Experimental Procedures) and genes with significant changes in both cell types are colored. Also indicated are gene names of HSF1-dependent genes (HDGs) defined by additional experiments and analyses (see Figures 4B and 4C and Experimental Procedures). (B) Wild-type (WT) and hsf1−/− mESCs and MEFs were heat shocked (42°C for 60′) and then analyzed by RNA-seq. Shown is a gene scatter plot of mRNA abundances in heat shocked WT versus hsf1−/− cells for each cell type. For definition of dark lines and gene names see part (A). (C) Venn diagram comparing genes in mESC and MEFs that are significantly induced by heat shock in WT cells (purple) with genes whose expression is significantly reduced during heat shock in hsf1−/− vs. WT cells. HDGs are defined as genes in the 4-way intersection and their names indicated. (D) and (E) Mammalian and yeast HDG protein-protein interaction network (see Experimental Procedures). (F) Wild-type, hsf1−/−, hsf2−/−, and hsf1−/− hsf2−/− MEFs were cultured at 37°C prior to harvesting for RNA-seq analysis. Shown are mRNA abundances for HDGs in each cell line. See also Figure S4 and Table S4.
Hsf1 is the sole factor in yeast that controls both basal and heat-induced expression of HDGs. By contrast, mammalian HSF1 is dispensable for high basal expression of HDGs. We wondered if HSF2, the only other HSF paralog with detectable expression in both the mESCs and the MEFs was responsible. Thus, we generated hsf2−/− MEFs and hsf1−/− hsf2−/− double mutant MEFs and measured mRNA abundance by RNA-seq in unstressed cells in comparison wild type and hsf1−/− MEFs. In the absence of HSF2, mRNA levels for 3 HDGs—two Hsp70 homologs (HSPA1A and HSPA1B) and one Hsp90 homolog (HSP90AB1)—were significantly up-regulated (p-value < 10−4) (Figure S4D), but we found that the basal expression of all HDGs in hsf1−/− hsf2−/− MEFs was similar to wild-type (Figure 4F and S4E). Taken together, these data strongly argue that the HSF family does not control basal expression of chaperones in MEFs.
A synthetic transcriptional program bypasses Hsf1’s essential function
In mammalian cells, high HSF1-independent basal expression of chaperone genes may explain why HSF1 is not essential in the absence of stress. This prompted us to ask if the essential function of yeast Hsf1 can be obviated by constitutive expression of HDGs from Hsf1-independent promoters. To this end we constructed four plasmids carrying in total 15 HDG ORFs—we excluded SSA1, HSP82, and HCH1 because they are redundant with their paralogs (SSA2, HSC82 and AHA1, respectively)—under the control of promoters from highly expressed housekeeping genes (Figures 5A and S5A). We termed these “synthetic HDGs” (synHDGs). Consistent with the idea that HDG expression provides negative Hsf1 feedback to maintain proteostasis, we found that Hsf1-AA cells expressing synHDGs had reduced Hsf1 basal activity (Figure S5B). Strikingly, even in the presence of rapamycin these cells continued to robustly proliferate (Figure 5B) and no longer formed visible mCherry-ubc9ts aggregates (Figures 5C and S5C). To test if constitutive expression of chaperones can enable cells to live in the complete absence of Hsf1, we introduced synHDG expression plasmids into hsf1Δ cells carrying an Hsf1 expression plasmid. Following plasmid shuffling, we found that hsf1Δ cells expressing synHDGs grew comparably to cells with a second Hsf1 expression plasmid (Figure S5D). Further, RNA-seq analysis did not reveal any coherent secondary changes to gene expression between these strains (Figure S5E).
Figure 5. A synthetic transcriptional program reveals the essential function of Hsf1.

(A) Schematic of promoter swapping strategy for constitutive expression of Hsf1 targets from strong Hsf1-independent promoters. See Supplementary Figure 4A for details. (B) Hsf1-AA cells with indicated plasmids were spotted at two concentrations onto rapamycin or mock (carrier-only) plates. Shown are images of plates incubated for 5 days at 30°C. (C) Representative confocal micrographs of Hsf1-AA cells expressing plasmid-borne mCherry-ubc9ts and indicated plasmids taken after treatment with rapamycin (360′ at 30°C). (D) Hsf-AA cells with indicated transgenes were spotted at two concentrations onto rapamycin or mock plates, which also contained β-estradiol to drive synHsp expression from β-estradiol-dependent promoters (see Experimental Procedures for details). Shown are images of plates incubated for 5 days at 30°C. See also Figure S5 and Table S5.
The essential function of Hsf1 is to drive high basal co-expression of Hsp70 and Hsp90
To determine if all 15 synHDGs were necessary to bypass the essential function of Hsf1, we transformed Hsf1-AA cells with subsets of the four expression plasmids and found that only two were necessary (Figure S5F): the plasmid containing SSA2 (Hsp70) along with three other synHDGs, and the plasmid containing HSC82 (Hsp90) and two other synHDGs. By deleting individual synHDGs on these two plasmids, we found that that only SSA2 and HSC82 were indispensable for Hsf1 bypass (Figure S5G). Impressively, co-expression of just SSA2 and HSC82—but neither gene expressed alone—enabled Hsf1-AA cells to robustly grow without apparent mCherry-ubc9ts aggregation in the presence of rapamycin (Figures 5D and S5H). These data demonstrate that the minimal essential function of Hsf1 is to drive high basal gene expression of Hsp70 and Hsp90. While it may not be surprising that cells require high levels of Hsp70 and Hsp90 to live, it is remarkable that Hsf1’s required contribution to cell viability can be pared down to the expression of only two chaperone genes.
Hsf1 activity is adjusted to the state of protein folding in the cell (Anckar and Sistonen, 2011; Wu, 1995). By contrast, Hsf1-AA cells expressing synHDGs lack this transcriptional feedback and should be susceptible to proteotoxic stress in the presence of rapamycin. Consistent with this notion, we found that Hsf1-AA cells expressing the synHDGs showed marked growth impairment on rapamycin plates at the elevated temperature of 37°C (Figure S5I). As an attempt to improve growth at 37°C, we introduced additional copies of synSSA2 and synHSC82 but observed only a modest improvement (Figure S5I). These data argue that coordinated homeostatic control of HDG expression by Hsf1 is required for fitness at elevated temperature.
DISCUSSION
Heat shock factors make up one of the most conserved families of sequence-specific transcription factors (Wu, 1995). In all eukaryotes that have been examined they have been implicated in the induction of heat shock protein (HSP) genes as a response to proteotoxic stress. However, mechanistic dissection of this deeply conserved core function has been challenging because members of this family have been deployed differentially through evolution to perform specific functions. In particular, yeast Hsf1 performs an essential function in all conditions, while mammalian HSF1 is dispensable for normal cell growth but becomes essential during oncogenic transformation (Dai et al., 2007). Here we used comparative genomics to understand how yeast Hsf1 and mammalian HSF1 have diverged from each other.
Lack of a suitable tool for acutely inactivating yeast Hsf1 has been a major obstacle to defining its function. Here we used “Anchor Away” (AA) to induce rapid Hsf1 nuclear export following rapamycin addition (Hsf1 AA, for short) and studied the immediate and long-term transcriptional consequences at the physiological temperature. By combining information from three independent genome-wide approaches (NET-seq, RNA-seq, ChIP-seq), we were able to define an unexpectedly small number of HDGs. We found that corroboration of putative target genes by independent genomic approaches yielded a great increase in specificity and reproducibility, without any apparent decrease in sensitivity, even when the number of targets defined by each approach differed significantly (Figure S2J). Our study should serve as a roadmap for revisiting other transcription factor target lists, as well as defining the gene targets of poorly characterized transcription factors.
We also found that prolonged Hsf1 AA triggered a global change in gene expression resembling the heat shock response. Controlled heat shock experiments and a chemical genetics approach that mimics heat as a stimulus revealed the heat shock response remains largely intact upon Hsf1 AA. Thus, the majority of the yeast heat shock response is the job of distinct general stress responsive transcription factors. Retrospectively, our study illustrates how earlier genomic approaches to dissect out Hsf1 gene targets from the complexities of the heat shock response have obscured the simplicity of Hsf1’s role in maintaining proteostasis: Hsf1 tunes expression of a compact regulon according to the protein folding needs of the cell. Our work also provides a complementary perspective to an earlier yeast proteomic analysis of proteins induced by cytosolic expression of a misfolded mutant protein, which post-hoc analysis revealed are strongly enriched for HDGs (12/27 are HDGs; p-value < 10−21) (Geiler-Samerotte et al., 2011). Although Hsf1’s contribution to proteotoxic stress is limited, it is critical, as cells we engineered to bypass Hsf1’s essential function with constitutive basal expression of synHDGs have diminished fitness at elevated temperature. This now provides a starting platform for introducing negative transcriptional feedback control of synHDGs to enable full functional replacement of Hsf1 with a synthetic proteostasis circuit.
Yeast Hsf1 is part of an essential transcriptional feedback loop that operates even at the physiological temperature. By contrast, we found that the mammalian HSF family is dispensable for cell growth in the absence of stress in two independent mouse cell types. The HSF1 core transcriptional program comprised 9 genes and was only revealed following heat shock. A complementary recent study has ascribed a similarly restricted role for HSF1 in the mammalian heat shock response (Mahat et al., 2016). Remarkably, 8/9 genes encoded chaperones that are organized into a strikingly similar functional network as the yeast HDGs (Figure 4 D, E). This provides a satisfying denouement for the previous seminal discovery that a constitutively active mutant of human HSF1 enables yeast cells to live without Hsf1 (Liu et al., 1997). We speculate that HSF1 is dispensable in unstressed mammalian cells because a distinct transcription factor or factors maintain high basal expression of HSPs. While we do not have any experimental evidence for the identity of this factor or factors, preliminary analysis of the promoters of the mammalian HDGs revealed enrichment for the E-box motif that is bound by the Myc/Max heterodimer (Desbarats et al., 1996). Myc is a potent, pro-growth transcriptional activator of ribosome biogenesis and many cancers depend on Myc oncogenic activity. Perhaps Myc enables coordination between chaperone protein gene expression and transcriptional control of ribosome biogenesis to provide sufficient chaperones for newly synthesized proteins. This raises the interesting possibility that multi-cellular organisms evolved a larger chaperone buffer under basal conditions by uncoupling expression of HSPs from HSF1 while still maintaining HSP expression control by HSF1 under extreme conditions. Malignant growth is a case in point because in this context HSF1 inhibition results in cell proliferation arrest (Dai et al., 2007) and the accumulation of toxic protein aggregates (Tang et al., 2015). Surprisingly, HSF1 appears to drive a transcriptional program distinct from heat shock in cancer cells (Mendillo et al., 2012). Establishing the oncogenic contribution of HSF1 core gene targets defined by our work versus the cancer-specific gene targets is an important future goal. By demonstrating the superior contribution of Hsp70 and Hsp90 to Hsf1-mediated proteostasis in yeast, our work also provides the impetus for testing whether Hsp70 and Hsp90 inhibitors have synergistic anti-neoplastic effects on HSF1-dependent cancers.
EXPERIMENTAL PROCEDURES
Yeast strains and plasmids
Yeast strains used in this work are detailed in Table S5, and plasmids used as well as growth conditions are described in detail (see Supplemental Experimental Procedures).
Anchor Away Experiments
Unless otherwise indicated, 1 μM rapamycin was used to anchor away Hsf1-FRB in a rapamycin-resistant background (TOR1-1 fpr1Δ) (see Supplemental Experimental Procedures for detailed experimental conditions for microscopy, flow cytometry and Western blotting).
NET-seq, RNA-seq and ChIP-seq Experiments
Genome-wide next-generation sequencing experiments and analysis are described in detail along with statistical and bioinformatics methods (see Supplemental Experimental Procedures).
Mammalian cell lines and genetic manipulation
Wild-type murine embryonic fibroblasts (MEFs) and embryonic stem cells (mESCs) were obtained from Jackson Laboratories (Immortalized MEFs: CBA316; mESCs: 129X1/SvJ strain). CRISPR/Cas9 procedures and validation are described in detail along with immunofluorescence and Western blotting procedures (see Supplemental Experimental Procedures).
Supplementary Material
HIGHLIGHTS.
Yeast Hsf1 prevents toxic protein aggregation even in the absence of heat stress.
Basal and heat-induced expression of a small chaperone network is Hsf1-dependent.
Mammalian Hsf1 drives chaperone gene expression during heat stress only.
Rescuing Hsp70 and Hsp90 expression prevents cell toxicity due to Hsf1 ablation.
Acknowledgments
We thank S. Churchman and B. Tye for reagents and assistance with NET-seq experiments and data analysis; J. Krakowiak for technical assistance with RNA preps; A. S. Hansen for TPKas strains and 1-NM-PP1; W. Reilly for generating HDG knockout plasmids; J. Offermann for reagents; P. Arvidson for administrative support; P. Stoddard, Q. Justman, B. Zid and R. Subramaniam for useful discussions; P. Rogers for flow cytometry and general support; Members of the Denic and Lindquist labs for comments and suggestions; C. Chan for help with figures; S. Churchman, A. Murray, J. Taunton, L. Whitesell, B. Stern, J. Stewart-Ornstein, B. Tye, C. Chan and C. Shoemaker for helpful comments on the manuscript. This work was supported by the National Institute on Aging R21 (R21 AG050134-01 to V.D.) and Kirschstein NRSA diversity fellowship (F31 AG044967-03 to E.S.), a NIH Early Independence Award (DP5 OD017941-01 to D.P.), and a Cancer Research Fellowship from the Alexander and Margaret Stewart Trust (D.P.).
Footnotes
Supplemental information incudes Supplemental Discussion, Supplemental Experimental Procedures, five figures and six tables.
AUTHOR CONTRIBUTIONS
V.D., D.P. and E.S. conceived of the project and designed the experiments. E.S. performed the cloning and yeast strain construction; E.S. developed the quantitative microscopy, flow cytometry and genetics assays and performed these experiments with the Hsf1-AA strain, with help from D.P. for live-cell confocal microscopy; V.D. performed the NET-seq experiments; X.Z. performed the ChIP-seq experiments; E.S and D.P. prepared yeast samples, which D.P. purified RNA from, for RNA-seq; J.P., D.J. and P.G. developed the CRISPR/Cas9 genome editing protocol; J.P. and D.J. generated the hsf1 knockout cell lines; J.P. prepared mammalian cell samples for RNA-seq. E.S., D.P. and V.D. analyzed the data with help from E.A.; E.S., D.P. and V.D. made the figures and wrote the paper. All authors edited the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akerfelt M, Morimoto RI, Sistonen L. Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol. 2010;11:545–555. doi: 10.1038/nrm2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anckar J, Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2011;80:1089–1115. doi: 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]
- Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- Boy-Marcotte E, Lagniel G, Perrot M, Bussereau F, Boudsocq A, Jacquet M, Labarre J. The heat shock response in yeast: differential regulations and contributions of the Msn2p/Msn4p and Hsf1p regulons. Molecular Microbiology. 1999;33:274–283. doi: 10.1046/j.1365-2958.1999.01467.x. [DOI] [PubMed] [Google Scholar]
- Brandman O, Stewart-Ornstein J, Wong D, Larson A, Williams CC, Li GW, Zhou S, King D, Shen PS, Weibezahn J, Dunn JG, Rouskin S, Inada T, Frost A, Weissman JS. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell. 2012;151:1042–1054. doi: 10.1016/j.cell.2012.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson JM, Chakravarty A, DeZiel CE, Gross RH. SCOPE: a web server for practical de novo motif discovery. Nucleic Acids Research. 2007;35:W259–64. doi: 10.1093/nar/gkm310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Causton HC, Ren B, Koh SS, Harbison CT, Kanin E, Jennings EG, Lee TI, True HL, Lander ES, Young RA. Remodeling of yeast genome expression in response to environmental changes. Mol Biol Cell. 2001;12:323–337. doi: 10.1091/mbc.12.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2011;469:368–373. doi: 10.1038/nature09652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clos J, Westwood JT, Becker PB, Wilson S, Lambert K, Wu C. Molecular cloning and expression of a hexameric Drosophila heat shock factor subject to negative regulation. Cell. 1990;63:1085–1097. doi: 10.1016/0092-8674(90)90511-c. [DOI] [PubMed] [Google Scholar]
- Dai C, Whitesell L, Rogers AB, Lindquist S. Heat Shock Factor 1 Is a Powerful Multifaceted Modifier of Carcinogenesis. Cell. 2007;130:1005–1018. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer CG, Hughes TR. YeTFaSCo: a database of evaluated yeast transcription factor sequence specificities. Nucleic Acids Res. 2011;40:gkr993–D179. doi: 10.1093/nar/gkr993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desbarats L, Gaubatz S, Eilers M. Discrimination between different E-box-binding proteins at an endogenous target gene of c-myc. Genes & development. 1996;10:447–460. doi: 10.1101/gad.10.4.447. [DOI] [PubMed] [Google Scholar]
- Eastmond DL, Nelson HCM. Genome-wide analysis reveals new roles for the activation domains of the Saccharomyces cerevisiae heat shock transcription factor (Hsf1) during the transient heat shock response. J Biol Chem. 2006;281:32909–32921. doi: 10.1074/jbc.M602454200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkine AM, Adams CC, Diken T, Gross DS. Heat shock factor gains access to the yeast HSC82 promoter independently of other sequence-specific factors and antagonizes nucleosomal repression of basal and induced transcription. Mol Cell Biol. 1996;16:7004–7017. doi: 10.1128/mcb.16.12.7004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M, Takaki E, Takii R, Tan K, Prakasam R, HAYASHIDA N, Iemura SI, Natsume T, Nakai A. RPA Assists HSF1 Access to Nucleosomal DNA by Recruiting Histone Chaperone FACT. Mol Cell. 2012;48:182–194. doi: 10.1016/j.molcel.2012.07.026. [DOI] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiler-Samerotte KA, Dion MF, Budnik BA, Wang SM, Hartl DL, Drummond DA. Misfolded proteins impose a dosage-dependent fitness cost and trigger a cytosolic unfolded protein response in yeast. Proc Natl Acad Sci USA. 2011;108:680–685. doi: 10.1073/pnas.1017570108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- Görner W, Durchschlag E, Martinez-Pastor MT, Estruch F, Ammerer G, Hamilton B, Ruis H, Schüller C. Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes & development. 1998;12:586–597. doi: 10.1101/gad.12.4.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross DS, Adams CC, Lee S, Stentz B. A critical role for heat shock transcription factor in establishing a nucleosome-free region over the TATA-initiation site of the yeast HSP82 heat shock gene. EMBO J. 1993;12:3931. doi: 10.1002/j.1460-2075.1993.tb06071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn JS, Hu Z, Thiele DJ, Iyer VR. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol Cell Biol. 2004;24:5249–5256. doi: 10.1128/MCB.24.12.5249-5256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao N, O’Shea EK. Signal-dependent dynamics of transcription factor translocation controls gene expression. Nat Struct Mol Biol. 2012;19:31–39. doi: 10.1038/nsmb.2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, Laemmli UK. The Anchor-Away Technique: Rapid, Conditional Establishment of Yeast Mutant Phenotypes. Mol Cell. 2008;31:925–932. doi: 10.1016/j.molcel.2008.07.020. [DOI] [PubMed] [Google Scholar]
- Jakobsen BK, Pelham HR. A conserved heptapeptide restrains the activity of the yeast heat shock transcription factor. EMBO J. 1991;10:369–375. doi: 10.1002/j.1460-2075.1991.tb07958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsen BK, Pelham HR. Constitutive binding of yeast heat shock factor to DNA in vivo. Mol Cell Biol. 1988;8:5040–5042. doi: 10.1128/mcb.8.11.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaganovich D, Kopito R, Frydman J. Misfolded proteins partition between two distinct quality control compartments. Nature. 2008;454:1088–1095. doi: 10.1038/nature07195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002 doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- Liu XD, Liu PCC, Santoro N, Thiele DJ. Conservation of a stress response: human heat shock transcription factors functionally substitute for yeast HSF. EMBO J. 1997;16:6466–6477. doi: 10.1093/emboj/16.21.6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahat DB, Salamanca HH, Duarte FM, Danko CG, Lis JT. Mammalian Heat Shock Response and Mechanisms Underlying Its Genome-wide Transcriptional Regulation. Mol Cell. 2016;62:63–78. doi: 10.1016/j.molcel.2016.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIsaac RS, Silverman SJ, McClean MN, Gibney PA, Macinskas J, Hickman MJ, Petti AA, Botstein D. Fast-acting and nearly gratuitous induction of gene expression and protein depletion in Saccharomyces cerevisiae. Mol Biol Cell. 2011;22:4447–4459. doi: 10.1091/mbc.E11-05-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan DR, Xiao X, Shao L, Graves K, Benjamin IJ. Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J Biol Chem. 1998;273:7523–7528. doi: 10.1074/jbc.273.13.7523. [DOI] [PubMed] [Google Scholar]
- Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, Tamimi RM, Fraenkel E, Ince TA, Whitesell L, Lindquist S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell. 2012;150:549–562. doi: 10.1016/j.cell.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morano KA, Santoro N, Koch KA, Thiele DJ. A trans-activation domain in yeast heat shock transcription factor is essential for cell cycle progression during stress. Mol Cell Biol. 1999;19:402–411. doi: 10.1128/mcb.19.1.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls S, Leach MD, Priest CL, Brown AJP. Role of the heat shock transcription factor, Hsf1, in a major fungal pathogen that is obligately associated with warm-blooded animals. Molecular Microbiology. 2009;74:844–861. doi: 10.1111/j.1365-2958.2009.06883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabindran SK, Giorgi G, Clos J, Wu C. Molecular cloning and expression of a human heat shock factor, HSF1. Proc Natl Acad Sci USA. 1991;88:6906–6910. doi: 10.1073/pnas.88.16.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter K, Haslbeck M, Buchner J. The Heat Shock Response: Life on the Verge of Death. Mol Cell. 2010;40:253–266. doi: 10.1016/j.molcel.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Sakurai H, Ota A. Regulation of chaperone gene expression by heat shock transcription factor in Saccharomyces cerevisiae: Importance in normal cell growth, stress resistance, and longevity. FEBS Letters. 2011 doi: 10.1016/j.febslet.2011.07.041. [DOI] [PubMed] [Google Scholar]
- Sarge KD, Murphy SP, Morimoto RI. Activation of heat shock gene transcription by heat shock factor 1 involves oligomerization, acquisition of DNA-binding activity, and nuclear localization and can occur in the absence of stress. Mol Cell Biol. 1993;13:1392–1407. doi: 10.1128/mcb.13.3.1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharf KD, Rose S, Zott W, Schöffl F, Nover L, Schöff F. Three tomato genes code for heat stress transcription factors with a region of remarkable homology to the DNA-binding domain of the yeast HSF. EMBO J. 1990;9:4495–4501. doi: 10.1002/j.1460-2075.1990.tb07900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AP, McEntee K. Msn2p, a zinc finger DNA-binding protein, is the transcriptional activator of the multistress response in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1996;93:5777–5782. doi: 10.1073/pnas.93.12.5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A, Ward MP, Garrett S. Yeast PKA represses Msn2p/Msn4p-dependent gene expression to regulate growth, stress response and glycogen accumulation. EMBO J. 1998;17:3556–3564. doi: 10.1093/emboj/17.13.3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorger PK, Pelham HR. Yeast heat shock factor is an essential DNA-binding protein that exhibits temperature-dependent phosphorylation. Cell. 1988;54:855–864. doi: 10.1016/s0092-8674(88)91219-6. [DOI] [PubMed] [Google Scholar]
- Sorger PK, Pelham HR. Purification and characterization of a heat-shock element binding protein from yeast. EMBO J. 1987;6:3035–3041. doi: 10.1002/j.1460-2075.1987.tb02609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Dai S, He Y, Doty RA, Shultz LD, Sampson SB, Dai C. MEK Guards Proteome Stability and Inhibits Tumor-Suppressive Amyloidogenesis via HSF1. Cell. 2015;160:729–744. doi: 10.1016/j.cell.2015.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevelein JM, de Winde JH. Novel sensing mechanisms and targets for the cAMP–protein kinase A pathway in the yeast Saccharomyces cerevisiae. Molecular Microbiology. 1999 doi: 10.1046/j.1365-2958.1999.01538.x. [DOI] [PubMed] [Google Scholar]
- Torres FAG, Bonner JJ. Genetic Identification of the Site of Dna Contact in the Yeast Heat-Shock Transcription Factor. Mol Cell Biol. 1995;15:5063–5070. doi: 10.1128/mcb.15.9.5063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treger JM, Schmitt AP, Simon JR, McEntee K. Transcriptional factor mutations reveal regulatory complexities of heat shock and newly identified stress genes in Saccharomyces cerevisiae. J Biol Chem. 1998;273:26875–26879. doi: 10.1074/jbc.273.41.26875. [DOI] [PubMed] [Google Scholar]
- Trinklein ND, Murray JI, Hartman SJ, Botstein D, Myers RM. The role of heat shock transcription factor 1 in the genome-wide regulation of the mammalian heat shock response. Mol Biol Cell. 2004;15:1254–1261. doi: 10.1091/mbc.E03-10-0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. Heat Shock Transcription Factors: Structure and Regulation. Annu Rev Cell Dev Biol. 1995;11:441–469. doi: 10.1146/annurev.cb.11.110195.002301. [DOI] [PubMed] [Google Scholar]
- Zarzov P, Boucherie H, Mann C. A yeast heat shock transcription factor (Hsf1) mutant is defective in both Hsc82/Hsp82 synthesis and spindle pole body duplication. J Cell Sci. 1997;110(Pt 16):1879–1891. doi: 10.1242/jcs.110.16.1879. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Huang L, Zhang J, Moskophidis D, Mivechi NF. Targeted disruption of hsf1 leads to lack of thermotolerance and defines tissue-specific regulation for stress-inducible Hsp molecular chaperones. J Cell Biochem. 2002;86:376–393. doi: 10.1002/jcb.10232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.