

Abstract
Mutations in the gene encoding leucine-rich repeat kinase 2 (LRRK2) can cause Parkinson's disease (PD), and the most common disease-associated mutation, G2019S, increases kinase activity. Because LRRK2 expression levels rise during synaptogenesis and are highest in dorsal striatal spiny projection neurons (SPNs), we tested the hypothesis that the LRRK2–G2019S mutation would alter development of excitatory synaptic networks in dorsal striatum. To circumvent experimental confounds associated with LRRK2 overexpression, we used mice expressing LRRK2–G2019S or D2017A (kinase-dead) knockin mutations. In whole-cell recordings, G2019S SPNs exhibited a fourfold increase in sEPSC frequency compared with wild-type SPNs in postnatal day 21 mice. Such heightened neural activity was increased similarly in direct- and indirect-pathway SPNs, and action potential-dependent activity was particularly elevated. Excitatory synaptic activity in D2017A SPNs was similar to wild type, indicating a selective effect of G2019S. Acute exposure to LRRK2 kinase inhibitors normalized activity, supporting that excessive neural activity in G2019S SPNs is mediated directly and is kinase dependent. Although dendritic arborization and densities of excitatory presynaptic terminals and postsynaptic dendritic spines in G2019S SPNs were similar to wild type, G2019S SPNs displayed larger spines that were matched functionally by a shift toward larger postsynaptic response amplitudes. Acutely isolating striatum from overlying neocortex normalized sEPSC frequency in G2019S mutants, supporting that abnormal corticostriatal activity is involved. These findings indicate that the G2019S mutation imparts a gain-of-abnormal function to SPN activity and morphology during a stage of development when activity can permanently modify circuit structure and function.
SIGNIFICANCE STATEMENT Mutations in the kinase domain of leucine-rich repeat kinase 2 (LRRK2) follow Parkinson's disease (PD) heritability. How such mutations affect brain function is poorly understood. LRRK2 expression levels rise after birth at a time when synapses are forming and are highest in dorsal striatum, suggesting that LRRK2 regulates development of striatal circuits. During a period of postnatal development when activity plays a large role in permanently shaping neural circuits, our data show how the most common PD-causing LRRK2 mutation dramatically alters excitatory synaptic activity and the shape of postsynaptic structures in striatum. These findings provide new insight into early functional and structural aberrations in striatal connectivity that may predispose striatal circuitry to both motor and nonmotor dysfunction later in life.
Keywords: activity, dorsal striatum, G2019S, LRRK2, spine morphology, synapse
Introduction
Parkinson's disease (PD) is a progressive neurological disorder characterized by impairments in motor and nonmotor functions of the dorsal striatum (Shiba et al., 2000; Goedert, 2001; Aarsland et al., 2009; Gerfen and Surmeier, 2011; Erro et al., 2012). The majority of inherited forms of PD are caused by mutations in the gene encoding leucine-rich repeat kinase 2 (LRRK2), most of which reside in the GTPase or kinase domains of LRRK2 (Paisán-Ruiz et al., 2004; Zimprich et al., 2004; Di Fonzo et al., 2005; Khan et al., 2005; Ozelius et al., 2006; Cookson, 2010). Although it is not clear how mutations in different enzymatic domains can give rise to PD, many serve to increase kinase activity (West et al., 2005, 2007; Greggio et al., 2006; Smith et al., 2006). The LRRK2–G2019S mutation is the most prevalent, estimated to cause up to 42% of familial PD and 10% of sporadic PD (Healy et al., 2008; Paisán-Ruiz et al., 2013), and it increases kinase activity by twofold to threefold (West et al., 2005; Jaleel et al., 2007). Disease progression in LRRK2–G2019S carriers is clinically indistinguishable from idiopathic cases (Taylor et al., 2006; Healy et al., 2008; Cookson, 2010), suggesting common disease mechanisms and highlighting the need to identify and understand the neurobiological actions of LRRK2–G2019S.
Although the effect of LRRK2 mutation on normal neuronal function is poorly understood, recent research has provided important clues. fMRI data show altered functional connectivity between cortex and striatum in asymptomatic human LRRK2–G2019S carriers (Helmich et al., 2015), and although there is disagreement over mechanism, loss-of-function studies of LRRK2 or its orthologs in Caenorhabditis elegans, Drosophila, and mouse support that wild-type (WT) LRRK2 can regulate developing synapse function (Sakaguchi-Nakashima et al., 2007; Shin et al., 2008; Lee et al., 2010b; Piccoli et al., 2011; Matta et al., 2012; Beccano-Kelly et al., 2014; Parisiadou et al., 2014). In brain, LRRK2 is most highly expressed in striatal spiny projection neurons (SPNs). Its expression levels rise dramatically over a period of development that coincides with the ingrowth of corticostriatal inputs and a burst in excitatory synaptogenesis on SPNs (Tepper et al., 1998; Biskup et al., 2007; Westerlund et al., 2008; Giesert et al., 2013) and continues to rise as neural activity plays a prominent role in shaping persistent striatal circuit structure and function (Biskup et al., 2007; Westerlund et al., 2008; Gerfen and Surmeier, 2011; Kozorovitskiy et al., 2012). Together, these data suggest that LRRK2 plays an important role in establishing and shaping activity in corticostriatal circuits.
Based on the temporal and spatial expression patterns of LRRK2, we hypothesized that the LRRK2–G2019S mutation would alter development of excitatory synaptic networks in dorsal striatum. To test this, we used LRRK2 mutant knockin (KI) mice to compare gain- and loss-of-LRRK2 kinase function. This strategy circumvents confounding experimental factors, such as protein overexpression and mixed expression of WT and mutant LRRK2 proteins that have complicated interpretation of LRRK2 experiments performed in transgenic mouse models (Aarsland et al., 2009; Dächsel et al., 2010; Li et al., 2010; Skibinski et al., 2014). Our findings reveal that LRRK2–G2019S exerts a robustly abnormal effect on corticostriatal synapses at postnatal day 21 (P21). LRRK2–G2019S generates abnormally elevated excitatory activity and altered spine morphology in dorsal striatal SPNs that is kinase dependent, a gain-of-abnormal activity that is outside the normal role of LRRK2 function. Excessive activity is developmentally transient and can be normalized by acute exposure to LRRK2 kinase inhibitors or by removing cortical afferents. These data show a clear biological outcome selectively attributable to LRRK2 mutation and suggest that striatal circuits are fundamentally altered by LRRK2–G2019S at early postnatal ages.
Materials and Methods
Animals
Treatment and use of all animals conform to animal welfare protocols approved by the Mount Sinai Institutional Animal Care and Use Committee and are in strict accordance with National Institutes of Health guidelines. Animals of both sexes were used in all experiments. Three principal lines of mice were used in this study: (1) mice expressing a homozygous KI G2019S gain-of-kinase activity mutation (hereafter abbreviated GSKI; West et al., 2005; Jaleel et al., 2007); (2) mice expressing a homozygous KI D2017A mutation that renders LRRK2 kinase dead (hereafter abbreviated DAKI); and (3) WT mice. The effect of G2019S and D2017A mutations on LRRK2 are very well characterized. The G2019S mutation appears to lock the kinase into a more active conformation, increasing its activity by twofold to threefold (West et al., 2005; Liu et al., 2011). The D2017A mutation prevents Mg2+ binding, destabilizing interactions with ATP (Johnson et al., 1996; Nolen et al., 2004; Ray and Liu, 2012) but preserving all other functional domains of the protein (Jaleel et al., 2007; Ito et al., 2014). Kinase activating effects of G2019S and inactivating effects of D2017A have been widely tested and characterized (Jaleel et al., 2007; Nichols et al., 2009, 2010; Matta et al., 2012; Ito et al., 2014). All mice are congenic on a C57BL/6NTac background (Fig. 1A).
Figure 1.
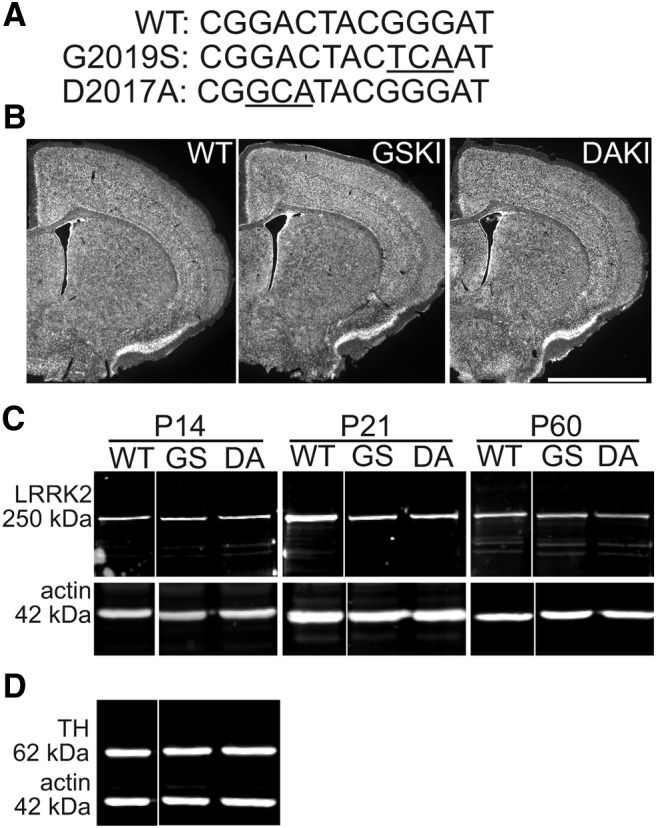
LRRK2 mutations and protein levels across genotypes. A, Nucleotide sequences from the WT LRRK2 gene and the corresponding sequences of the G2019S (GS) (Gly to Ser) and D2017A (DA) (Asp to Ala) mutations (underlined nucleotides) in exon 41. B, Nissl staining of coronal sections through striatum from P21 mice shows similar cytoarchitecture across genotypes. Scale bar, 2 mm. C, Western blotting for LRRK2 in striatal lysates taken from WT, GSKI, and DAKI mice at the indicated ages. Labeling with anti-LRRK2 C41-2 antibody shows a 250 kDa band present in WT, GSKI, and DAKI lanes. Labeling for actin (42 kDa) was used as loading control. There were no significant changes in LRRK2 expression levels across genotypes (see Results). D, Western blotting of P21 striatal lysates for TH shows similar levels across genotypes (see Results).
To generate G2019S and D2017A KI mice, targeting vectors generated using C57BL/6J BAC–DNA and having proximal loxP and frt-flanked PGK–neomycin cassettes were inserted into intron 41; a distal loxP site was inserted into intron 42 of the endogenous mouse LRRK2 gene. The vectors were introduced into C57BL/6NTAc (Taconic Biosciences)-derived embryonic stem (ES) cells, and successfully targeted ES cells were injected into blastocysts. Chimeric mice were bred with an Flp deleter strain to remove the neomycin cassette [C57BL/6NTac–Tg(CAG-Flpe)2Arte] and then bred with C57BL/6NTac to remove the Flp transgene. KI mice are crossed to WT C57BL/6NTac every fourth generation to guard against genetic drift.
Additional experiments were conducted on heterozygous GSKI mice expressing tdTomato under control of the dopamine receptor 1a promoter (GSKI-Drd1aT). These mice were the F2 progeny of crosses between homozygous GSKI mice and the reporter lines B6.Cg–Tg(Drd1a–tdTomato)6Calak/J (strain 016204; The Jackson Laboratory).
Genotype confirmation
The presence of GSKI and DAKI mutations were verified by PCR using the primers 5′-CTG TGT GTT AAA GCT CCA GTT GCC TAC-3′ (forward) and 5′-AAA CAG TAA CTA TTT CCG TCG TGA TCC G-3′ (reverse), gel extracted, and sequenced (Genewiz) using the forward primer to confirm mutation homozygosity in exon 41 (WT, CGGACTACGGGATC; G2019S, CGGACTACTCAATT; D2017A, CGGCATATGGGATC). Homozygosity was confirmed via analysis of the sequencing trace results.
The presence of the tdTomato or eGFP transgene was confirmed via PCR using the following primers: for tdTomato, forward, 5′-CTT CTG AGG CGG AAA GAA CC-3′ and reverse, 5′-TTT CTG ATT GAG AGC ATT CG-3′; for eGFP, forward, 5′-CCC GAA GCT TCT CGA GGC GCG CCC TGT GCG TCA GCA TTT GCA A-3′ and reverse, 5′-TCA GGG TCA GCT TGC CGT AGG-3′. Bands of 600 bp indicate the presence of the transgene.
Immunoblotting
Immunoblotting was performed on whole striatal lysates from GSKI, DAKI, or WT (n = 3 mice per genotype per age) or striatal synaptoneurosomes according to our previous descriptions (Nikitczuk et al., 2014). Briefly, synaptoneurosomes were prepared from striata that were homogenized in ice-cold SynPER lysis buffer (100 μl/100 mg tissue) containing phosphatase (Life Technologies) and protease inhibitors (Roche) via 3 × 5 s pulses with a Kontes pestal gun. Samples were then centrifuged at 4°C at 1500 × g for 10 min. The supernatant was extracted and centrifuged at 4°C at 17,000 × g for 20 min, and the pellet was resuspended in RIPA buffer with protease inhibitors (Roche). Protein concentration was determined using a Bradford assay (Bio-Rad Laboratories). Protein (25 μg) from each animal was separated via 8% SDS-PAGE and transferred to Immune-Blot-F PVDF membranes (Millipore) by wet electroblotting at 4°C overnight (25 V). Membranes were rinsed in PBS (5 min), air dried (5 min), and blocked with LI-COR Odyssey Blocking Buffer for 45 min at room temperature (RT) and then incubated with primary antibodies for 1–2 h. The primary antibodies were directed against the following: LRRK2 (c41-2, 1:1000; Epitomics), PSD95 (catalog #MA1045, 1:2000; Thermo Fisher Scientific), actin (catalog #MAB1501, 1:4000; Millipore), VGluT1 (AB5905, 1:1000; Millipore), VGluT2 (AB2251, 1:1000; Millipore), and tyrosine hydroxylase (TH; catalog #22941, 1:1000; ImmunoStar). All were diluted in LI-COR Odyssey Blocking Buffer containing 0.1% Tween 20 (Thermo Fisher Scientific). Membranes were washed and incubated for 45 min with fluorophore-conjugated secondary antibodies (DyLight 800 by Cell Signaling Technology and DyLight 680 by Pierce) diluted in PBS with 50% Odyssey Blocking Buffer and 0.1% Tween 20, washed again, and visualized with a LI-COR Odyssey CLX imager (LI-COR Biosciences). Protein levels were determined via film densitometry (NIH ImageJ) in which band intensity of the protein of interest was normalized to an actin loading control within the same lane. This was conducted three to five separate times. Data were compared using one-way ANOVA with Dunnett's post hoc tests; a p value <0.05 was considered statistically significant.
Immunofluorescence and histology
Mice (aged P21) of each genotype were perfused transcardially with 4% paraformaldehyde, and their brains were cut on a freezing sliding microtome into a 35-μm-thick series of coronal sections through the dorsal striatum. One set was stained with NeuroTrace 500/525 (Invitrogen) and imaged on an Olympus MVX10 Stereoscope using a Hamamatsu C8484 camera. The others were processed for immunofluorescence using methods that have been detailed previously (Nikitczuk et al., 2014). Sections were immunolabeled with antibodies directed against one of the following: PSD95 (catalog #MA1045, 1:1000; Thermo Fisher Scientific), VGluT1 (AB5905, 1:1000; Millipore), and VGluT2 (AB2251, 1:1000; Millipore). Antibody binding was visualized by fluorophore-conjugated secondary antibodies (Alexa Fluor 488 or Alexa Fluor 654; Abcam). Five mice per genotype per time point were sectioned, and three single optical images per section per mouse were imaged (1024 × 1024 pixels, 16 bit) across dorsal striatum with a 100 × objective on a Zeiss LSM 780 confocal microscope. Immunolabeled synaptic-like puncta were quantified with NIH ImageJ using a thresholding function to capture for analysis all immunolabeled puncta; subsequent comparisons of puncta size or density across genotypes were made using a one-way ANOVA with Dunnett's post hoc test; a p value <0.05 was considered statistically significant. All image acquisition and quantification was conducted by an investigator blinded to genotype.
For DiI labeling, slices were prepared as for electrophysiology (see below) and fixed in 4% paraformaldehyde, and DiI crystals were placed into the dorsal striatum using a glass micropipette. After several weeks of incubation, labeling was imaged using a Zeiss AxioImager.Z2 and a Zeiss MRM camera.
Electrophysiology
Slice preparation.
Mice were deeply anesthetized with isoflurane and decapitated. Brains were rapidly removed in ice-cold sucrose–artificial CSF (aCSF) consisting of the following (in mm): 233.7 sucrose, 26 NaHCO3, 3 KCl, 8 MgCl2, 0.5 CaCl2, 20 glucose, and 0.4 ascorbic acid. Coronal slices (350 μm) were cut with a Leica VT1000S vibratome and allowed to equilibrate in recording aCSF at RT for ∼1 h before being transferred to the recording chamber. The aCSF comprised the following (in mm): 117 NaCl, 4.7 KCl, 1.2 MgSO4, 2.5 CaCl2, 1.2 NaH2PO4, 24.9 NaHCO3, and 11.5 glucose. During recording, slices were maintained at 31°C and perfused (1.5 ml/min) with oxygenated aCSF (95% O2–5% CO2) in an immersion chamber containing the GABAA receptor antagonist gabazine (GBZ; 10 μm). For some experiments (see Results), immediately after vibratome slicing, the dorsal striatum on one side was surgically isolated from surrounding neocortex using a scalpel. After isolation, sections were maintained in oxygenated, ice-cold sucrose–aCSF and then allowed to equilibrate at RT for 45 min before recording.
Whole-cell patch-clamp recordings.
Dorsal striatal SPNs were visualized on an upright epifluorescence microscope (BX50WI; Olympus) with a 40 × water-immersion lens and an IR-1000 infrared CCD monochrome video camera (Dage-MTI). Whole-cell recordings from SPNs were performed with glass micropipettes pulled from borosilicate glass capillaries using a P-87 micropipette puller (Sutter Instruments). The pipette resistance was 2–4 MΩ. To record spontaneous EPSCs (sEPSCs; no TTX in the bath), electrodes were filled with an intracellular solution containing the following: 124 mm K-gluconate, 10 mm HEPES, 10 mm phosphocreatine di(Tris), 0.2 mm EGTA, 4 mm Mg2ATP, 0.3 mm Na2GTP, and 0.3% biocytin. To isolate miniature EPSCs (mEPSCs), electrodes were filled with similar K-gluconate-based intracellular solution to which was added QX-314 (5 mm), whereas TTX (3 μm) was added to the bath. Osmolarity of the intracellular solution was adjusted to 280–290 mOsm, and pH was adjusted to 7.3. Recordings were made using a Multiclamp 200B (Molecular Devices) in both voltage- and current-clamp mode. Analog signals were low-pass filtered at 2 kHz and digitized at 5 kHz with the use of a Digidata 1440A interface and pClamp10 software (Molecular Devices). Gigaseal and additional access to the intracellular neuronal compartment was achieved in voltage-clamp mode, with the holding potential set at −70 mV. Soon after rupturing the membrane, the intracellular neuronal fluid reached equilibrium with the pipette solution without significant changes in either series resistance or membrane capacitance values. Membrane voltage was kept at −70 mV through all voltage-clamp experiments (sEPSC and mEPSC). Fast compensation was used to maintain the series resistance values below 12 MΩ. To electrophysiologically identify SPNs after recording sEPSC activity in voltage-clamp mode (5–10 min), we switched to current-clamp mode and applied a series of depolarizing current pulses (10 s interpulse intervals, 500-ms-long pulses, 20 pA current intervals). This protocol allowed us to confirm SPN identity electrophysiologically by characteristic action potential firing patterns described previously (Fino et al., 2007). To confirm that EPSC responses were glutamatergic, we applied NMDA and AMPA/kainate receptor antagonists 2-amino-5-phosphovalerate (APV; 40 μm) and 6-cyano-7-nitroquinoaxaline-2,3-dione (CNQX; 10 μm), respectively, to the bath solution to block fast glutamatergic activity, followed by washout of the glutamate receptor blockers and a return of normal EPSCs within 10–20 min. In some experiments, the specific LRRK2-kinase inhibitor GNE-7915 (Estrada et al., 2012) was diluted in DMSO and then added to the aCSF bath to a final concentration of 50 nm 30 min before recording. Control slices were exposed to equivalent concentrations of the diluent (DMSO) alone. Applied drugs were administered to the slice via a peristaltic pump (Cole-Parmer), and pump speed was adjusted so that solutions reached the slice 1.5 min after the start of application. All drugs were purchased from Sigma-Aldrich except for the LRRK2 kinase inhibitor, which was purchased from ChemiTek.
Identification of direct- and indirect-pathway SPNs.
Mice (n = 3–4 per injection site) aged P18 were deeply anesthetized with isoflurane and secured in a stereotaxic frame (David Kopf Instruments). Cholera toxin subunit B conjugated to Alexa Fluor 488 (0.1% in PBS; Invitrogen) was injected into either the substantia nigra pars reticulata [anteroposterior (AP), −3.0 mm; mediolateral (ML), +1.4 mm; dorsoventral (DV), −4.1 mm], the target of direct-pathway SPNs, or the external segment of the globus pallidus (AP, −0.7 mm; ML, +2.0 mm; DV, −3.2 mm), the target of indirect-pathway SPNs, with a 32 gauge Hamilton syringe as described previously (Friedman et al., 2015). Mice were returned to their home cage for 72 h to allow for retrograde transport of the fluorescent tracer and then killed for electrophysiological recording as described above. Labeled cells were positively identified as direct- or indirect-pathway SPNs on the basis of retrogradely transported fluorescence and targeted for whole-cell recording.
Data analysis.
Offline analysis was performed using Clampfit (Molecular Devices) and Mini Analysis software (Synaptosoft). Only neurons whose action potential firing patterns resembled those of typical SPNs were analyzed (long depolarizing ramp before action potential threshold; Dong et al., 2006). sEPSCs and mEPSCs were analyzed using Mini Analysis with thresholds set at 6 pA for amplitude and 15 pA for area. For all datasets, Bartlett's test for equal variances was performed; one-way or two-way ANOVA was used when appropriate to compare more than two groups with equal variances (p > 0.05), whereas sample groups with significantly different sample variances (p < 0.05) were further analyzed using linear mixed effects models with random animal effect to account for repeated measures within an animal. Log transformations were performed on the outcome variables to stabilize the variance. The Holm step-down procedure was applied to control type I error rate in experiments with multiple comparisons between groups. A p value <0.05 was considered statistically significant. Analyses used SAS statistical software (version 9.4) or GraphPad Software Prism (version 5.01). When comparing two groups, a two-tailed Student's t test was performed. Data are presented as mean ± SEM. When relevant, numbers used for individual experiments is listed as n = numbers of cells (numbers of mice). Cumulative probability distributions were compared using Kolmogorov–Smirnov tests; p < 0.05 was considered significant. Two-way ANOVA was used to analyze the effect of decortication on sEPSCs to compare equal numbers of cells from equal numbers of animals across conditions. Statistical tests and values are provided in Data Dryad: http://dx.doi.org/10.5061/dryad.j5n50.
Dendrite imaging and analysis
All recorded SPNs were filled with biocytin contained within the patch pipette. After recording, slices were immersed in 4% paraformaldehyde overnight at 4°C, permeabilized with Triton X-100 (0.1%) for 2 h at RT, and labeled with streptavidin-conjugated Alexa Fluor 594 (1:300; Jackson ImmunoResearch) at 4°C. Intracellularly filled SPNs were first imaged on a confocal LSM 780 (Zeiss) using a 20× objective and Nyquist sampling criteria to generate 3D renderings of dendritic arbors and then with a 63× objective using Nyquist sampling criteria to visualize dendritic protrusions. Z-stacks were deconvolved using AutoDeblur/AutoVisualize X (MediaCybernetics). Dendrites were traced using Neurolucida and analyzed using NeuroExplorer (both from MBF Bioscience); protrusion density and morphology were assessed using NeuronStudio (Rodriguez et al., 2006). Dendritic protrusions were categorized as filopodia if they were longer than 2 μm and thinner than 300 nm; those that were shorter than 2 μm and wider than 300 nm in diameter were categorized as spines (Yuste and Bonhoeffer, 2004). Groups were compared using two-tailed t tests; a p value <0.05 was considered statistically significant. Spine-head diameter cumulative frequency distributions were compared using two-sample Kolmogorov–Smirnov tests.
Results
LRRK2 gain- or loss-of-kinase activity mutations do not affect cytoarchitecture or LRRK2 protein levels
To evaluate the effect of the PD-related LRRK2–G2019S mutation on striatal synaptic circuit development, we used two KI mouse lines that were generated at Eli Lilly laboratories: one carrying the GSKI mutation (gain-of-kinase function; Fig. 1A) and the other carrying the DAKI mutation (that renders LRRK2 kinase dead; Fig. 1A). Data from several laboratories concur that the G2019S mutation increases kinase activity twofold to threefold (West et al., 2005; Jaleel et al., 2007), and a recent study using these GSKI mice demonstrated decreased levels of some phospho-Rabs, putative physiological targets of LRRK2, during in vivo administration of a LRRK2 inhibitor (Steger et al., 2016). The D2017A mutation eliminates LRRK2 kinase activity by interfering with obligatory interactions with Mg2+ (Jaleel et al., 2007; Ito et al., 2014). The presence of the gene mutations was confirmed by sequencing. Mutant mice appeared grossly normal and exhibited normal litter sizes and growth rates compared with WT mice. Nissl-stained sections showed no notable differences between genotypes in cytoarchitecture of neocortex or striatum (Fig. 1B). A previous study of a different kinase-dead point mutation suggested that LRRK2 kinase-dead mutations may destabilize LRRK2 protein in adulthood (Herzig et al., 2011). To test whether LRRK2 destabilization occurred in our KI mice, we compared levels of mutant and WT LRRK2 in striatal lysates but found no significant differences in LRRK2 levels between groups (Fig. 1C; ANOVA, p = 0.5784, F = 0.601, n = 3 animals each). Comparing LRRK2 protein levels in whole-brain lysates across genotypes yielded similar outcomes (data not shown). Additionally, there were no significant differences across genotypes in striatal levels of TH (Fig. 1D; ANOVA, p = 0.9948, F = 0.005, n = 3 animals each).
LRRK2–G2019S mutation significantly and transiently increases frequency of sEPSCs in SPNs
To compare glutamatergic EPSCs across genotypes, we used whole-cell patch-clamp recordings from dorsal striatal SPNs in acutely prepared coronal slices (Fig. 2A). In mice, LRRK2 expression levels are similar in striosomes and matrix (Davies et al., 2013; West et al., 2014), so we did not distinguish between these compartments. SPNs were identified visually by their somal size and confirmed electrophysiologically by their characteristic firing patterns in response to depolarizing current steps (Fino et al., 2007; Fig. 2B). All recorded cells were filled with biocytin, and SPN identity was confirmed by post hoc visualization of dendritic spines (Fig. 2C). EPSCs were isolated by bath application of GBZ, which blocks fast GABAA receptor-mediated currents, and confirmed to be glutamate receptor mediated because they were eliminated entirely by bath application of the AMPA/kainate and NMDA receptor antagonists CNQX and APV, respectively (Fig. 2D).
Figure 2.

G2019S mutation increases sEPSC frequency in dorsal striatal SPNs. A, Schematic of a coronal cross-section showing approximate locations of recording pipettes in dorsal striatum. B, Example of current-clamp traces showing patterns of action potentials elicited by depolarizing current steps that are characteristic of SPNs. C, Confocal image of biocytin-filled SPN after whole-cell recording. Inset shows dendritic spines characteristic of SPNs. D, Continuous voltage-clamp traces (holding at −70 mV) showing sEPSCs recorded from WT and GSKI SPNs in the presence of GBZ (10 μm; top pair of traces). All sEPSCs were blocked by glutamate receptor antagonists [middle pair of traces; APV (10 μm) and CNQX (40 μm)] and recovered after washout (bottom pair of traces). E, Mixed bar graphs/scatter plots showing average frequencies of sEPSCs at P21 from WT (n = 15 SPNs, 5 mice), GSKI (n = 12 SPNs, 6 mice; ***p = 0.0001), and DAKI (n = 10 SPNs, 5 mice; p > 0.05), F = 14.93. F, Mixed bar graphs/scatter plots showing average amplitudes of sEPSCs. There were no significant differences across genotypes. p > 0.05, F = 0.01. In E and F, linear mixed effects models were used to compare groups (see Materials and Methods). G, Sample traces of sEPSCs recorded from WT, GSKI, and DAKI. H, Cumulative probability distribution of IEIs of WT, GSKI, and DAKI sEPSCs. ***Kolmogorov–Smirnov test, WT versus GSKI, p = 0.0001 and WT versus DAKI, p = 0.45. Scale bar (in C): top, 25 μm; bottom, 7 μm.
We focused first on the effect of LRRK2 kinase activity on action potential-dependent sEPSCs (no TTX in the bath) by comparing responses across GSKI, DAKI, and WT SPNs at P14, P21, and P60. At P14, which follows ingrowth of corticostriatal axons (Sharpe and Tepper, 1998; Sohur et al., 2014), there were no significant differences across genotypes in frequency (Table 1). In contrast, at P21, a time point that follows a burst in corticostriatal synaptogenesis (Tepper et al., 1998), GSKI SPNs displayed a significant fourfold increase in frequency of sEPSCs compared with WT or DAKI SPNs (Fig. 2E,G; Table 1) and a significantly shorter interevent interval (IEI) between sEPSCs (Fig. 2H). There were no differences across genotypes in average sEPSC amplitudes (Fig. 2F,G).
Table 1.
Postnatal time course of average sEPSC frequency across genotypes
| Genotypes compared | Average sEPSC frequency (Hz) | p value | F value |
|---|---|---|---|
| P14 | |||
| WT versus GSKI | 0.87 ± 0.10 versus 0.91 ± 0.16 | 0.28 | 2.36 |
| WT versus DAKI | 0.87 ± 0.10 versus 0.52 ± 0.24 | 0.14 | |
| P21 | |||
| WT versus GSKI | 0.52 ± 0.17 versus 1.93 ± 0.24 | <0.0001*** | 14.93 |
| WT versus DAKI | 0.52 ± 0.17 versus 0.59 ± 0.25 | 0.65 | |
| P60 | |||
| WT versus GSKI | 0.90 ± 0.21 versus 0.74 ± 0.33 | 0.46 | 0.71 |
| WT versus DAKI | 0.90 ± 0.21 versus 0.88 ± 0.31 | 0.62 |
Values are means ± SE.
***p < 0.0001.
The sEPSC frequencies observed in GSKI SPNs also showed a wider range compared with WTs. We asked whether the wider range in frequencies reflected differential effects of the GSKI mutation on direct- and indirect-pathway SPNs. To test this, fluorescent retrograde labeling from the substantia nigra or globus pallidus was used to identify and record from direct- or indirect-pathway GSKI SPNs, respectively. There were no significant differences in sEPSC frequency between direct- and indirect-pathway identified GSKI SPNs [p = 0.6639, F = 1.739, Student's t test, n = 8 (3) direct-pathway SPNs and 9 (3) indirect-pathway SPNs], indicating that the abnormally elevated EPSC frequency caused by the G2019S mutation is not pathway specific. As a second test, GSKI mice were crossed to a line of mice expressing tdTomato in Drd1R (direct-pathway) SPNs, and sEPSCs were recorded from tdTomato-positive (D1R) and tdTomato-negative (presumptive D2R) SPNs. Here also, there were no differences in sEPSC frequency between tdTomato-positive and tdTomato-negative SPNs (p = 0.9850, F = 2.627, Student's t test, n = 4 tomato-positive D1 SPNs and n = 3 tomato-negative SPNs). We confirmed that heterozygous GSKI SPNs exhibited the same elevated frequency of sEPSCs compared with homozygous GSKI SPNs [2.080 ± 0.3379 Hz (heterozygous; n = 12 [3]) and 1.926 ± 0.2902 Hz (homozygous; n = 12 [6]), p = 0.7329, F = 1.356, Student's t test], which was expected because in humans, the G2019S mutation is autosomal dominant (Zimprich et al., 2004). Together, these data demonstrate that the effects of LRRK2–G2019S do not differentially affect SPN subtypes.
By P60, sEPSC frequencies were similar across all genotypes (Table 1), similar to what has been observed in adult mice overexpressing LRRK2–G2019S (Beccano-Kelly et al., 2015). Our data suggest that developmental abnormalities in synaptic neurotransmission at P21 in GSKI mice may trigger compensatory mechanisms as circuits mature.
The abnormally elevated sEPSC frequency in GSKI SPNs is driven by the excessive kinase activity of the G2019S mutation
The G2019S mutation increases LRRK2 kinase activity (West et al., 2005; Jaleel et al., 2007), suggesting that the effects on EPSCs were kinase driven. It was not clear whether the mutation exaggerates a normal effect of LRRK2 on activity or whether the excessive activity represents a gain-of-abnormal function. To distinguish between these possibilities, we used DAKI mutant mice to compare sEPSCs between kinase-dead DAKI SPNs and WT SPNs at P21. We found no significant differences in sEPSC frequency, IEI, or amplitude between DAKI and WT SPNs (Fig. 2E–H), indicating that LRRK2 kinase activity is not necessary for normal levels of spontaneous action potential-driven activity. Moreover, the analysis demonstrates that the elevated sEPSC frequency observed in G2019S SPNs is a gain-of-abnormal function driven by excessive kinase activity.
Aberrant SPN activity in G2019S mutants could result from ongoing and excessive kinase activity, but it is also possible that the mutation sets in motion a series of earlier developmental events that indirectly regulate activity. To test whether the effects of the kinase are acute, we exposed GSKI SPNs to the LRRK2 kinase inhibitor GNE-7915 (Estrada et al., 2012). Acute treatment of striatal slices with GNE-7915 (50 nm) effectively reduced GSKI sEPSC frequency to levels indistinguishable from WT (Fig. 3A,B) but had no effect on activity in WT or DAKI mutant SPNs (Fig. 3A,B). When coupled with the observation that sEPSCs did not differ between DAKI and WT SPNs, the data indicate that, under normal, nonpathophysiological conditions, the kinase activity of LRRK2 is dispensable for spontaneous synaptic neurotransmission and that the elevated frequency of sEPSCs in GSKI SPNs represents a gain-of-abnormal function by the G2019S mutation.
Figure 3.
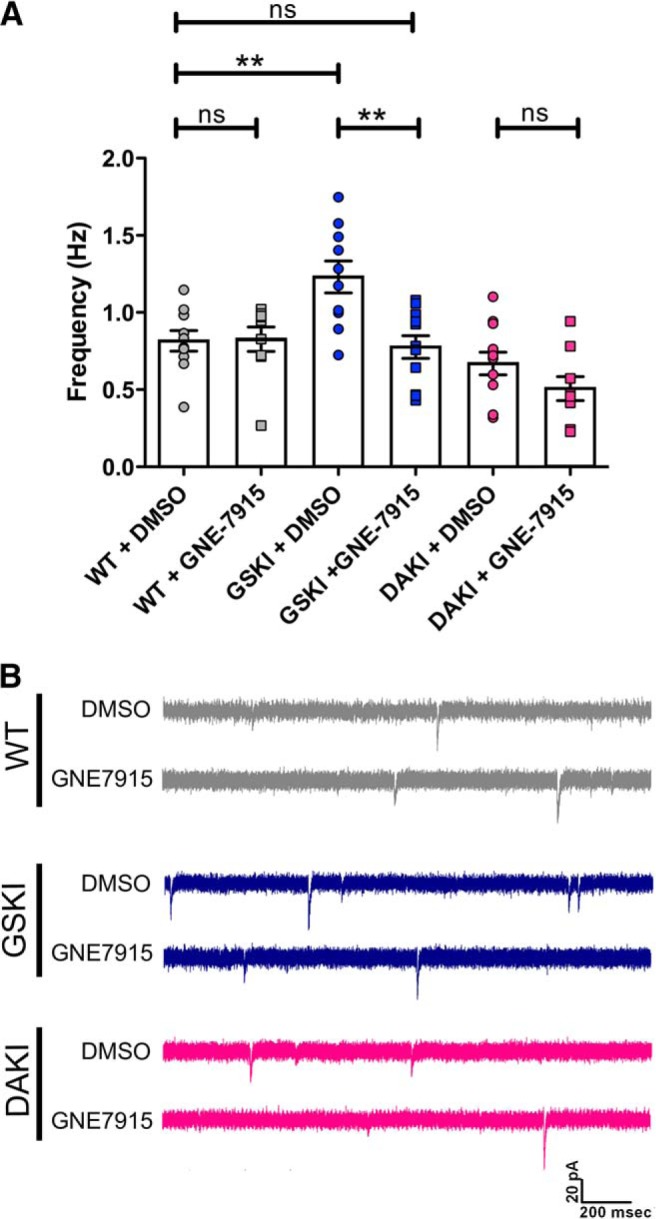
LRRK2 kinase inhibitor blocks abnormal activity in GSKI SPNs. A, Mixed bar graphs/scatter plots showing frequency of sEPSCs in WT (gray), GSKI (blue), and DAKI (pink) SPNs in the presence of the diluent DMSO (circles) or GNE-7915 (50 nm) (squares). The inhibitor significantly decreased the frequency of sEPSCs in GSKI SPNs to WT levels (**p = 0.001) such that there was no significant difference in sEPSC frequency between DMSO-treated WT and GNE-7915-treated GSKI SPNs (p > 0.05). GNE-7915 had no effect on sEPSC frequencies of WT SPNs or DAKI SPNs. A one-way ANOVA with Bonferroni's post hoc tests was used to compare groups, F = 8.755. B, Sample traces showing WT (gray, top), GSKI (blue, middle), and DAKI (pink, bottom) sEPSCs in the presence of DMSO (top trace of each pair) or GNE-7915 (50 nm, bottom trace of each pair).
Action potential-dependent synaptic activity is particularly affected by the G2019S mutation
sEPSCs include both action potential-generated synaptic activity and spontaneous fusion of synaptic vesicles (mEPSCs). Action potential-dependent vesicle fusion (sEPSCs) and spontaneous vesicle fusion (mEPSCs) can involve different vesicle pools (Sara et al., 2005; Kavalali, 2015) and produce distinct postsynaptic consequences (Sutton et al., 2006, 2007), raising the possibility that G2019S could be differentially affecting one or the other modes of vesicle release. To examine specifically effects of the G2019S mutation on spontaneous presynaptic vesicle release, we recorded mEPSCs at P21 after the addition of TTX (3 μm) into the bath to eliminate all action potential-mediated presynaptic activity. Although we found a slight elevation in frequency of mEPSCs in GSKI SPNs compared with the other genotypes, the difference was not significant (Fig. 4A,C). Moreover, there were no significant differences across genotypes in mEPSC amplitudes, consistent with the sEPSC data (Fig. 4B,C). Thus, these data indicate that the G2019S mutant LRRK2 affects action potential-generated activity in particular, with only slight and nonsignificant effects on spontaneous vesicle fusion.
Figure 4.
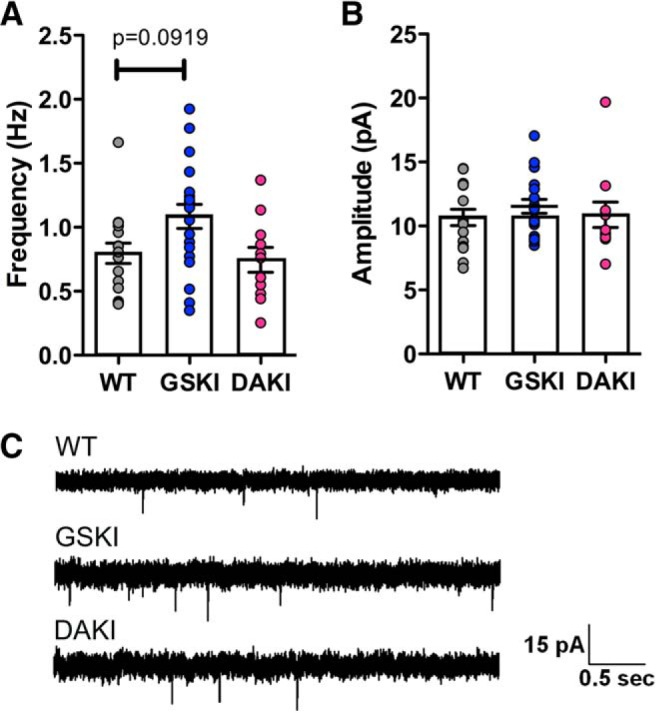
mEPSCs are not significantly altered in GSKI SPNs. Mixed bar graphs/scatter plots of averaged mEPSC frequencies (A) and amplitudes (B) at P21 from WT [n = 16 (7)] versus GSKI [n = 20 (8)] and DAKI [n = 12(4)] SPNs. There was a slight, but nonsignificant, increase in GSKI mEPSC frequency compared with WT (p = 0.0919, F = 2.87). There was no difference between DAKI and WT frequencies (p = 0.5095). B, No significant differences in mEPSC amplitudes were evident (WT vs GSKI, p = 0.3386; WT vs DAKI, p = 0.9183, F = 0.66). In A and B, a linear mixed effect model was used to compare groups (see Materials and Methods). C, Sample traces of mEPSCs recorded from SPNs of the indicated genotypes.
Corticostriatal afferents underlie elevated frequency of sEPSCs in GSKI mutants
We next sought to identify the source of the overly active glutamatergic afferents contributing to the elevated frequency of sEPSCs in the G2019S mutants. The vast majority of extrinsic glutamatergic inputs to striatum derive from neocortex and thalamus (Wilson et al., 1983; Gerfen, 1992), with a smaller glutamatergic contribution via corelease from intrinsic cholinergic neurons (Higley et al., 2011). In coronal slices, thalamostriatal inputs originate posteriorly and are removed, but some intact corticostriatal inputs are preserved (Centonze et al., 2004), which we confirmed by backlabeling cortical projection neurons after DiI placement in dorsal striatum (Fig. 5A). We first ruled out differences between genotypes in intrinsic excitability of SPNs; there were no differences between WT and GSKI SPNs in the number of action potentials elicited by depolarizing current steps (Fig. 5B). Additionally, there were no significant differences between genotypes in membrane capacitance [WT, μ = 21.73 ± 0.954 pF; GSKI, μ = 22.00 ± 1.210 pF, Student's t test, p = 0.860, F = 1.169; WT, n = 11 (3); GSKI, n = 9 (3)], resting membrane potential [WT, μ = −60.43 ± 1.131 mV; GSKI, μ = −61.57 ± 1.412 mV, Student's t test, p = 0.539, F = 1.559; WT, n = 7 (2); GSKI, n = 7 (2)], or action potential threshold [WT, μ = −27.9 ± 2.689 mV; GSKI, μ = −30.17 ± 1.237 mV, Student's t test, p = 0.756, F = 3.126; WT, n = 10 (6); GSKI, n = 7 (4)]. We next tested whether the elevated frequency of glutamatergic synaptic events in GSKI striatum reflected elevated activity of cortical inputs. We recorded sEPSCs from GSKI and WT SPNs in P21 acute coronal slices in which the striatum of one side was surgically isolated from the overlying cortex (Fig. 5C), thereby acutely disrupting cortical input as described previously (Smeal et al., 2007). sEPSC frequency recorded from GSKI SPNs on the surgically isolated side was lowered to WT levels and was significantly lower than that recorded from GSKI SPNs on the contralateral (intact) side of the same slices (Fig. 5D,E). In contrast, there were no differences in sEPSC frequency between the intact and isolated sides in WT slices, indicating that effects seen in the mutants were not attributable to an injury artifact (Fig. 5D,E). Together, these data suggest that corticostriatal activity is abnormally high in GSKI mice and contributes to the significantly elevated frequency of sEPSCs recorded from SPNs.
Figure 5.

Isolating dorsal striatum from cortex normalizes excessive frequency of sEPSCs in GSKI SPNs. A, Schematic showing the distribution of corticostriatal neurons (boxed region; yellow dots in the expanded view) retrogradely labeled by DiI crystals placed into dorsal striatum in coronal slices. Layers are indicated by dashed lines and labeled by roman numerals at the bottom. Photomicrograph of DiI backlabeled neurons is shown in inset (scale bar, 40 μm). B, Graph showing action potential firing properties of WT and GSKI SPNs in response to increasing current steps. There were no significant differences in numbers of action potentials elicited between WT (n = 8) and GSKI (n = 4) SPNs (two-way ANOVA, p = 0.467, F = 0.54 for genotype; p = 0.959, F = 0.20 for genotype and injected current interaction). C, Schematic showing scalpel cuts (dotted red lines) used to acutely isolate the striatum of one side (isolated side) from the overlying neocortex. The contralateral side was left intact (intact side); whole-cell recordings were made from both sides (position indicated by pipettes). D, Mixed bar graph/scatter plots showing average frequency of sEPSCs from WT and GSKI SPNs in isolated and intact sides. Two-way ANOVA, **p = 0.0143, F = 7.68 for genotype interaction in isolated versus intact; ***p = 0.0005, F = 19.48 for decortication effect, GSKI versus WT, n = 9 (4) per genotype (WT and GSKI) and condition (isolated and intact). E, Sample traces showing WT and GSKI sEPSCs recorded from SPNs in intact or isolated sides.
Increased frequency of sEPSCs is not accounted for by changes in synapse or spine number
The increased frequency of sEPSCs in GSKI mice could reflect a greater number of excitatory synapses. We tested this possibility in three complementary ways. First, we compared the density of presynaptic and postsynaptic glutamatergic molecular markers in immunolabeled sections through dorsal striatum across all genotypes at P21. Immunolabeling for VGluT1 was used to label corticostriatal terminals, VGluT2 to label thalamostriatal terminals (Fujiyama et al., 2004; Raju et al., 2006), and PSD95 to label all glutamatergic postsynaptic sites. There were no significant differences in the density of VGluT1-, VGluT2-, or PSD95-labeled puncta or in the average puncta area across genotypes (Fig. 6A). Second, we evaluated protein levels of these synaptic molecular markers by Western blot of synaptoneurosomes prepared from P21 dorsal striatum. Consistent with the immunolabeling data, we found no changes in levels of these synaptic proteins across genotypes (Fig. 6B). Third, because corticostriatal afferents predominantly target dendritic spines (Kreitzer and Malenka, 2008), we compared between GSKI and WT mice at P21 the density of protrusions (spines and filopodia) on SPNs after biocytin filling (Fig. 6C). Spines were distinguished from filopodia based on length and width (protrusions were classified as spines if they were <2 μm in length and >300 nm in width; Yuste and Bonhoeffer, 2004). In accordance with our immunohistochemical and biochemical findings, there were no differences between genotypes in the overall density of dendritic spines (Fig. 6C, left) or filopodia (Fig. 6C, right). Additionally, there were no differences in average total dendritic length per cell between genotypes (Fig. 6D). Together, these data indicate that there are no changes in synapse number in GSKI SPNs.
Figure 6.

G2019S SPNs exhibit altered spine morphology and distribution of postsynaptic response amplitudes but no change in synapse/spine density. A, Representative images of sections through P21 dorsal striatum immunofluorescently labeled for synaptic markers PSD95 (top, green), VGluT1 (middle, blue), and VGluT2 (bottom, red) taken from the indicated genotypes. Scale bar, 10 μm. Bar graphs of puncta area and density for each marker. There were no significant differences between genotypes, one-way ANOVA, n = 3–5 animals per genotype, p > 0.05. B, Western blots of synaptoneurosomes prepared from P21 striatum showing protein levels of PSD95, VGluT1, and VGluT2 from the indicated genotypes and actin, which was used as a loading control. There were no significant differences between genotypes. One-way ANOVA, n = 3 mice per genotype, p > 0.05 for all proteins. C, Bar graphs show average spine density (left) or average filopodia density (right). For classification of spines and filopodia, see Results. There were no significant differences in density between WT and GSKI SPNs [spines and filopodia, Student's t test, p = 0.214 (F = 12.19) and p = 0.460 (F = 2.632), respectively, n = 7 (5) for WT and 8 (3) for GSKI]. D, Bar graph of average total dendrite length per cell in WT and GSKI SPNs. There were no significant differences; Student's t test, p = 0.56, F = 1.275, n = 12 cells for WT and 10 cells for GSKI. E, Bar graph of average spine-head widths show significantly increased width in GSKI neurons; Student's t test, **p = 0.006, F = 3.154, n = 7 WT neurons and 8 GSKI neurons. At right are examples of deconvolved confocal image Z-stacks of biocytin-filled, Alexa Fluor 594-labeled GSKI and WT SPN dendrite segments; scale bar, 4 μm. F, Cumulative probability distributions of spine-head widths in SPNs from WT (gray line) and GSKI (blue line) mice. Rightward shift is significant, ***Kolmogorov–Smirnov test, p = 0.0001; n = 7 (5) WT and 8 (3) GSKI. Inset, Binned data of spine-head widths. G, Cumulative probability distributions of sEPSC amplitudes of the first 50 events per cell from WT (gray) or GSKI (blue) SPNs. Rightward shift is significant, ***Kolmogorov–Smirnov test, p = 0.0001, n = 15 (5) for WT and 12(6) for GSKI. Inset, Binned data of all of the events.
Dendritic spine-head size and distribution of postsynaptic responses are abnormal in GSKI mutants
Strikingly, however, the G2019S mutation significantly increased postsynaptic dendritic spine-head size compared with WT (Fig. 6E,F). Histograms and cumulative probability analysis demonstrated that GSKI SPNs display dendritic spine-head widths that were consistently and significantly shifted toward larger values compared with WT SPNs (Fig. 6F). Because excitatory postsynaptic responses are proportional to spine-head size (Harris and Stevens, 1989; Takumi et al., 1999; Matsuzaki et al., 2004; Segal, 2005), we asked whether there was any functional correlation between abnormal spine size and excitatory responses in GSKI SPNs. Comparisons of histograms and cumulative probability plots of sEPSC amplitudes also showed a rightward shift in GSKI SPNs compared with WT responses (Fig. 6G), indicating an increased probability of larger current amplitudes in G2019S mutant SPNs. Together, these data reveal coordinated presynaptic and postsynaptic structural and functional changes to developing G2019S striatal circuits.
Discussion
Here we provide evidence for robust gain-of-abnormal function phenotypes early in postnatal striatal circuit development caused by the PD-associated G2019S mutation in LRRK2. These abnormalities affect developing neural circuits that participate in the generation of PD-related symptoms later in life. Three principal findings emerged. First, in G2019S mutant SPNs, there is an abnormal and highly significant LRRK2 kinase-dependent increase in frequency of EPSCs. Action potential-dependent activity is particularly high, appears to arise principally from corticostriatal afferents, and peaks at P21. Second, dendritic spine-head sizes are larger in G2019S mutant SPNs, a morphological change that is correlated with a shift toward larger postsynaptic responses. In contrast, synapse density and dendritic length in GSKI mice are unchanged. Third, endogenous LRRK2 kinase activity per se is not necessary for normal spontaneous excitatory synaptic neurotransmission, because there was no effect on sEPSCs in SPNs when kinase activity of LRRK2 was ablated genetically or inhibited pharmacologically. Together, our data indicate that the G2019S mutation imparts a unique gain-of-abnormal function to striatal excitatory circuits during a critical stage of development when heightened activity can permanently affect striatal circuit structure and function (Andersen, 2003; Kozorovitskiy et al., 2012; Molero et al., 2016; Peixoto et al., 2016).
The abnormally elevated frequency of EPSCs observed in GSKI SPNs at P21 reflects mostly action potential-dependent events (sEPSCs) rather than mEPSCs. This distinction is revealing because action potential-driven events and spontaneous miniature events (minis) can involve different pools of presynaptic vesicles (Andreae et al., 2012; Bal et al., 2013), activate different postsynaptic targets (Atasoy et al., 2008), and produce distinct signaling outcomes at postsynaptic sites (Sutton et al., 2006, 2007; Kavalali, 2015). Action potential-driven fusion demands a rapid and steep rise in calcium coupled to the actions of the canonical SNARE complex generated by Synaptobrevin 2, Synaptotagmin 1, and synaptosomal-associated protein of 25 kDa (SNAP25; Kavalali, 2015), outlining a restricted mechanistic framework in which LRRK2–G2019S is likely to act.
Our data indicate that the abnormally elevated sEPSC frequency in GSKI SPNs is caused by enhanced kinase activity. Kinase-dead DAKI SPNs displayed normal sEPSC frequency, and a LRRK2 kinase inhibitor normalized the excessive EPSC frequency in GSKI neurons. The effect is most likely mediated presynaptically, because acutely removing cortical afferents normalized postsynaptic responses and GSKI SPNs displayed normal densities of dendritic spines and synaptic molecular markers. There is a strong precedent for LRRK2 to regulate presynaptic terminal function; results from LRRK2 deletion, knockdown, or loss-of-function studies collectively support that the entire multifunctional LRRK2 protein can regulate synaptic vesicle endocytosis and/or exocytosis (Shin et al., 2008; Matta et al., 2012; Yun et al., 2013; Piccoli et al., 2014; Arranz et al., 2015). However, there is no consensus on the general mechanism(s) of action, and even less clear are the relevant molecular substrates for normal or mutant (G2019S) LRRK2 kinase activity because the majority of targets identified in vitro have not been confirmed as targets under physiological conditions. A recent report indicates that LRRK2 can phosphorylate N-ethylmaleimide-sensitive factor (NSF) in vitro (Belluzzi et al., 2016), an event that would be expected to promote SNARE dissociation after fusion and one that would be consistent with the increased glutamatergic activity seen in GSKI SPNs. Conversely, similarly convincing in vitro data have shown that LRRK2 can phosphorylate SNAPIN (SNAP associated protein), a protein that sequesters SNAP25 and negatively regulates fusion (Yun et al., 2013). Neither NSF nor SNAPIN has been shown to be phosphorylated by overexpressed or endogenously expressed LRRK2 in cells. Currently, the most plausible presynaptic candidate substrate for mediating the effects shown here is Rab3a, a GTPase that purifies with synaptic vesicles and promotes exocytosis (Pavlos et al., 2010). Although Rab3a has not yet been shown to be a bona fide substrate of endogenously expressed LRRK2 or LRRK2–G2019S, recent work shows that Rab3a is a substrate for LRRK2 in cells and in vitro (Steger et al., 2016) and provides strong evidence that similar Thr sites in Rab8 and Rab10 are phosphorylated by LRRK2 in an endogenous context (Steger et al., 2016). Significantly, Rab3a is expressed in a subpopulation of synaptic vesicles (Pavlos et al., 2010) and can selectively modulate action potential-driven vesicle release (Feliciano et al., 2013). Thus, G2019S may be affecting action potential-dependent activity in particular through Rab3a.
Our undercut experiments suggest that corticostriatal afferents are the likely source of elevated synaptic activity in GSKI SPNs, although other sources of glutamatergic input (e.g., thalamus) cannot be ruled out ex vivo. Cortical neurons in layer 5, the source of the majority of corticostriatal inputs (Wise and Jones, 1977; Shepherd, 2013; Sohur et al., 2014), also express LRRK2 (Taymans et al., 2006; Davies et al., 2013; Giesert et al., 2013; West et al., 2014), suggesting that abnormal kinase activity within corticostriatal projection neurons acts to increase their synaptic drive onto SPNs. Consistent with this idea, dissociated cortical neurons cultured from G2019S–KI or G2019S–BAC transgenic mice show elevated spontaneous activity (Beccano-Kelly et al., 2014; Belluzzi et al., 2016).
The data also show a significant postsynaptic consequence of G2019S expression in SPNs. Mutant SPNs display significantly larger spine-head widths and sEPSC magnitudes compared with WT SPNs. These data are consistent with the wider range of action potential-driven activity on GSKI SPNs and indicate that there is both cellular and synaptic heterogeneity in G2019S-mediated outcomes. It remains to be determined whether such aberrant spine morphology reflects direct postsynaptic effects of mutant LRRK2, but a postsynaptic locus of LRRK2 activity would be consistent with anatomical studies in mouse and monkey striatum showing LRRK2 protein localization in cell bodies and dendrites (Aarsland et al., 2009; Lee et al., 2010a; West et al., 2014) and increased proportions of thin compared with mushroom-shaped dendritic protrusions in LRRK2 knock-out mice at P15 (Parisiadou et al., 2014). Spine/synapse size and synaptic response magnitude are tightly correlated but independently regulated (Shi et al., 1999; Zito et al., 2004; Yang et al., 2008), and LRRK2 G2019S could conceivably be regulating either or both pathways. LRRK2 could control spine shape via its interactions with the ERM family, cofilin, or other actin regulatory proteins suspected to be substrates of LRRK2 (Jaleel et al., 2007; Parisiadou et al., 2009, 2014; Meixner et al., 2011). It could also affect postsynaptic receptor recycling via Rab10, a confirmed LRRK2 substrate (Steger et al., 2016) that regulates AMPA receptor recycling in C. elegans (Glodowski et al., 2007). Consistent with this idea, a recent study showed that LRRK2–G2019S overexpression disrupts hippocampal LTD by a mechanism that may involve impaired GluR recycling (Sweet et al., 2015). Alternatively, altered spine shape and amplitude in the GSKI mutants could be an indirect effect of the abnormally elevated excitatory synaptic activity, because synaptic activity generally plays a large role in determining spine shape (Yuste and Bonhoeffer, 2001).
Although spine morphology was altered in G2019S SPNs, there were no differences between G2019S and WT SPNs in dendritic length or glutamatergic synapse density. Several laboratories have shown that overexpression of LRRK2–G2019S can reduce neurite growth (MacLeod et al., 2006; Parisiadou et al., 2009; Dächsel et al., 2010; Ramonet et al., 2011; Winner et al., 2011; Sepulveda et al., 2013). However, this phenotype appears to be attributable at least in part to overexpression (Skibinski et al., 2014) because changes in neither dendrite length nor synapse density have been reported in cultured hippocampal or cortical neurons expressing physiological levels of LRRK2–G2019S (Dächsel et al., 2010; Beccano-Kelly et al., 2014), consistent with results reported here.
Our data reveal clearly abnormal neurobiological outcomes selectively attributable to a prevalent PD-related LRRK2 mutation early in postnatal life. The abnormally elevated synaptic activity seen at P21 in G2019S SPNs was not yet evident at P14. Differences in maturation between cortical and thalamic innervation may account for this because thalamic afferents are present at birth (Vercelli et al., 2003), whereas cortical inputs, which grow into striatum in early postnatal life (Sohur et al., 2014), are still immature, both structurally and functionally, at P14 (Peixoto et al., 2016). High levels of synaptic activity observed at P21 were also not sustained into adulthood. There is a strong propensity for neural circuits to undergo homeostatic adjustments to normalize activity levels, thereby tuning neuronal responses to activity patterns (Turrigiano, 2008). Such mechanistic adjustments are likely involved here at later ages. Nevertheless, abnormal timing or patterns of activity early in developing striatal, visual cortical, and other circuits profoundly disrupt circuit organization, synapse plasticity, and behaviors later in life (Andersen, 2003; Ehlers and Criado, 2010; Marco et al., 2011; Kozorovitskiy et al., 2012; Archer and Garcia, 2015; Greenhill et al., 2015; Molero et al., 2016; Peixoto et al., 2016) and are consistent with recent fMRI studies showing that presymptomatic human LRRK2 G2019S carriers display reorganized prefrontal–striatal circuits (Helmich et al., 2015), changes in task-related activity during cognitive testing (Thaler et al., 2013), and decreased executive function suggested by impaired performance on the Stroop test compared with noncarrier controls recruited from the same families (Thaler et al., 2016). Additional investigation will reveal precisely how the dramatic but transient alterations in excitatory circuitry observed in G2019S SPNs have a lasting effect on circuit function and striatally based behaviors.
Footnotes
This work was supported by National Institute of Mental Health Grant MH103455, Predoctoral T32 Training Grant MH096678 (B.A.M.-A.), and Fellowship F31NS096892 (B.A.M.-A.); the Michael J. Fox Foundation; the Tisch Cancer Institute at Mount Sinai (Cancer Center Support Grant P30 CA196521); and a postdoctoral fellowship from the American Parkinson's Disease Association (N.K.). We thank Chenel Morrison and Yue Zhong for their excellent technical assistance, Drs. Kalpana Merchant and Patrick May for their efforts and initiative in the early phases of this project, Casey Lardner for her editorial assistance, and Crystal Pristell and Dr. Nikolaos Tzavaras of the Microscopy Core at Mount Sinai for imaging and analysis support. Last, we thank the Clinical and Translational Science Award funded (UL1TR001433) Biostatistics, Epidemiology, and Research Design Program at Mount Sinai for assistance in statistical analysis.
The authors declare no competing financial interests.
References
- Aarsland D, Brønnick K, Larsen JP, Tysnes OB, Alves G. Cognitive impairment in incident, untreated Parkinson disease: the Norwegian ParkWest study. Neurology. 2009;72:1121–1126. doi: 10.1212/01.wnl.0000338632.00552.cb. [DOI] [PubMed] [Google Scholar]
- Andersen SL. Trajectories of brain development: point of vulnerability or window of opportunity? Neurosci Biobehav Rev. 2003;27:3–18. doi: 10.1016/S0149-7634(03)00005-8. [DOI] [PubMed] [Google Scholar]
- Andreae LC, Fredj NB, Burrone J. Independent vesicle pools underlie different modes of release during neuronal development. J Neurosci. 2012;32:1867–1874. doi: 10.1523/JNEUROSCI.5181-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer T, Garcia D. Attention-deficit/hyperactivity disorder: focus upon aberrant N-methyl-d-aspartate receptors systems. Curr Top Behav Neurosci. 2015 doi: 10.1007/7854_2015_415. doi: 10.1007/7854_2015_415. Advance online publication. Retrieved June 1, 2015. [DOI] [PubMed] [Google Scholar]
- Arranz AM, Delbroek L, Van Kolen K, Guimarães MR, Mandemakers W, Daneels G, Matta S, Calafate S, Shaban H, Baatsen P, De Bock PJ, Gevaert K, Vanden Berghe P, Verstreken P, De Strooper B, Moechars D. LRRK2 functions in synaptic vesicle endocytosis through a kinase-dependent mechanism. J Cell Sci. 2015;128:541–552. doi: 10.1242/jcs.158196. [DOI] [PubMed] [Google Scholar]
- Atasoy D, Ertunc M, Moulder KL, Blackwell J, Chung C, Su J, Kavalali ET. Spontaneous and evoked glutamate release activates two populations of NMDA receptors with limited overlap. J Neurosci. 2008;28:10151–10166. doi: 10.1523/JNEUROSCI.2432-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal M, Leitz J, Reese AL, Ramirez DM, Durakoglugil M, Herz J, Monteggia LM, Kavalali ET. Reelin mobilizes a VAMP7-dependent synaptic vesicle pool and selectively augments spontaneous neurotransmission. Neuron. 2013;80:934–946. doi: 10.1016/j.neuron.2013.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beccano-Kelly DA, Kuhlmann N, Tatarnikov I, Volta M, Munsie LN, Chou P, Cao LP, Han H, Tapia L, Farrer MJ, Milnerwood AJ. Synaptic function is modulated by LRRK2 and glutamate release is increased in cortical neurons of G2019S LRRK2 knock-in mice. Front Cell Neurosci. 2014;8:301. doi: 10.3389/fncel.2014.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beccano-Kelly DA, Volta M, Munsie LN, Paschall SA, Tatarnikov I, Co K, Chou P, Cao LP, Bergeron S, Mitchell E, Han H, Melrose HL, Tapia L, Raymond LA, Farrer MJ, Milnerwood AJ. LRRK2 overexpression alters glutamatergic presynaptic plasticity, striatal dopamine tone, postsynaptic signal transduction, motor activity and memory. Hum Mol Genet. 2015;24:1336–1349. doi: 10.1093/hmg/ddu543. [DOI] [PubMed] [Google Scholar]
- Belluzzi E, Gonnelli A, Cirnaru MD, Marte A, Plotegher N, Russo I, Civiero L, Cogo S, Carrion MP, Franchin C, Arrigoni G, Beltramini M, Bubacco L, Onofri F, Piccoli G, Greggio E. LRRK2 phosphorylates pre-synaptic N-ethylmaleimide sensitive fusion (NSF) protein enhancing its ATPase activity and SNARE complex disassembling rate. Mol Neurodegener. 2016;11:1. doi: 10.1186/s13024-015-0066-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biskup S, Moore DJ, Rea A, Lorenz-Deperieux B, Coombes CE, Dawson VL, Dawson TM, West AB. Dynamic and redundant regulation of LRRK2 and LRRK1 expression. BMC Neurosci. 2007;8:102. doi: 10.1186/1471-2202-8-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze D, Usiello A, Costa C, Picconi B, Erbs E, Bernardi G, Borrelli E, Calabresi P. Chronic haloperidol promotes corticostriatal long-term potentiation by targeting dopamine D2L receptors. J Neurosci. 2004;24:8214–8222. doi: 10.1523/JNEUROSCI.1274-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson's disease. Nat Rev Neurosci. 2010;11:791–797. doi: 10.1038/nrn2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dächsel JC, Behrouz B, Yue M, Beevers JE, Melrose HL, Farrer MJ. A comparative study of Lrrk2 function in primary neuronal cultures. Parkinsonism Relat Disord. 2010;16:650–655. doi: 10.1016/j.parkreldis.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies P, Hinkle KM, Sukar NN, Sepulveda B, Mesias R, Serrano G, Alessi DR, Beach TG, Benson DL, White CL, Cowell RM, Das SS, West AB, Melrose HL. Comprehensive characterization and optimization of anti-LRRK2 (leucine-rich repeat kinase 2) monoclonal antibodies. Biochem J. 2013;453:101–113. doi: 10.1042/BJ20121742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fonzo A, Rohé CF, Ferreira J, Chien HF, Vacca L, Stocchi F, Guedes L, Fabrizio E, Manfredi M, Vanacore N, Goldwurm S, Breedveld G, Sampaio C, Meco G, Barbosa E, Oostra BA, Bonifati V. A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson's disease. Lancet. 2005;365:412–415. doi: 10.1016/S0140-6736(05)17829-5. [DOI] [PubMed] [Google Scholar]
- Dong Y, Green T, Saal D, Marie H, Neve R, Nestler EJ, Malenka RC. CREB modulates excitability of nucleus accumbens neurons. Nat Neurosci. 2006;9:475–477. doi: 10.1038/nn1661. [DOI] [PubMed] [Google Scholar]
- Ehlers CL, Criado JR. Adolescent ethanol exposure: does it produce long-lasting electrophysiological effects? Alcohol. 2010;44:27–37. doi: 10.1016/j.alcohol.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erro R, Santangelo G, Picillo M, Vitale C, Amboni M, Longo K, Costagliola A, Pellecchia MT, Allocca R, De Rosa A, De Michele G, Santoro L, Barone P. Link between non-motor symptoms and cognitive dysfunctions in de novo, drug-naive PD patients. J Neurol. 2012;259:1808–1813. doi: 10.1007/s00415-011-6407-0. [DOI] [PubMed] [Google Scholar]
- Estrada AA, Liu X, Baker-Glenn C, Beresford A, Burdick DJ, Chambers M, Chan BK, Chen H, Ding X, DiPasquale AG, Dominguez SL, Dotson J, Drummond J, Flagella M, Flynn S, Fuji R, Gill A, Gunzner-Toste J, Harris SF, Heffron TP, Kleinheinz T, et al. Discovery of highly potent, selective, and brain-penetrable leucine-rich repeat kinase 2 (LRRK2) small molecule inhibitors. J Med Chem. 2012;55:9416–9433. doi: 10.1021/jm301020q. [DOI] [PubMed] [Google Scholar]
- Feliciano P, Andrade R, Bykhovskaia M. Synapsin II and Rab3a cooperate in the regulation of epileptic and synaptic activity in the CA1 region of the hippocampus. J Neurosci. 2013;33:18319–18330. doi: 10.1523/JNEUROSCI.5293-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Glowinski J, Venance L. Effects of acute dopamine depletion on the electrophysiological properties of striatal neurons. Neurosci Res. 2007;58:305–316. doi: 10.1016/j.neures.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Friedman LG, Riemslagh FW, Sullivan JM, Mesias R, Williams FM, Huntley GW, Benson DL. Cadherin-8 expression, synaptic localization, and molecular control of neuronal form in prefrontal corticostriatal circuits. J Comp Neurol. 2015;523:75–92. doi: 10.1002/cne.23666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiyama F, Kuramoto E, Okamoto K, Hioki H, Furuta T, Zhou L, Nomura S, Kaneko T. Presynaptic localization of an AMPA-type glutamate receptor in corticostriatal and thalamostriatal axon terminals. Eur J Neurosci. 2004;20:3322–3330. doi: 10.1111/j.1460-9568.2004.03807.x. [DOI] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 1992;15:133–139. doi: 10.1016/0166-2236(92)90355-C. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giesert F, Hofmann A, Bürger A, Zerle J, Kloos K, Hafen U, Ernst L, Zhang J, Vogt-Weisenhorn DM, Wurst W. Expression analysis of Lrrk1, Lrrk2 and Lrrk2 splice variants in mice. PLoS One. 2013;8:e63778. doi: 10.1371/journal.pone.0063778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glodowski DR, Chen CC, Schaefer H, Grant BD, Rongo C. RAB-10 regulates glutamate receptor recycling in a cholesterol-dependent endocytosis pathway. Mol Biol Cell. 2007;18:4387–4396. doi: 10.1091/mbc.E07-05-0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M. Parkinson's disease and other alpha-synucleinopathies. Clin Chem Lab Med. 2001;39:308–312. doi: 10.1515/CCLM.2001.047. [DOI] [PubMed] [Google Scholar]
- Greenhill SD, Juczewski K, de Haan AM, Seaton G, Fox K, Hardingham NR. Neurodevelopment. Adult cortical plasticity depends on an early postnatal critical period. Science. 2015;349:424–427. doi: 10.1126/science.aaa8481. [DOI] [PubMed] [Google Scholar]
- Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Harris KM, Stevens JK. Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J Neurosci. 1989;9:2982–2997. doi: 10.1523/JNEUROSCI.09-08-02982.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy DG, Falchi M, O'Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S, Ferreira JJ, Tolosa E, Kay DM, Klein C, Williams DR, Marras C, Lang AE, Wszolek ZK, Berciano J, Schapira AH, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol. 2008;7:583–590. doi: 10.1016/S1474-4422(08)70117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmich RC, Thaler A, van Nuenen BF, Gurevich T, Mirelman A, Marder KS, Bressman S, Orr-Urtreger A, Giladi N, Bloem BR, Toni I. Reorganization of corticostriatal circuits in healthy G2019S LRRK2 carriers. Neurology. 2015;84:399–406. doi: 10.1212/WNL.0000000000001189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig MC, Kolly C, Persohn E, Theil D, Schweizer T, Hafner T, Stemmelen C, Troxler TJ, Schmid P, Danner S, Schnell CR, Mueller M, Kinzel B, Grevot A, Bolognani F, Stirn M, Kuhn RR, Kaupmann K, van der Putten PH, Rovelli G, Shimshek DR. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum Mol Genet. 2011;20:4209–4223. doi: 10.1093/hmg/ddr348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, Gittis AH, Oldenburg IA, Balthasar N, Seal RP, Edwards RH, Lowell BB, Kreitzer AC, Sabatini BL. Cholinergic interneurons mediate fast VGluT3-dependent glutamatergic transmission in the striatum. PLoS One. 2011;6:e19155. doi: 10.1371/journal.pone.0019155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito G, Fujimoto T, Kamikawaji S, Kuwahara T, Iwatsubo T. Lack of correlation between the kinase activity of LRRK2 harboring kinase-modifying mutations and its phosphorylation at Ser910, 935, and Ser955. PLoS One. 2014;9:e97988. doi: 10.1371/journal.pone.0097988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaleel M, Nichols RJ, Deak M, Campbell DG, Gillardon F, Knebel A, Alessi DR. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J. 2007;405:307–317. doi: 10.1042/BJ20070209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LN, Noble ME, Owen DJ. Active and inactive protein kinases: structural basis for regulation. Cell. 1996;85:149–158. doi: 10.1016/S0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- Kavalali ET. The mechanisms and functions of spontaneous neurotransmitter release. Nat Rev Neurosci. 2015;16:5–16. doi: 10.1038/nrn3875. [DOI] [PubMed] [Google Scholar]
- Khan NL, Jain S, Lynch JM, Pavese N, Abou-Sleiman P, Holton JL, Healy DG, Gilks WP, Sweeney MG, Ganguly M, Gibbons V, Gandhi S, Vaughan J, Eunson LH, Katzenschlager R, Gayton J, Lennox G, Revesz T, Nicholl D, Bhatia KP, et al. Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson's disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain. 2005;128:2786–2796. doi: 10.1093/brain/awh667. [DOI] [PubMed] [Google Scholar]
- Kozorovitskiy Y, Saunders A, Johnson CA, Lowell BB, Sabatini BL. Recurrent network activity drives striatal synaptogenesis. Nature. 2012;485:646–650. doi: 10.1038/nature11052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Striatal plasticity and basal ganglia circuit function. Neuron. 2008;60:543–554. doi: 10.1016/j.neuron.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Melrose HL, Yue M, Pare JF, Farrer MJ, Smith Y. Lrrk2 localization in the primate basal ganglia and thalamus: a light and electron microscopic analysis in monkeys. Exp Neurol. 2010a;224:438–447. doi: 10.1016/j.expneurol.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Liu HP, Lin WY, Guo H, Lu B. LRRK2 kinase regulates synaptic morphology through distinct substrates at the presynaptic and postsynaptic compartments of the Drosophila neuromuscular junction. J Neurosci. 2010b;30:16959–16969. doi: 10.1523/JNEUROSCI.1807-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Patel JC, Wang J, Avshalumov MV, Nicholson C, Buxbaum JD, Elder GA, Rice ME, Yue Z. Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson's disease mutation G2019S. J Neurosci. 2010;30:1788–1797. doi: 10.1523/JNEUROSCI.5604-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Kang S, Ray S, Jackson J, Zaitsev AD, Gerber SA, Cuny GD, Glicksman MA. Kinetic, mechanistic, and structural modeling studies of truncated wild-type leucine-rich repeat kinase 2 and the G2019S mutant. Biochemistry. 2011;50:9399–9408. doi: 10.1021/bi201173d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Marco EM, Macrì S, Laviola G. Critical age windows for neurodevelopmental psychiatric disorders: evidence from animal models. Neurotox Res. 2011;19:286–307. doi: 10.1007/s12640-010-9205-z. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta S, Van Kolen K, da Cunha R, van den Bogaart G, Mandemakers W, Miskiewicz K, De Bock PJ, Morais VA, Vilain S, Haddad D, Delbroek L, Swerts J, Chávez-Gutiérrez L, Esposito G, Daneels G, Karran E, Holt M, Gevaert K, Moechars DW, De Strooper B, Verstreken P, et al. LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron. 2012;75:1008–1021. doi: 10.1016/j.neuron.2012.08.022. [DOI] [PubMed] [Google Scholar]
- Meixner A, Boldt K, Van Troys M, Askenazi M, Gloeckner CJ, Bauer M, Marto JA, Ampe C, Kinkl N, Ueffing M. A QUICK screen for Lrrk2 interaction partners—leucine-rich repeat kinase 2 is involved in actin cytoskeleton dynamics. Mol Cell Proteomics. 2011;10:M110.001172. doi: 10.1074/mcp.M110.001172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molero AE, Arteaga-Bracho EE, Chen CH, Gulinello M, Winchester ML, Pichamoorthy N, Gokhan S, Khodakhah K, Mehler MF. Selective expression of mutant huntingtin during development recapitulates characteristic features of Huntington's disease. Proc Natl Acad Sci U S A. 2016;113:5736–5741. doi: 10.1073/pnas.1603871113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols RJ, Dzamko N, Hutti JE, Cantley LC, Deak M, Moran J, Bamborough P, Reith AD, Alessi DR. Substrate specificity and inhibitors of LRRK2, a protein kinase mutated in Parkinson's disease. Biochem J. 2009;424:47–60. doi: 10.1042/BJ20091035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols RJ, Dzamko N, Morrice NA, Campbell DG, Deak M, Ordureau A, Macartney T, Tong Y, Shen J, Prescott AR, Alessi DR. 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson's disease-associated mutations and regulates cytoplasmic localization. Biochem J. 2010;430:393–404. doi: 10.1042/BJ20100483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikitczuk JS, Patil SB, Matikainen-Ankney BA, Scarpa J, Shapiro ML, Benson DL, Huntley GW. N-cadherin regulates molecular organization of excitatory and inhibitory synaptic circuits in adult hippocampus in vivo. Hippocampus. 2014;24:943–962. doi: 10.1002/hipo.22282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolen B, Taylor S, Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell. 2004;15:661–675. doi: 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Ozelius LJ, Senthil G, Saunders-Pullman R, Ohmann E, Deligtisch A, Tagliati M, Hunt AL, Klein C, Henick B, Hailpern SM, Lipton RB, Soto-Valencia J, Risch N, Bressman SB. LRRK2 G2019S as a cause of Parkinson's disease in Ashkenazi Jews. N Engl J Med. 2006;354:424–425. doi: 10.1056/NEJMc055509. [DOI] [PubMed] [Google Scholar]
- Paisán-Ruiz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, López de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Martí-Massó JF, Pérez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Paisán-Ruiz C, Lewis PA, Singleton AB. LRRK2: cause, risk, and mechanism. J Parkinsons Dis. 2013;3:85–103. doi: 10.3233/JPD-130192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisiadou L, Xie C, Cho HJ, Lin X, Gu XL, Long CX, Lobbestael E, Baekelandt V, Taymans JM, Sun L, Cai H. Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J Neurosci. 2009;29:13971–13980. doi: 10.1523/JNEUROSCI.3799-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisiadou L, Yu J, Sgobio C, Xie C, Liu G, Sun L, Gu XL, Lin X, Crowley NA, Lovinger DM, Cai H. LRRK2 regulates synaptogenesis and dopamine receptor activation through modulation of PKA activity. Nat Neurosci. 2014;17:367–376. doi: 10.1038/nn.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlos NJ, Grønborg M, Riedel D, Chua JJ, Boyken J, Kloepper TH, Urlaub H, Rizzoli SO, Jahn R. Quantitative analysis of synaptic vesicle Rabs uncovers distinct yet overlapping roles for Rab3a and Rab27b in Ca2+-triggered exocytosis. J Neurosci. 2010;30:13441–13453. doi: 10.1523/JNEUROSCI.0907-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto RT, Wang W, Croney DM, Kozorovitskiy Y, Sabatini BL. Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B(−/−) mice. Nat Neurosci. 2016;19:716–724. doi: 10.1038/nn.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccoli G, Condliffe SB, Bauer M, Giesert F, Boldt K, De Astis S, Meixner A, Sarioglu H, Vogt-Weisenhorn DM, Wurst W, Gloeckner CJ, Matteoli M, Sala C, Ueffing M. LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. J Neurosci. 2011;31:2225–2237. doi: 10.1523/JNEUROSCI.3730-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccoli G, Onofri F, Cirnaru MD, Kaiser CJ, Jagtap P, Kastenmüller A, Pischedda F, Marte A, von Zweydorf F, Vogt A, Giesert F, Pan L, Antonucci F, Kiel C, Zhang M, Weinkauf S, Sattler M, Sala C, Matteoli M, Ueffing M, Gloeckner CJ. Leucine-rich repeat kinase 2 binds to neuronal vesicles through protein interactions mediated by its C-terminal WD40 domain. Mol Cell Biol. 2014;34:2147–2161. doi: 10.1128/MCB.00914-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju DV, Shah DJ, Wright TM, Hall RA, Smith Y. Differential synaptology of vGluT2-containing thalamostriatal afferents between the patch and matrix compartments in rats. J Comp Neurol. 2006;499:231–243. doi: 10.1002/cne.21099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramonet D, Daher JP, Lin BM, Stafa K, Kim J, Banerjee R, Westerlund M, Pletnikova O, Glauser L, Yang L, Liu Y, Swing DA, Beal MF, Troncoso JC, McCaffery JM, Jenkins NA, Copeland NG, Galter D, Thomas B, Lee MK, Dawson TM, Dawson VL, Moore DJ. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One. 2011;6:e18568. doi: 10.1371/journal.pone.0018568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray S, Liu M. Current understanding of LRRK2 in Parkinson's disease: biochemical and structural features and inhibitor design. Future Med Chem. 2012;4:1701–1713. doi: 10.4155/fmc.12.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Ehlenberger DB, Hof PR, Wearne SL. Rayburst sampling, an algorithm for automated three-dimensional shape analysis from laser scanning microscopy images. Nat Protoc. 2006;1:2152–2161. doi: 10.1038/nprot.2006.313. [DOI] [PubMed] [Google Scholar]
- Sakaguchi-Nakashima A, Meir JY, Jin Y, Matsumoto K, Hisamoto N. LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr Biol. 2007;17:592–598. doi: 10.1016/j.cub.2007.01.074. [DOI] [PubMed] [Google Scholar]
- Sara Y, Virmani T, Deák F, Liu X, Kavalali ET. An isolated pool of vesicles recycles at rest and drives spontaneous neurotransmission. Neuron. 2005;45:563–573. doi: 10.1016/j.neuron.2004.12.056. [DOI] [PubMed] [Google Scholar]
- Segal M. Dendritic spines and long-term plasticity. Nat Rev Neurosci. 2005;6:277–284. doi: 10.1038/nrn1649. [DOI] [PubMed] [Google Scholar]
- Sepulveda B, Mesias R, Li X, Yue Z, Benson DL. Short- and long-term effects of LRRK2 on axon and dendrite growth. PLoS One. 2013;8:e61986. doi: 10.1371/journal.pone.0061986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe NA, Tepper JM. Postnatal development of excitatory synaptic input to the rat neostriatum: an electron microscopic study. Neuroscience. 1998;84:1163–1175. doi: 10.1016/S0306-4522(97)00583-6. [DOI] [PubMed] [Google Scholar]
- Shepherd GM. Corticostriatal connectivity and its role in disease. Nat Rev Neurosci. 2013;14:278–291. doi: 10.1038/nrn3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- Shiba M, Bower JH, Maraganore DM, McDonnell SK, Peterson BJ, Ahlskog JE, Schaid DJ, Rocca WA. Anxiety disorders and depressive disorders preceding Parkinson's disease: a case-control study. Mov Disord. 2000;15:669–677. doi: 10.1002/1531-8257(200007)15:4<669::AID-MDS1011>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Shin N, Jeong H, Kwon J, Heo HY, Kwon JJ, Yun HJ, Kim CH, Han BS, Tong Y, Shen J, Hatano T, Hattori N, Kim KS, Chang S, Seol W. LRRK2 regulates synaptic vesicle endocytosis. Exp Cell Res. 2008;314:2055–2065. doi: 10.1016/j.yexcr.2008.02.015. [DOI] [PubMed] [Google Scholar]
- Skibinski G, Nakamura K, Cookson MR, Finkbeiner S. Mutant LRRK2 toxicity in neurons depends on LRRK2 levels and synuclein but not kinase activity or inclusion bodies. J Neurosci. 2014;34:418–433. doi: 10.1523/JNEUROSCI.2712-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeal RM, Gaspar RC, Keefe KA, Wilcox KS. A rat brain slice preparation for characterizing both thalamostriatal and corticostriatal afferents. J Neurosci Methods. 2007;159:224–235. doi: 10.1016/j.jneumeth.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9:1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- Sohur US, Padmanabhan HK, Kotchetkov IS, Menezes JR, Macklis JD. Anatomic and molecular development of corticostriatal projection neurons in mice. Cereb Cortex. 2014;24:293–303. doi: 10.1093/cercor/bhs342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, Wachter S, Lorentzen E, Duddy G, Wilson S, Baptista MA, Fiske BK, Fell MJ, Morrow JA, Reith AD, Alessi DR, Mann M. Phosphoproteomics reveals that Parkinson's disease kinase LRRK2 regulates a subset of Rab GTPases. Elife. 2016:5. doi: 10.7554/eLife.12813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Taylor AM, Ito HT, Pham A, Schuman EM. Postsynaptic decoding of neural activity: eEF2 as a biochemical sensor coupling miniature synaptic transmission to local protein synthesis. Neuron. 2007;55:648–661. doi: 10.1016/j.neuron.2007.07.030. [DOI] [PubMed] [Google Scholar]
- Sweet ES, Saunier-Rebori B, Yue Z, Blitzer RD. The Parkinson's disease-associated mutation LRRK2–G2019S impairs synaptic plasticity in mouse hippocampus. J Neurosci. 2015;35:11190–11195. doi: 10.1523/JNEUROSCI.0040-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takumi Y, Ramírez-León V, Laake P, Rinvik E, Ottersen OP. Different modes of expression of AMPA and NMDA receptors in hippocampal synapses. Nat Neurosci. 1999;2:618–624. doi: 10.1038/10172. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Mata IF, Farrer MJ. LRRK2: a common pathway for parkinsonism, pathogenesis and prevention? Trends Mol Med. 2006;12:76–82. doi: 10.1016/j.molmed.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Taymans JM, Van den Haute C, Baekelandt V. Distribution of PINK1 and LRRK2 in rat and mouse brain. J Neurochem. 2006;98:951–961. doi: 10.1111/j.1471-4159.2006.03919.x. [DOI] [PubMed] [Google Scholar]
- Tepper JM, Sharpe NA, Koós TZ, Trent F. Postnatal development of the rat neostriatum: electrophysiological, light- and electron-microscopic studies. Dev Neurosci. 1998;20:125–145. doi: 10.1159/000017308. [DOI] [PubMed] [Google Scholar]
- Thaler A, Mirelman A, Helmich RC, van Nuenen BF, Rosenberg-Katz K, Gurevich T, Orr-Urtreger A, Marder K, Bressman S, Bloem BR, Giladi N, Hendler T. Neural correlates of executive functions in healthy G2019S LRRK2 mutation carriers. Cortex. 2013;49:2501–2511. doi: 10.1016/j.cortex.2012.12.017. [DOI] [PubMed] [Google Scholar]
- Thaler A, Helmich RC, Or-Borichev A, van Nuenen BF, Shapira-Lichter I, Gurevich T, Orr-Urtreger A, Marder K, Bressman S, Bloem BR, Giladi N, Hendler T, Mirelman A. Intact working memory in non-manifesting LRRK2 carriers—an fMRI study. Eur J Neurosci. 2016;43:106–112. doi: 10.1111/ejn.13120. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercelli A, Marini G, Tredici G. Anatomical organization of the telencephalic connections of the parafascicular nucleus in adult and developing rats. Eur J Neurosci. 2003;18:275–289. doi: 10.1046/j.1460-9568.2003.02743.x. [DOI] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- West AB, Cowell RM, Daher JP, Moehle MS, Hinkle KM, Melrose HL, Standaert DG, Volpicelli-Daley LA. Differential LRRK2 expression in the cortex, striatum, and substantia nigra in transgenic and nontransgenic rodents. J Comp Neurol. 2014;522:2465–2480. doi: 10.1002/cne.23583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerlund M, Belin AC, Anvret A, Bickford P, Olson L, Galter D. Developmental regulation of leucine-rich repeat kinase 1 and 2 expression in the brain and other rodent and human organs: implications for Parkinson's disease. Neuroscience. 2008;152:429–436. doi: 10.1016/j.neuroscience.2007.10.062. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Chang HT, Kitai ST. Origins of post synaptic potentials evoked in spiny neostriatal projection neurons by thalamic stimulation in the rat. Exp Brain Res. 1983;51:217–226. doi: 10.1007/BF00237197. [DOI] [PubMed] [Google Scholar]
- Winner B, Melrose HL, Zhao C, Hinkle KM, Yue M, Kent C, Braithwaite AT, Ogholikhan S, Aigner R, Winkler J, Farrer MJ, Gage FH. Adult neurogenesis and neurite outgrowth are impaired in LRRK2 G2019S mice. Neurobiol Dis. 2011;41:706–716. doi: 10.1016/j.nbd.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise SP, Jones EG. Cells of origin and terminal distribution of descending projections of the rat somatic sensory cortex. J Comp Neurol. 1977;175:129–157. doi: 10.1002/cne.901750202. [DOI] [PubMed] [Google Scholar]
- Yang Y, Wang XB, Frerking M, Zhou Q. Spine expansion and stabilization associated with long-term potentiation. J Neurosci. 2008;28:5740–5751. doi: 10.1523/JNEUROSCI.3998-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun HJ, Park J, Ho DH, Kim H, Kim CH, Oh H, Ga I, Seo H, Chang S, Son I, Seol W. LRRK2 phosphorylates Snapin and inhibits interaction of Snapin with SNAP-25. Exp Mol Med. 2013;45:e36. doi: 10.1038/emm.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat Rev Neurosci. 2004;5:24–34. doi: 10.1038/nrn1300. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Müller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Zito K, Knott G, Shepherd GM, Shenolikar S, Svoboda K. Induction of spine growth and synapse formation by regulation of the spine actin cytoskeleton. Neuron. 2004;44:321–334. doi: 10.1016/j.neuron.2004.09.022. [DOI] [PubMed] [Google Scholar]