

Abstract
The cellular processes that cause high caloric diet (HCD)-induced infertility are poorly understood but may involve upregulation of suppressor of cytokine signaling (SOCS-3) proteins that are associated with hypothalamic leptin resistance. Deletion of SOCS-3 from brain cells is known to protect mice from diet-induced obesity, but the effects on HCD-induced infertility are unknown. We used neuron-specific SOCS3 knock-out mice to elucidate this and the effects on regional hypothalamic leptin resistance. As expected, male and female neuron-specific SOCS3 knock-out mice were protected from HCD-induced obesity. While female wild-type mice became infertile after 4 months of HCD feeding, infertility onset in knock-out females was delayed by 4 weeks. Similarly, knock-out mice had delayed leptin resistance development in the medial preoptic area and anteroventral periventricular nucleus, regions important for generation of the surge of GnRH and LH that induces ovulation. We therefore tested whether the suppressive effects of HCD on the estradiol-induced GnRH/LH surge were overcome by neuron-specific SOCS3 knock-out. Although only 20% of control HCD-mice experienced a preovulatory-like LH surge, LH surges could be induced in almost all neuron-specific SOCS3 knock-out mice on this diet. In contrast to females, HCD-fed male mice did not exhibit any fertility decline compared with low caloric diet-fed males despite their resistance to the satiety effects of leptin. These data show that deletion of SOCS3 delays the onset of leptin resistance and infertility in HCD-fed female mice, but given continued HCD feeding this state does eventually occur, presumably in response to other mechanisms inhibiting leptin signal transduction.
SIGNIFICANCE STATEMENT Obesity is commonly associated with infertility in humans and other animals. Treatments for human infertility show a decreased success rate with increasing body mass index. A hallmark of obesity is an increase in circulating leptin levels; despite this, the brain responds as if there were low levels of leptin, leading to increased appetite and suppressed fertility. Here we show that leptin resistant infertility is caused in part by the leptin signaling molecule SOCS3. Deletion of SOCS3 from brain neurons delays the onset of diet-induced infertility.
Keywords: GnRH, high caloric diet, high fat diet, leptin, SOCS3
Introduction
Mammalian fertility is governed by the hypothalamic gonadotropin-releasing hormone (GnRH) neuronal network. The activity of GnRH neurons is regulated by metabolic factors, in particular the adipocyte-derived hormone leptin, which is permissive for fertility (Barash et al., 1996; Chehab et al., 1996; Chua et al., 1996; Donato et al., 2011a). Although leptin acts centrally to modulate pulsatile GnRH release (Lebrethon et al., 2000; Nagatani et al., 2000), this occurs indirectly via neurons afferent to GnRH cells because they do not contain leptin receptors (Quennell et al., 2009). Potential upstream sites of action for leptin's effects include the ventral premammillary nucleus (PMV), where leptin receptors have been shown to be sufficient for fertility in female mice (Donato et al., 2011b), the arcuate nucleus (ARC), where many of the leptin-responsive GABAergic neurons that have been shown to be required for normal fertility in mice (Zuure et al., 2013) are located, and the anteroventral periventricular nucleus (AVPV) and surrounding medial preoptic area (mPOA). Estradiol acts on neurons within the AVPV during the period immediately preceding ovulation to induce a switch from negative to positive estrogenic feedback (Le et al., 1997; Smith et al., 2006), causing a surge of GnRH and consequently luteinizing hormone (LH) from the anterior pituitary gland, followed by ovulation (Homburg et al., 1976; Franz, 1988). Leptin may regulate estrous cyclicity and LH release in mice via mPOA neuronal nitric oxide-producing cells (Bellefontaine et al., 2014).
Obesity is associated with a deregulation of the hypothalamo-pituitary-gonadal axis. While exogenous gonadotropin stimulation can improve fertility in chronically high caloric diet (HCD)-fed mice (Tortoriello et al., 2004), treatments for human infertility show a decreased success rate with increasing body mass index (Dokras et al., 2006; Koning et al., 2010; Souter et al., 2011). A hallmark of obesity is an increase in circulating leptin concentration (van Dielen et al., 2002; Tortoriello et al., 2004). Despite this, the brain responds as if there were low levels of leptin, leading to increased appetite and suppressed fertility, implying central leptin resistance (Wauman and Tavernier, 2011). In animals models of diet-induced obesity, leptin resistance can be quantified by the loss of a signaling response to exogenous leptin treatment (Münzberg et al., 2004), but recent experiments using leptin receptor antagonists imply that endogenous leptin sensitivity may be maintained (Ottaway et al., 2015).
Chronic HCD feeding leads to hypothalamic upregulation of suppressor of cytokine signaling 3 (SOCS3) (Münzberg et al., 2004). SOCS3 acts as a feedback inhibitor of leptin signaling via the JAK (Janus kinase)/STAT (signal transducer and activator of transcription) pathway as well as of insulin signaling by binding to the insulin receptor and by degrading insulin receptor substrates (Howard and Flier, 2006). Brain-specific SOCS3 deletion protects mice from diet-induced obesity (Mori et al., 2004), but the effects on HCD-induced infertility have yet to be established. We used neuron-specific SOCS3 knock-out mice, expressed on a background strain prone to diet-induced infertility, to elucidate whether SOCS3 mediates the effects of high dietary fat on the onset of infertility and regional hypothalamic leptin resistance.
Materials and Methods
Animals and diets.
Wild-type and transgenic mice (DBA/2J background strain) were obtained from the University of Otago animal breeding facility and housed under conditions of controlled lighting (lights on from 0600 to 1800 h) and temperature (22 ± 1°C). Except for overnight fasting as described, they had free access from the date of weaning to either standard low fat and sucrose (4.8% fat, 0% sucrose by weight, Specialty Feeds; designated LCD) or high fat and sucrose (23% fat, 42% sucrose by weight, SF03–020, Specialty Feeds; designated HCD) diets that were matched for protein, fiber, vitamin, and mineral content. The University of Otago Animal Ethics Committee approved all animal experimental protocols.
CamKinaseIIα Cre and SOCS3-flox transgenic mice were backcrossed onto a DBA/2J inbred mouse strain five times from a C57BL/6J strain because pilot studies in our laboratory showed that female DBA/2J mice suffer a 90% increase in the duration between litters in response to chronic HCD feeding compared with LCD-fed littermates, whereas HCD is without effect on the fertility of C57BL/6J mice. Similar findings have been reported by others (Tortoriello et al., 2004). Neuron-specific SOCS3 knock-out mice were bred as follows. Homozygous female Socs3 flox mice (Socs3fl/fl; loxP sites flanking Socs3 coding exon 2) (Croker et al., 2003) were bred to male CamKIIα-iCre BAC (referred to as CamKIIα-Cre) mice (Casanova et al., 2001). CamKIIα is expressed around birth in almost all forebrain neurons but not in glial cells (Ouimet et al., 1984; Burgin et al., 1990). The resulting female Socs3fl/+,CamKIIα-Cre mice were backcrossed to male Socs3fl/fl mice to generate Socs3fl/fl,CamKIIα-Cre conditional knock-out mice (referred to as neuron-specific SOCS3 knock-out mice). Socs3fl/fl littermates served as controls. Transgenic mice were identified by PCR analysis of genomic DNA isolated from tail biopsies using the following primer sets, at an annealing temperature of 62°C. For CamKIIα-Cre identification: GGT TCT CCG TTT GCA CTC AGG A, CCT GTT GTT CAG CTT GCA CCA G and CTG CAT GCA CGG GAC AGC TCT (290 bp product indicates the wild-type gene and 345 bp product indicates the Cre-expressing gene), for Socs3fl identification: TCT TGT GTC TCT CCC CAT CC and TGA CGC TCA ACG TGA AGA AG (∼650 bp product indicates the wild-type gene and ∼800 bp product indicates the floxed gene), and to detect Socs excision: TCT TGT GTC TCT CCC CAT CC and ACG TCT GTG ATG CTT TGC TG (∼375 bp product indicates excised gene). PCR analysis of experimental versus control tissues showed that Cre-mediated flox excision of the Socs3 gene was specific to Cre-expressing mice and to brain tissue (Fig. 1). All mice were weighed every 2 weeks, except for female mice when they were paired with males for fertility analysis at 75 d of age.
Figure 1.

Representative examples of agarose gel bands indicating brain-specific Socs3 deletion in neuron-specific SOCS3 knock-out mice (right 3 lanes), but not control mice (left 3 lanes). Middle lane contains a 50 base pair ladder.
Characterization of HCD-induced leptin resistance in DBA/2J mice.
To characterize the development of obesity in the DBA/2J mouse strain, 6 male and female wild-type DBA/2J mice, raised on a LCD until 12 weeks of age, were fed either an LCD or HCD for a further 12 weeks. Energy consumption and body weights were monitored. At 9–10 weeks on the experimental diets, mice were fasted for 12 h and then subjected to either a leptin (1 mg/kg i.p. recombinant mouse leptin; National Hormone and Pituitary Program) or vehicle (0.1 m PBS) challenge at 1700 h. The respective diets were returned 30 min after the start of the dark phase (1830 h). Food intake was measured at 3 h after return of food (2130 h). A 7 d recovery period was given between vehicle and leptin challenges. After 12 weeks on the experimental diets, mice were fasted overnight and terminal trunk blood samples were collected for measurement of serum leptin concentration. Inguinal, retroperitoneal, and gonadal fat pads were collected and weighed.
Effect of neuron-specific SOCS3 deletion on puberty onset, estrous cyclicity, and fertility in LCD- and HCD-fed male and female mice.
Female neuron-specific SOCS3 knock-out and control mice were weaned onto either an LCD or HCD (n = 7 or 8 per group) and monitored for puberty onset by daily examination for vaginal opening and subsequently first estrus, based on vaginal cytology. At 60 d of age, estrous cyclicity was monitored for 10 consecutive days by vaginal cytology. Between 75 and 200 d of age, they were paired with wild-type C57BL/6J male mice for fertility analysis, maintaining their respective diets. The use of a mouse strain known to remain fully fertile on an HCD (i.e., C57BL/6J) as the mating partner in all mating studies enabled any effects of diet to be linked to the experimental (DBA/2J) mice (G. M. Anderson, unpublished data; Tortoriello et al., 2004). Cages were checked daily for the presence of litters, the date and size of the litter recorded, and the pups were immediately removed and culled. Estrous cyclicity was again monitored for 10 d at the end of this period of fertility testing (215-d-old, after 195 d on the diets).
Male neuron-specific SOCS3 knock-out and control mice (n = 7 per group) were paired with adult wild-type C57BL/6J female mice between 35 and 180 d of age; cages were checked daily for litters as above. Date of puberty was measured as date of first successful mating, determined by subtracting 20 d of gestation from the date of birth of the first litter. Mice were weighed every 2 weeks; for females, a separate cohort was used to monitor body weight to avoid the confounding effects of pregnancy. Weight at puberty was calculated using the measured body weights either side of the date of puberty, assuming a linear growth rate. For glucose tolerance tests, fasted (8 h) tail tip blood glucose concentrations were measured and animals then injected with a bolus of 1.5 g/kg intraperitoneal glucose in 100 μl physiological saline. Blood glucose concentrations were measured at 15, 30, 45, 60, and 90 min thereafter using an automatic glucose monitor (Roche; Accu-Chek Performa).
Effect of neuron-specific SOCS3 deletion on the ability of estradiol to induce a preovulatory-like GnRH/LH surge in LCD- and HCD-fed female mice.
Female neuron-specific SOCS3 knock-out and control mice were fed an HCD from weaning for 110 d (n = 5 or 6 per group) or 225 d (n = 7–10 per group). They were then subjected to a preovulatory LH surge induction model as described previously (Wintermantel et al., 2006; Zuure et al., 2013). Briefly, mice were ovariectomized and had an estradiol-filled silicone rubber capsule, 1.0 mm internal, 2.1 mm external diameter (Dow Corning), filled with medical-grade silicone rubber adhesive (Dow Corning) containing 17-β estradiol (0.1 mg/ml adhesive, Sigma-Aldrich) implanted subcutaneously. Each mouse was given a 1-cm-long capsule (containing 1 μg estradiol) per 20 g body weight. Six days after ovariectomy, mice received a subcutaneous injection of estradiol benzoate (1 μg/20 g body weight, Sigma-Aldrich) at 0900 h to induce an LH surge. At 1800 h on the following day, the surge peak time (Wintermantel et al., 2006), a blood sample was collected via heart puncture and the mice were perfused via the heart with 20 ml of 4% PFA in PBS, pH 7.4. The serum was harvested and stored at −20°C for LH radioimmunoassay. Brains were collected and cryoprotected in 30% sucrose for later sectioning and dual immunohistochemistry for cFos and GnRH (110 d group only).
Effect of neuron-specific SOCS3 deletion on hypothalamic leptin sensitivity in LCD- and HCD-fed female mice.
Leptin-induced phosphorylation of signal transducer and activator of transcription (pSTAT3) was used to assess hypothalamic leptin sensitivity. Female neuron-specific SOCS3 knock-out and control mice were fed either a LCD or HCD from weaning and subjected to a leptin challenge to determine hypothalamic leptin responsiveness after either 110 or 225 d on the diets (n = 5–10 mice per group). These times were chosen based on the times that female control and neuron-specific SOCS3 knock-out mice became infertile in response to the HCD in the present study. All mice were fasted overnight to reduce the concentration of endogenous circulating leptin. Recombinant mouse leptin (1 mg/kg) (National Hormone and Peptide Program) was injected intraperitoneally, and mice were perfused 2 h later via the heart with 20 ml of 4% PFA. Brains were collected and cryoprotected in 30% sucrose for later sectioning and immunohistochemistry for pSTAT3.
Immunohistochemical staining for pSTAT3, GnRH, and cFos.
Coronal (30-μm-thick) sections throughout the septal-preoptic area (sections collected for GnRH+cFos dual immunohistochemistry) or the entire hypothalamus (sections collected for pSTAT3 immunohistochemistry) were cut on a sliding microtome with a freezing stage to provide three sets of consecutive sections each (90 μm apart). The primary antibodies used were as follows: monoclonal rabbit anti-pSTAT3 (Tyr705, D3A7 XP; Cell Signaling Technology), polyclonal rabbit anti cFos (AB5; Calbiochem), and polyclonal guinea pig anti GnRH antibody (GA02) (Rizwan et al., 2012). Omission of any of the primary antibodies resulted in a complete absence of staining.
Before staining, sections were incubated for 30 min in 1% H2O2 and 40% methanol to quench endogenous peroxidase activity. For pSTAT3 staining, the sections were then incubated in 1 mm EDTA, pH 8.0, at 90°C for 15 min to unmask the antigen. The rabbit anti-pSTAT3 (1:2000 dilution) or cFos (1:20,000 dilution) was diluted in a blocking solution of 0.05 m TBS, 0.25% BSA, 2% normal rabbit serum, and 0.1% Triton X-100 and incubated for 48 h at 4 C. This was followed by a 60 min incubation in biotinylated goat anti-rabbit IgGs (1:1000; Vector Laboratories), a 60 min incubation in Vector Elite ABC solution (1:200, Vector Laboratories), and a brief incubation in nickel-enhanced (0.8% Ni(NH4)2SO4) diaminobenzidine solution (Sigma-Aldrich) until blue-black nuclear staining was clearly visible. For GnRH and cFos dual immunohistochemistry, the tissues were next incubated for 48 h in guinea pig anti-GnRH primary antibody (1:3000 dilution in the blocking solution described above), then for 60 min in HRP-conjugated rabbit anti-guinea pig IgGs (1:500; Dako) and then briefly in unenhanced diaminobenzidine solution until a light brown cytoplasmic staining was visible. For dilution of secondary antibodies and washing the sections between each step, 0.5 m TBS was used. Sections were mounted on (3-aminopropyl)-trithoxyl-silane-coated slides, dried, hydrated in water, dehydrated in graded series of alcohols, and then cleared in xylene before coverslipping with dibutylphthalate polystyrene xylene. Staining was observed using an Olympus AX70 Provis light microscope at 100× and 200× magnification.
GnRH neurons were counted as cFos-positive if they had a distinct blue-black-stained nucleus surrounded by brown cytoplasmic staining. All GnRH neurons in three sections per animal, which included the medial septum and region around the organum vasculosum of the lamina terminalis (0.4–0.7 mm anterior to bregma), as shown by Franklin and Paxinos (2008), were counted by an operator blind to the treatment groups. For pSTAT3, numbers of immunoreactive cells were counted using the image analysis software ImageJ (National Institutes of Health) to identify and count darkly stained nuclei within uniformly sized hypothalamic regions and the Franklin and Paxinos (2008) mouse brain atlas to define these regions. The regions analyzed and their coordinates anterior to bregma were the organum vasculosum of the lamina terminalis (OVLT, 0.4–0.5 mm), the mPOA and the AVPV (both 0.3–0.4 mm), the ARC (−1.5 to −2.0 mm), and the PMV (−2.5 to −2.6 mm). Images were photographed at 100× magnification. For each region, two or three sections were counted and averaged to provide a single data point (counts per region per section) for each animal.
Blood serum immunoassay for leptin, LH, estradiol, and testosterone concentrations.
Serum leptin concentration was assayed in 10 μl aliquots using a commercial ELISA kit (Millipore EZML-82K), according to the manufacturer's instructions. The sensitivity of the assay (95% CI at 0 ng/ml on the standard curve) was 0.1 ng/ml. For a serum pool falling in the middle of the standard curve, the intra-assay CV was 8%.
Serum LH concentration was measured in 25 μl aliquots by radioimmunoassay. Values are expressed in terms of the rat standard NIDDK-rat LH-RP-3. Highly purified iodinated hormone (NIDDK-rat LH-I-10) was used as tracer, and the primary antiserum was NIDDK-rabbit anti-rat LH-S11 (final dilution 1:500,000). The sensitivity of the assay was 0.05 ng/ml. For a serum pool falling in the middle of the standard curve, the intra-assay CV was 9%.
Serum total estradiol concentration was assayed in 25 μl aliquots using a commercial ELISA kit (Calbiotech, Mouse/Rat Estradiol ELISA ES180S-100), according to the manufacturer's instructions. The sensitivity of the assay was 2.5 pg/ml, and the intra-assay CV was 5%. Serum total testosterone concentration was assayed in 10 μl aliquots using a commercial ELISA kit (LDN Labor Diagnostika Testosterone Rat/Mouse ELISA AR E-8000), according to the manufacturer's instructions. The sensitivity of the assay was 10 pg/ml, and the intra-assay CV was 11%.
Data analysis.
All 2 × 2 factorial data were analyzed using two-way ANOVA with diet and genotype or diet and leptin/vehicle treatment as the factors for comparison. Post hoc analysis was performed using the Holm–Sidak multiple-comparisons test. A preovulatory-like LH surge was determined to have occurred if the plasma LH concentration was >2 ng/ml. The proportion of mice exhibiting a preovulatory-like LH surge was analyzed using a χ2 test. All other data were analyzed using Student's t tests or Mann–Whitney U tests. Results are presented as mean ± SEM, and differences were considered significant at p < 0.05.
Results
Characterization of HCD-induced leptin resistance in DBA/2J mice
Male and female wild-type DBA/2J mice fed an HCD for 10 weeks had a 33%–50% higher caloric intake compared with LCD-fed controls (t(10) = 5.81 [male] and 6.71 [female], p < 0.001), despite an 8%–16% reduced food intake on a mass basis (Fig. 2A). When fasted for 24 h and treated with 1 mg/kg leptin, LCD-fed male and female mice consumed significantly less food over the first 3 h of refeeding compared with saline-treated controls (t(4) = 5.49 [male] and 4.42 [female], p < 0.01), whereas HCD-fed mice showed no anorexigenic response to leptin (Fig. 2B), indicating the development of central leptin resistance in both sexes. In male HCD-fed mice serum leptin concentration (Fig. 2C) and gonadal, inguinal, and retroperitoneal fat pad masses (Fig. 2D) were elevated 3- to 10-fold compared with LCD-fed controls (t(10) = 10.53 [gonadal], 3.53 [inguinal], and 9.20 [retroperitoneal], p < 0.005); in females, the same responses of fat mass to HCD feeding occurred but were less pronounced (t(10) = 3.40 [gonadal], 2.87 [inguinal], and 7.22 [retroperitoneal], p < 0.01) (Fig. 2C,D).
Figure 2.

Characterization of the effects of high (23%) versus low (<5%) fat diets on energy consumption, leptin resistance, and adiposity in male and female DBA/2J mice. A, Daily energy consumption (right bars), calculated over a 2 week period, was significantly elevated despite a reduced food intake by mass (left bars). B, In 12 h fasted LCD-fed mice, leptin (1 mg/kg i.p.) significantly reduced 3 h food intake; whereas in HCD-fed mice, there was no response to leptin, indicating that they had become resistant to this hormone. Serum leptin concentration (C) and regional (gonadal, inguinal, and retroperitoneal) fat pad masses (D) were markedly increased in male, and to a lesser extent in female, mice in response to the HCD. Mice (n = 6 per group) were 5–6 months old and were fed the treatment diets for 12 weeks. *p < 0.05 versus LCD controls. **p < 0.01 versus LCD controls.
In male (Fig. 3A) and female (Fig. 3B) control mice, the HCD significantly increased body weight; these mice were 30%–65% heavier than LCD-fed mice from ∼6 weeks of age. As expected based on previously published data (Mori et al., 2004), neuron-specific SOCS3 knock-out completely protected the male mice from HCD-induced obesity compared with their control littermates (two-way repeated-measures ANOVA on body weight: F(1,96) = 42.38, p < 0.001). Post hoc analysis revealed that the HCD-fed control male mice were significantly heavier than their SOCS3 knock-out counterparts from 15 weeks of age, respectively (Fig. 3A). The effect of SOCS3 knock-out was far less pronounced in females; although there was a significantly lower overall body weight in the HCD-fed knock-out mice compared with the HCD-fed control mice (main group effect: F(1,15) = 5.09, p < 0.05), post hoc analysis at specific time points revealed few significant differences (Fig. 3A). Glucose tolerance was tested after 120 d of HCD treatment. Female HCD-fed SOCS3 knock-out and control mice both exhibited impaired glucose clearance in response to a 1.5 g/kg intraperitoneal glucose challenge compared with their LCD-fed knock-out and control counterparts (35% higher area under the glucose response curve; two-way ANOVA main effect of diet: F(1,28) = 4.86, p < 0.05). After 225 d of HCD treatment, total abdominal fat mass was similarly elevated in females HCD-fed SOCS3 knock-out and control mice (2.72 ± 0.44 and 2.57 ± 0.29 g, respectively) compared with their LCD-fed knock-out and control counterparts (0.10 ± 0.03 and 0.14 ± 0.14 g; two-way ANOVA main effect of diet: F(1,28) = 87.05, p < 0.001).
Figure 3.

Effects of HCD and forebrain neuronal SOCS3 deletion growth profiles and onset of reproductive maturity. In male (A, n = 7 per group) and female (B, n = 7–8 per group) mice, HCD significantly increased body weight gain, and this effect was prevented by neuronal SOCS3 deletion. Insets, Age of first successful mating (A, males) and vaginal opening (B, females). HCD feeding advanced the date of first mating relative to LCD-fed mice regardless of genotype. The body weight at which the first fertile mating in HCD-fed males (C) and the first estrus in females fed either diet (D) occurred was advanced in neuron-specific SOCS3 knock-out mice compared with controls. *p < 0.05 versus LCD controls. **p < 0.01 versus LCD controls.
In males, HCD feeding also advanced the age of first mating relative to LCD-fed mice, regardless of genotype (two-way ANOVA: F(1,28) = 18.28, p < 0.001). Because the HCD-fed neuron-specific SOCS3 knock-out mice had a normal body weight, this meant that they became fertile at a lower weight than all other mice (Fig. 3C; two-way ANOVA interaction of body weight and diet: F(1,28) = 4.31, p < 0.05). In females, there was no significant effect of diet (which had only been initiated ∼10 d previously) or of genotype on age of first estrus; however, the body weight at which this event occurred was lower (by ∼2 g) in neuron-specific SOCS3 knock-out females fed either diet compared with controls (Fig. 3D; two-way ANOVA: F(1,28) = 4.31, p < 0.01).
Adult fecundity in males was not affected by either HCD or genotype. All males remained able to sire litters at regular intervals of ∼28 d (number of litters born over 145 d of mating: LCD control, 4.3 ± 0.5; LCD SOCS3 KO, 4.7 ± 0.2; HCD control, 5.0 ± 0.4; HCD SOCS3 KO, 4.8 ± 0.5) until the end of the experiment (age when last litter was born: LCD control, 147 ± 8 d; LCD SOCS3 KO, 165 ± 7 d; HCD control, 160 ± 9 d; HCD SOCS3 KO, 159 ± 9 d). In marked contrast, the HCD markedly attenuated the period of adult fertility in female mice (two-way ANOVA for age at birth of last litter: F(1,17) = 56.15, p < 0.001; number of litters delivered: F(1,17) = 77.87, p < 0.001). While LCD-fed females of both genotypes remained fertile until the end of the experiment (5.9 litters born over the mating study; average interlitter interval 22 d), HCD-fed control mice averaged only 2.2 litters per dam and gave birth to their final litter at an average of 45 d into the mating study. In contrast to this, neuron-specific SOCS3 knock-out prevented the abrupt loss of fertility at this time in HCD-fed mice. The rescue from HCD-induced infertility in response to SOCS3 disruption was limited to ∼4 weeks duration, however; following 84 d of mating and having given birth to 3.8 litters on average, neuron-specific SOCS3 knock-out mice also succumbed to HCD-induced fertility (Fig. 4A–C), significantly later than their control counterparts but earlier than the LCD-fed groups (two-way ANOVA interaction of genotype and diet for age at birth of last litter: F(1,17) = 5.26, p < 0.05; interaction of genotype and diet for number of litters delivered: F(1,17) = 6.09, p < 0.05). The eventual loss of fertility in both HCD-fed groups was reflected in the pattern of estrous cycles at the beginning and end of the study. While mice in all four groups exhibited regular cycles at 8 weeks of age as evidenced by their vaginal cytology, at the end of the study the frequency of estrous smears was significantly reduced (from 28% at 8 weeks to 12% at 30 weeks) in both the HCD-fed groups (two-way ANOVA: F(1,17) = 20.10, p < 0.001), and diestrous-like cytological patterns predominated instead (data not shown). There were no diet or genotype effects on plasma estradiol and total testosterone concentrations after 120 d of HCD treatment in female mice.
Figure 4.

A, Timelines showing fecundity over 4 months for individual neuron-specific SOCS3 knock-out and control female mice fed either an LCD or HCD and paired with wild-type mates. Triangles represent birth of individual litters. Bold vertical lines indicate the average day that the last litter was born for each group. Shaded region represents the period for which neuron-specific SOCS3 knock-out was able to rescue mice from HCD-induced infertility. B, Comparison of the total number of litters born. C, Mean age at which the last litter was born (n = 4–6 per group). Recordings ceased after 180 d on the experimental diets (200 d of age). *p < 0.05 versus control females.
To test whether the HCD was able to inhibit the estradiol-induced preovulatory GnRH/LH surge via a SOCS3-dependent mechanism, we next subjected additional neuron-specific SOCS3 knock-out mice and control littermates, fed the HCD from weaning for 110 d, to an exogenous estradiol treatment protocol that reliably causes activation of GnRH neurons and a preovulatory-like surge of LH (Wintermantel et al., 2006; Zuure et al., 2013). This age and duration of HCD exposure were chosen because it falls within the window of time identified in the fertility experiment when neuron-specific SOCS3 knock-out mice are protected from HCD-induced infertility (Fig. 4A). Only 20% of control HCD-fed mice experienced a preovulatory-like LH surge (serum LH concentration >2.0 ng/ml), implying that the HCD suppressed the neuroendocrine control of ovulation at this time. In marked contrast, robust LH surges could be induced in almost all (83%) of neuron-specific SOCS3 knock-out mice on this diet (see Fig. 6A; χ(1,N=11)2 = 11) = 4.41, p < 0.05; mean serum LH concentration: U(9) = 4, p < 0.05). There was a nonsignificant trend (p = 0.095) for an increased percentage of GnRH neurons activated by estradiol (i.e., coespressing cFos) in the neuron-specific SOCS3 knock-out mice (Fig. 5A; for representative examples for GnRH and cFos immunreactivity, see Fig. 5B). When the ability of this estradiol treatment to induce a preovulatory-like LH surge was tested after 225 d of HCD feeding, when all mice in the fecundity test had become infertile, both control and SOCS3 KO mice exhibited low plasma LH concentrations (mean <1.8 ng/ml, and <20% surging) (Fig. 5C). In a group of similarly aged LCD-fed mice tested at the same time, 75% of mice surged with mean LH levels of 5.8 ng/ml.
Figure 6.

Neuron-specific SOCS3 knock-out delays the onset of HCD-induced leptin resistance in hypothalamic regions in female mice. Graphs represent quantification of pSTAT3-immunoreactive cell numbers in female mice (n = 5–10 per group) perfused 2 h following a leptin injection (1 mg/kg). Mice were maintained on an HCD for either 110 d (A–E, sufficient to induce infertility in controls but not knock-outs) or 225 d (F–J, sufficient to induce infertility in both controls and knock-outs). K–O, Representative examples of leptin-induced pSTAT3 immunoreactivity in hypothalamic regions in neuron-specific SOCS3 KO mice fed an HCD for 110 d (left images) or 225 d (right images). Scale bar, 200 μm. *p < 0.05 versus LCD.
Figure 5.
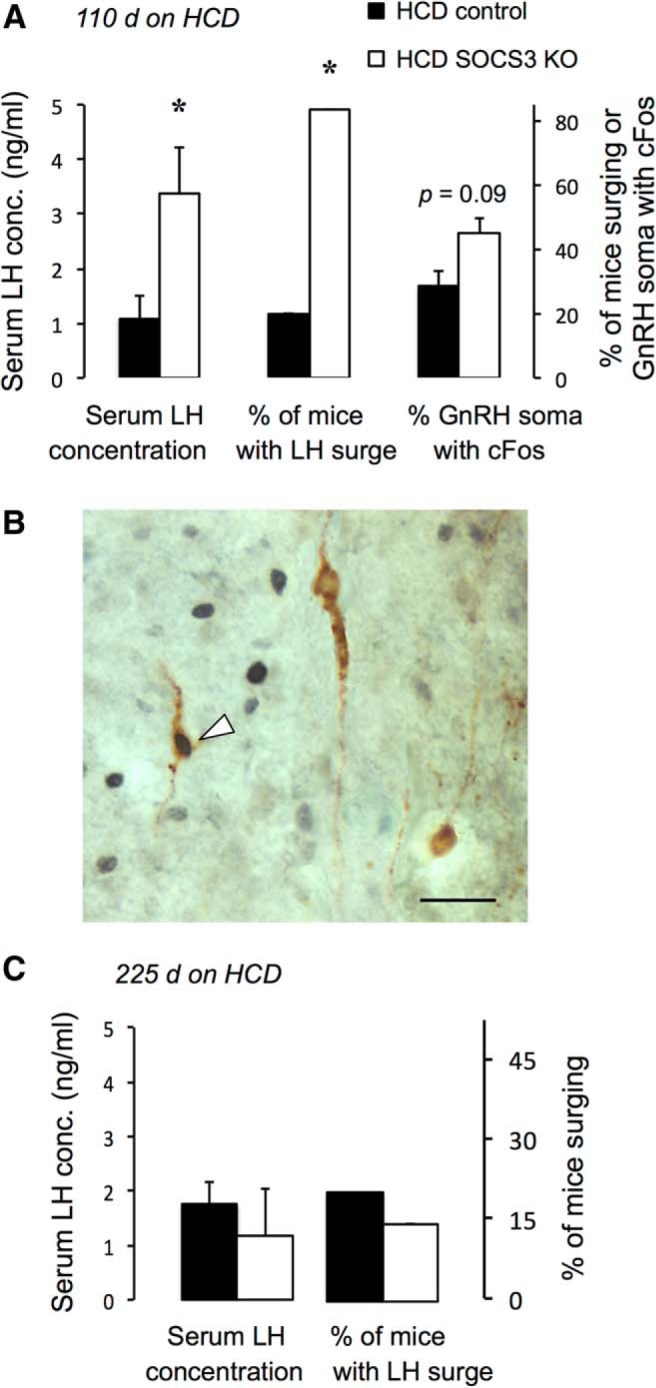
Neuron-specific SOCS3 knock-out prevents HCD-induced suppression of the estradiol-induced LH surge in female mice. A, Serum LH concentration at the time of surge occurrence (left bars), percentage of mice exhibiting an LH surges (serum concentration >2 ng/ml) (middle bars), and percentage of activated GnRH neurons (i.e., coexpressing cFos) (right bars) after 120 d of HCD feeding (n = 5 or 6 per group). B, Representative example of GnRH neurons (brown unenhanced DAB cytosolic staining) that are either negative or positive (indicated by arrowhead) for cFos immunoreactivity (black nickel-enhanced DAB nuclear staining). Control and SOCS3 knock-out mice were fed an HCD from weaning for 110 d (sufficient to induce infertility in controls but not knock-outs). They were then ovariectomized and subjected to an estradiol-induced preovulatory surge induction model. C, When the ability of this estradiol to induce an LH surge was tested after 225 d of HCD feeding, both control and SOCS3 KO mice exhibited low plasma LH concentrations (n = 7–10 per group). Scale bar, 20 μm. *p < 0.05 versus controls.
Finally, we questioned whether these effects of HCD to cause infertility and their prevention by SOCS3 knock-out were associated with altered leptin responsiveness in physiological systems and in specific hypothalamic nuclei. For example, the AVPV is a region known to be critical for generation of the preovulatory GnRH/LH surge in mice surge (Le et al., 2001). Leptin acts in this nucleus and in the surrounding medial preoptic area (Quennell et al., 2009; Scott et al., 2009) and, given the effects of HCD and SOCS3 knock-out on surge generation described above, might be expected to show altered leptin signaling in response to these treatments. Indeed, at 110 d of HCD feeding during the previously identified window of time when neuron-specific SOCS3 knock-out mice are protected from HCD-induced infertility, significantly reduced leptin-induced pSTAT3 (indicative of leptin resistance) was observed in the mPOA and AVPV of female HCD-fed control mice compared with LCD-fed mice, whereas leptin responsiveness was maintained at normal levels in neuron-specific SOCS3 knock-out mice in these regions (Fig. 6B,C; two-way ANOVA interaction of genotype and diet for pSTAT3 in mPOA: F(1,24) = 6.71, p < 0.01; interaction of genotype and diet for pSTAT3 in AVPV: F(1,22) = 4.32, p < 0.05). The same effect, albeit at a lower magnitude, was observed in the ARC (Fig. 6D; two-way ANOVA interaction of genotype and diet: F(1,28) = 6.43, p < 0.01). In the OVLT and PMV, leptin sensitivity was unaffected by HCD or SOCS3 knock-out at this time (Fig. 6A,E). At 8 months of age, however, leptin-induced pSTAT3 immunoreactivity was reduced in all HCD-fed in the OVLT, AVPV, and ARC relative to LCD mice regardless of genotype (Fig. 6F,H,I; two-way ANOVA for pSTAT3 in OVLT: F(1,17) = 13.16, p < 0.01; two-way ANOVA for pSTAT3 in AVPV: F(1,17) = 55.6, p < 0.001; two-way ANOVA for pSTAT3 in ARC: F(1,17) = 105.1, p < 0.001), and in the mPOA even LCD mice displayed low pSTAT3 levels at this time (Fig. 6G). Only the PMV maintained a robust response to leptin in all groups, regardless of diet or treatment, at this age (Fig. 6J). Representative examples of pSTAT3 immunostaining depicting the regional loss of leptin responsiveness in neuron-specific SOCS3 knock-out mice between 110 and 225 d of HCD treatment are shown in Figure 6K–O.
Leptin responsiveness was also measured after 160 d of HCD in two systems known to be affected by this hormone: suppression of voluntary food intake and stimulation of circulating LH levels. As expected, fasted female LCD-fed mice showed a robust anorexigenic response to 1 mg/kg leptin, administered as described earlier (two-way ANOVA main effect of leptin treatment: F(1,18) = 8.94, p < 0.01). This response was completely suppressed in HFD-fed mice, regardless of genotype (Fig. 7A), suggesting onset of leptin resistance in appetite-modulation circuitry at this stage of the dietary regimen regardless of SOCS3 KO. A similar pattern was seen for leptin stimulation of LH secretion (measured under the same conditions as the food intake experiment). Although 1 mg/kg leptin-treated LCD-fed mice induced an increase in plasma LH concentration in tail tip blood after 20 min compared with saline-treated mice (two-way ANOVA main effect of leptin treatment: F(1,16) = 8.61, p < 0.01), this response was not seen in either HFD-fed group (Fig. 7B).
Figure 7.

Leptin responsiveness of voluntary food intake and circulating LH concentration is suppressed after 160 d of HCD feeding in female mice. A, Twelve hour fasted LCD-fed mice showed a robust anorexigenic response in 3 h food intake to leptin (1 mg/kg i.p.) compared with saline vehicle. This response was completely suppressed in both control and SOCS3 KO HFD-fed mice (n = 5–7 per group). B, Twelve hour fasted LCD-fed mice exhibited a stimulation of LH secretion 20 min following leptin treatment (1 mg/kg i.p.), and this response was also suppressed in both genotypes of HFD-fed mice. **p < 0.01 versus LCD controls.
Discussion
Although the correlation between obesity and infertility is widely recognized (Koning et al., 2010), the physiological causes of HCD-induced infertility are poorly understood. Central resistance to leptin, a hormone essential for fertility, is known to accompany this condition and is likely to be a major contributing factor. Upregulation of SOCS3 when fed an HCD is one mechanism proposed to cause central resistance to leptin (Bjørbaek et al., 1998; Münzberg et al., 2004) and insulin (Howard and Flier, 2006), of which the former is most important for reproductive activity (Quennell et al., 2009; Evans et al., 2015). Central deletion of SOCS3 protects against the development of obesity (Mori et al., 2004), but until now it has not been determined whether this is also the case in regards to fertility. Here we show that SOCS3 deletion is protective against HCD-induced infertility and leptin resistance in the mPOA, AVPV, and ARC of female mice but becomes ineffective in this regard if the diet is maintained for several months.
The role of SOCS3 in HCD-induced obesity has been demonstrated using brain-specific SOCS3 knock-out mice (Mori et al., 2004). Supporting the findings of that study, neuron-specific SOCS3 knock-out completely corrected the overweight phenotype exhibited by control male mice fed an HCD, although interestingly the protective effect of SOCS3 knock-out on the body weight and metabolic phenotype of females was considerably less pronounced than in males in our experiments, perhaps reflecting a difference in the mouse background strains used in the two studies or in the cell types targeted for Socs3 deletion (CamKinaseIIα vs nestin-expressing cells). It is interesting to note that even neuron-specific SOCS3 knock-out HCD-fed mice eventually developed resistance to leptin (defined as loss of leptin-induced STAT3 activation) in most hypothalamic regions. This suggests that mechanisms other than SOCS3 upregulation can eventually bring about hypothalamic leptin resistance. The key novel finding from this study is that female fertility when fed on an HCD is improved by the absence of SOCS3 in diet-induced infertility-prone mice. This was despite the relatively limited effects of SOCS3 knock-out in female metabolic function; therefore, their improved fertility does not appear to be secondary to reduced obesity. Female neuron-specific SOCS3 knock-out mice did not become infertile at the same time as their control counterparts, and this was associated with an absence of leptin resistance onset after 110 d of HCD-feeding in the mPOA and AVPV, where susceptibility to leptin resistance has not previously been reported (Münzberg et al., 2004). By comparison, control HCD-fed mice exhibited infertility and pronounced leptin resistance in these regions. The rodent AVPV contains neurons that are activated by the rising circulating estradiol concentration on the afternoon of proestrus (Le et al., 1997; Smith et al., 2006). Leptin acts on a small number of cells in this nucleus and to a much greater extent in the surrounding medial preoptic area (Fig. 7) (Quennell et al., 2009; Scott et al., 2009). Collectively, these results may point to HCD-induced infertility in female mice being caused by a failure of ovulation, which is in turn due to SOCS3-induced onset of leptin resistance in the AVPV. Such site specificity of the effects of leptin resistance would explain the lack of correlation between blood SOCS3 concentrations and infertility in women in a recent study (Ozkan et al., 2014).
To further examine this hypothesis, we tested whether the suppressive effects of an HCD on estradiol-induced GnRH/LH surge suppression were overcome by neuron-specific SOCS3 knock-out. Although only 20% of control mice fed an HCD experienced a preovulatory-like LH surge, LH surges could be induced in almost all neuron-specific SOCS3 knock-out mice on this diet. In contrast, after 225 d of HCD feeding, when all mice in the fecundity test had become infertile, the ability of estradiol to induce an LH surge was suppressed in both control and SOCS3 KO mice. It is important to note that, because the AVPV kisspeptin neurons which drive the preovulatory GnRH surge do not express leptin receptors or show leptin signaling responses (Cravo et al., 2011; Donato et al., 2011b; Louis et al., 2011; Quennell et al., 2011), the effects of leptin may instead be relayed to GnRH and/or kisspeptin neurons by nearby leptin receptor-expressing cells, such as neuronal nitric oxide-producing neurons in the mPOA (Donato et al., 2010). These neurons have recently been shown to be dependent in part on leptin for neuronal nitric oxide synthase activation, which is in turn required for leptin's stimulatory effects on LH release and the progression from diestrus to proestrus cycle stages (Bellefontaine et al., 2014).
Another recently proposed site of action for leptin's effects on fertility is the PMV, which contains neurons that express leptin receptors and project to GnRH soma (Rondini et al., 2004; Leshan et al., 2009; Louis et al., 2011) and to AVPV kisspeptin neurons and GnRH terminals in the median eminence (Donato et al., 2011b). Ablation of this nucleus disrupted estrous cyclicity in female rats (Donato et al., 2009). Selective PMV leptin receptor reexpression in leptin receptor null mice induced female puberty onset and some pregnancies but had no effect on males (Donato et al., 2011b). However, glutamatergic neurons, which are very highly expressed in the PMV, are not required as direct mediators of leptin actions on reproductive activity (Zuure et al., 2013). It was therefore very interesting to note that HCD-induced infertility occurred despite the PMV retaining normal leptin responsiveness throughout the study. Lack of leptin resistance in HCD-fed male C57BL/6J mice has previously been reported for the PMV (Münzberg et al., 2004).
Another region shown to be protected from HCD-induced leptin resistance by SOCS3 knock-out was the ARC. Although the effects of HCD on leptin signaling in this region were not as pronounced as those in the mPOA and AVPV at 110 d of HCD feeding, its likely importance as a site of leptin regulation of reproductive activity should not be overlooked. It contains numerous GABAergic neurons that have recently been shown to be required as conduits for leptin's permissive regulation of fertility in mice (Zuure et al., 2013; Zuure et al., 2016), among which agouti-related peptide/neuropeptide Y (Horvath et al., 1997; Israel et al., 2012; Roa and Herbison, 2012; Wu et al., 2012) and galanin-like peptide (Juréus et al., 2000; Takatsu et al., 2001) neurons may be important participants.
In contrast to the females, HCD-fed male mice did not exhibit any decline in fertility compared with LCD-fed males in this study, despite their markedly increased body weight. This is consistent with other studies (Tortoriello et al., 2004), although HCD-induced sperm defects have been reported (Bakos et al., 2010; Ghanayem et al., 2010; Fernandez et al., 2011). The sex specificity of HCD feeding on fertility lends further support to the idea that the AVPV and its neuroendocrine control of ovulation may be the principal targets for leptin resistant infertility.
Together with other data showing region-specific upregulation of Socs3 mRNA in response to HCD feeding (Münzberg et al., 2004), our data indicate that hypothalamic leptin resistance and consequent female infertility caused by an HCD may be initially induced by upregulation of SOCS3. Socs3 gene knock-out confers protection against these effects. However, other mechanisms are also involved in the generation of leptin resistance, and these mechanisms are likely to have been the cause of the eventual infertility exhibited by the neuron-specific SOCS3 knock-out mice after ∼5 months of HCD feeding. It is possible that other SOCS family proteins are upregulated to compensate for the lack of SOCS3. For example, SOCS1 levels are also increased along with SOCS3 in HCD-induced obesity (Peiser et al., 2000; Emanuelli et al., 2008). Knocking out SOCS1 does not protect against the development of obesity, however (Emanuelli et al., 2008), suggesting that SOCS3 is the major SOCS inhibitor of hypothalamic leptin signaling. HCD-induced long-form leptin receptor downregulation is also thought to contribute to leptin resistance, and this may provide an explanation for how site-specific leptin resistance can occur in the absence of SOCS3. This downregulation of leptin receptor in response to an HCD is apparent in some studies (Tortoriello et al., 2004; Wilsey and Scarpace, 2004), but not others (Münzberg et al., 2004; Jørgensen et al., 2006). Another potential mediator of SOCS3-independent leptin resistance is protein tyrosine phosphatase 1B (PTP1B). PTP1B inhibits both insulin and leptin signaling by dephosphorylating activated insulin receptor and insulin receptor substrates as well as JAK2 (Salmeen et al., 2000), and PTP1B knock-out mice have markedly reduced adiposity when fed an HCD compared with controls (Tsou et al., 2012). It would be interesting to test whether PTP1B knock-out mice would exhibit complete rescue of fertility when fed an HCD, or whether combined knock-out of both SOCS3 and PTP1B would be required for this.
SOCS3 also negatively regulates numerous other cytokine signaling pathways, including proinflammatory cytokines. SOCS3 is upregulated in the hypothalamus in response to inflammation following a high fat diet (Wu et al., 2014), and is important for restraining inflammation and allowing optimal levels of protective immune responses against infection (Carow and Rottenberg, 2014). Because central inflammation may itself be a cause rather than a consequence of leptin resistance and obesity (Wang et al., 2012), SOCS3 knock-out could have affected leptin responsiveness by this mechanism in the current study. However, the direction of the response we observed (higher leptin responsiveness in the SOCS3 knock-out mice) is not consistent with this. It remains possible, however, that inflammation was a cause of the eventual onset of hypothalamic leptin resistance and infertility after prolonged high calorie diet exposure.
In conclusion, deleting SOCS3 from forebrain neurons prevents HCD-induced suppression of the estradiol-induced GnRH/LH surge and fertility in female mice, as well as preventing HCD-induced leptin resistance from occurring in the mPOA and AVPV. However, this protective effect of SOCS3 deletion is not permanent, and it is likely that other longer-term mechanisms eventually bring about leptin resistance and consequent female infertility. Further work needs to be undertaken to determine how the different mechanisms that cause leptin resistance change between short- and long-term feeding of HCD, as well as determining how the PMV appears to remain entirely responsive to leptin under high caloric dietary conditions while the mPOA and AVPV become leptin resistant, and the sex-specific consequences of this.
Notes
Supplemental material for this article is available at http://hdl.handle.net/10523/6230. This material has not been peer reviewed.
Footnotes
This work was supported by the Health Research Council of New Zealand and New Zealand Lottery Health Research grants. We thank G. Schutz and W. Alexander for supplying transgenic mice.
The authors declare no competing financial interests.
References
- Bakos HW, Mitchell M, Setchell BP, Lane M. The effect of paternal diet-induced obesity on sperm function and fertilization in a mouse model. Int J Androl. 2011;34:402–410. doi: 10.1111/j.1365-2605.2010.01092.x. [DOI] [PubMed] [Google Scholar]
- Barash IA, Cheung CC, Weigle DS, Ren H, Kabigting EB, Kuijper JL, Clifton DK, Steiner RA. Leptin is a metabolic signal to the reproductive system. Endocrinology. 1996;137:3144–3147. doi: 10.1210/endo.137.7.8770941. [DOI] [PubMed] [Google Scholar]
- Bellefontaine N, Chachlaki K, Parkash J, Vanacker C, Colledge W, D'Anglemont De Tassigny X, Garthwaite J, Bouret SG, Prevot V. Leptin-dependent neuronal NO signaling in the preoptic hypothalamus facilitates reproduction. J Clin Invest. 2014;124:2550–2559. doi: 10.1172/JCI65928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell. 1998;1:619–625. doi: 10.1016/S1097-2765(00)80062-3. [DOI] [PubMed] [Google Scholar]
- Burgin KE, Waxham MN, Rickling S, Westgate SA, Mobley WC, Kelly PT. In situ hybridization histochemistry of Ca2+/calmodulin-dependent protein kinase in developing rat brain. J Neurosci. 1990;10:1788–1798. doi: 10.1523/JNEUROSCI.10-06-01788.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carow B, Rottenberg ME. SOCS3, a major regulator of infection and inflammation. Front Immunol. 2014;5:58. doi: 10.3389/fimmu.2014.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova E, Fehsenfeld S, Mantamadiotis T, Lemberger T, Greiner E, Stewart AF, Schütz G. A CamKIIalpha iCre BAC allows brain-specific gene inactivation. Genesis. 2001;31:37–42. doi: 10.1002/gene.1078. [DOI] [PubMed] [Google Scholar]
- Chehab FF, Lim ME, Lu R. Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin. Nat Genet. 1996;12:318–320. doi: 10.1038/ng0396-318. [DOI] [PubMed] [Google Scholar]
- Chua SC, Jr, Chung WK, Wu-Peng XS, Zhang Y, Liu SM, Tartaglia L, Leibel RL. Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor. Science. 1996;271:994–996. doi: 10.1126/science.271.5251.994. [DOI] [PubMed] [Google Scholar]
- Cravo RM, Margatho LO, Osborne-Lawrence S, Donato J, Jr, Atkin S, Bookout AL, Rovinsky S, Frazão R, Lee CE, Gautron L, Zigman JM, Elias CF. Characterization of Kiss1 neurons using transgenic mouse models. Neuroscience. 2011;173:37–56. doi: 10.1016/j.neuroscience.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Förster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW, Alexander WS. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003;4:540–545. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- Dokras A, Baredziak L, Blaine J, Syrop C, VanVoorhis BJ, Sparks A. Obstetric outcomes after in vitro fertilization in obese and morbidly obese women. Obstet Gynecol. 2006;108:61–69. doi: 10.1097/01.AOG.0000219768.08249.b6. [DOI] [PubMed] [Google Scholar]
- Donato J, Jr, Silva RJ, Sita LV, Lee S, Lee C, Lacchini S, Bittencourt JC, Franci CR, Canteras NS, Elias CF. The ventral premammillary nucleus links fasting-induced changes in leptin levels and coordinated luteinizing hormone secretion. J Neurosci. 2009;29:5240–5250. doi: 10.1523/JNEUROSCI.0405-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato J, Jr, Frazão R, Fukuda M, Vianna CR, Elias CF. Leptin induces phosphorylation of neuronal nitric oxide synthase in defined hypothalamic neurons. Endocrinology. 2010;151:5415–5427. doi: 10.1210/en.2010-0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato J, Jr, Cravo RM, Frazão R, Elias CF. Hypothalamic sites of leptin action linking metabolism and reproduction. Neuroendocrinology. 2011a;93:9–18. doi: 10.1159/000322472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato J, Jr, Cravo RM, Frazão R, Gautron L, Scott MM, Lachey J, Castro IA, Margatho LO, Lee S, Lee C, Richardson JA, Friedman J, Chua S, Jr, Coppari R, Zigman JM, Elmquist JK, Elias CF. Leptin's effect on puberty in mice is relayed by the ventral premammillary nucleus and does not require signaling in Kiss1 neurons. J Clin Invest. 2011b;121:355–368. doi: 10.1172/JCI45106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelli B, Macotela Y, Boucher J, Ronald Kahn C. SOCS-1 deficiency does not prevent diet-induced insulin resistance. Biochem Biophys Res Commun. 2008;377:447–452. doi: 10.1016/j.bbrc.2008.09.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MC, Rizwan MZ, Anderson GM. Insulin does not target CamkIIα neurons to critically regulate the neuroendocrine reproductive axis in mice. J Neuroendocrinol. 2015;27:899–910. doi: 10.1111/jne.12330. [DOI] [PubMed] [Google Scholar]
- Fernandez CD, Bellentani FF, Fernandes GS, Perobelli JE, Favareto AP, Nascimento AF, Cicogna AC, Kempinas WD. Diet-induced obesity in rats leads to a decrease in sperm motility. Reprod Biol Endocrinol. 2011;9:32. doi: 10.1186/1477-7827-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin BJ, Paxinos G. The mouse brain atlas in stereotaxic coordinates. New York: Elsevier; 2008. [Google Scholar]
- Franz WB., 3rd Basic review: endocrinology of the normal menstrual cycle. Prim Care. 1988;15:607–616. [PubMed] [Google Scholar]
- Ghanayem BI, Bai R, Kissling GE, Travlos G, Hoffler U. Diet-induced obesity in male mice is associated with reduced fertility and potentiation of acrylamide-induced reproductive toxicity. Biol Reprod. 2010;82:96–104. doi: 10.1095/biolreprod.109.078915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homburg R, Potashnik G, Lunenfeld B, Insler V. The hypothalamus as a regulator of reproductive function. Obstet Gynecol Surv. 1976;31:455–471. doi: 10.1097/00006254-197606000-00001. [DOI] [PubMed] [Google Scholar]
- Horvath TL, Bechmann I, Naftolin F, Kalra SP, Leranth C. Heterogeneity in the neuropeptide Y-containing neurons of the rat arcuate nucleus: GABAergic and non-GABAergic subpopulations. Brain Res. 1997;756:283–286. doi: 10.1016/S0006-8993(97)00184-4. [DOI] [PubMed] [Google Scholar]
- Howard JK, Flier JS. Attenuation of leptin and insulin signaling by SOCS proteins. Trends Endocrinol Metab. 2006;17:365–371. doi: 10.1016/j.tem.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Israel DD, Sheffer-Babila S, de Luca C, Jo YH, Liu SM, Xia Q, Spergel DJ, Dun SL, Dun NJ, Chua SC., Jr Effects of leptin and melanocortin signaling interactions on pubertal development and reproduction. Endocrinology. 2012;153:2408–2419. doi: 10.1210/en.2011-1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen EA, Vogelsang TW, Knigge U, Watanabe T, Warberg J, Kjaer A. Increased susceptibility to diet-induced obesity in histamine-deficient mice. Neuroendocrinology. 2006;83:289–294. doi: 10.1159/000095339. [DOI] [PubMed] [Google Scholar]
- Juréus A, Cunningham MJ, McClain ME, Clifton DK, Steiner RA. Galanin-like peptide (GALP) is a target for regulation by leptin in the hypothalamus of the rat. Endocrinology. 2000;141:2703–2706. doi: 10.1210/endo.141.7.7669. [DOI] [PubMed] [Google Scholar]
- Koning AM, Kuchenbecker WK, Groen H, Hoek A, Land JA, Khan KS, Mol BW. Economic consequences of overweight and obesity in infertility: a framework for evaluating the costs and outcomes of fertility care. Hum Reprod Update. 2010;16:246–254. doi: 10.1093/humupd/dmp053. [DOI] [PubMed] [Google Scholar]
- Le WW, Attardi B, Berghorn KA, Blaustein J, Hoffman GE. Progesterone blockade of a luteinizing hormone surge blocks luteinizing hormone-releasing hormone Fos activation and activation of its preoptic area afferents. Brain Res. 1997;778:272–280. doi: 10.1016/S0006-8993(97)00971-2. [DOI] [PubMed] [Google Scholar]
- Le WW, Wise PM, Murphy AZ, Coolen LM, Hoffman GE. Parallel declines in Fos activation of the medial anteroventral periventricular nucleus and LHRH neurons in middle-aged rats. Endocrinology. 2001;142:4976–4982. doi: 10.1210/endo.142.11.8470. [DOI] [PubMed] [Google Scholar]
- Lebrethon MC, Vandersmissen E, Gérard A, Parent AS, Bourguignon JP. Cocaine and amphetamine-regulated-transcript peptide mediation of leptin stimulatory effect on the rat gonadotropin-releasing hormone pulse generator in vitro. J Neuroendocrinol. 2000;12:383–385. doi: 10.1046/j.1365-2826.2000.00497.x. [DOI] [PubMed] [Google Scholar]
- Leshan RL, Louis GW, Jo YH, Rhodes CJ, Münzberg H, Myers MG., Jr Direct innervation of GnRH neurons by metabolic- and sexual odorant-sensing leptin receptor neurons in the hypothalamic ventral premammillary nucleus. J Neurosci. 2009;29:3138–3147. doi: 10.1523/JNEUROSCI.0155-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis GW, Greenwald-Yarnell M, Phillips R, Coolen LM, Lehman MN, Myers MG., Jr Molecular mapping of the neural pathways linking leptin to the neuroendocrine reproductive axis. Endocrinology. 2011;152:2302–2310. doi: 10.1210/en.2011-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, Torisu T, Chien KR, Yasukawa H, Yoshimura A. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med. 2004;10:739–743. doi: 10.1038/nm1071. [DOI] [PubMed] [Google Scholar]
- Münzberg H, Flier JS, Bjørbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- Nagatani S, Zeng Y, Keisler DH, Foster DL, Jaffe CA. Leptin regulates pulsatile luteinizing hormone and growth hormone secretion in the sheep. Endocrinology. 2000;141:3965–3975. doi: 10.1210/endo.141.11.7762. [DOI] [PubMed] [Google Scholar]
- Ottaway N, Mahbod P, Rivero B, Norman LA, Gertler A, D'Alessio DA, Perez-Tilve D. Diet-induced obese mice retain endogenous leptin action. Cell Metab. 2015;21:877–882. doi: 10.1016/j.cmet.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouimet CC, McGuinness TL, Greengard P. Immunocytochemical localization of calcium/calmodulin-dependent protein kinase II in rat brain. Proc Natl Acad Sci U S A. 1984;81:5604–5608. doi: 10.1073/pnas.81.17.5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozkan ZS, Deveci D, Kumbak B, Simsek M, Ilhan F, Sekercioglu S, Sapmaz E. What is the impact of Th1/Th2 ratio, SOCS3, IL17, and IL35 levels in unexplained infertility? J Reprod Immunol. 2014;103:53–58. doi: 10.1016/j.jri.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Peiser C, McGregor GP, Lang RE. Leptin receptor expression and suppressor of cytokine signaling transcript levels in high-fat-fed rats. Life Sci. 2000;67:2971–2981. doi: 10.1016/S0024-3205(00)00884-5. [DOI] [PubMed] [Google Scholar]
- Quennell JH, Mulligan AC, Tups A, Liu X, Phipps SJ, Kemp CJ, Herbison AE, Grattan DR, Anderson GM. Leptin indirectly regulates gonadotropin-releasing hormone neuronal function. Endocrinology. 2009;150:2805–2812. doi: 10.1210/en.2008-1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quennell JH, Howell CS, Roa J, Augustine RA, Grattan DR, Anderson GM. Leptin deficiency and diet-induced obesity reduce hypothalamic kisspeptin expression in mice. Endocrinology. 2011;152:1541–1550. doi: 10.1210/en.2010-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizwan MZ, Poling MC, Corr M, Cornes PA, Augustine RA, Quennell JH, Kauffman AS, Anderson GM. RFamide-related peptide-3 receptor gene expression on GnRH and kisspeptin neurons and GnRH-dependent mechanism of action. Endocrinology. 2012;153:3770–3779. doi: 10.1210/en.2012-1133. [DOI] [PubMed] [Google Scholar]
- Roa J, Herbison AE. Direct regulation of GnRH neuron excitability by arcuate nucleus POMC and NPY neuron neuropeptides in female mice. Endocrinology. 2012;153:5587–5599. doi: 10.1210/en.2012-1470. [DOI] [PubMed] [Google Scholar]
- Rondini TA, Baddini SP, Sousa LF, Bittencourt JC, Elias CF. Hypothalamic cocaine- and amphetamine-regulated transcript neurons project to areas expressing gonadotropin releasing hormone immunoreactivity and to the anteroventral periventricular nucleus in male and female rats. Neuroscience. 2004;125:735–748. doi: 10.1016/j.neuroscience.2003.12.045. [DOI] [PubMed] [Google Scholar]
- Salmeen A, Andersen JN, Myers MP, Tonks NK, Barford D. Molecular basis for the dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol Cell. 2000;6:1401–1412. doi: 10.1016/S1097-2765(00)00137-4. [DOI] [PubMed] [Google Scholar]
- Scott MM, Lachey JL, Sternson SM, Lee CE, Elias CF, Friedman JM, Elmquist JK. Leptin targets in the mouse brain. J Comp Neurol. 2009;514:518–532. doi: 10.1002/cne.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JT, Popa SM, Clifton DK, Hoffman GE, Steiner RA. Kiss1 neurons in the forebrain as central processors for generating the preovulatory luteinizing hormone surge. J Neurosci. 2006;26:6687–6694. doi: 10.1523/JNEUROSCI.1618-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souter I, Baltagi LM, Kuleta D, Meeker JD, Petrozza JC. Women, weight, and fertility: the effect of body mass index on the outcome of superovulation/intrauterine insemination cycles. Fertil Steril. 2011;95:1042–1047. doi: 10.1016/j.fertnstert.2010.11.062. [DOI] [PubMed] [Google Scholar]
- Takatsu Y, Matsumoto H, Ohtaki T, Kumano S, Kitada C, Onda H, Nishimura O, Fujino M. Distribution of galanin-like peptide in the rat brain. Endocrinology. 2001;142:1626–1634. doi: 10.1210/endo.142.4.8089. [DOI] [PubMed] [Google Scholar]
- Tortoriello DV, McMinn J, Chua SC. Dietary-induced obesity and hypothalamic infertility in female DBA/2J mice. Endocrinology. 2004;145:1238–1247. doi: 10.1210/en.2003-1406. [DOI] [PubMed] [Google Scholar]
- Tsou RC, Zimmer DJ, De Jonghe BC, Bence KK. Deficiency of PTP1B in leptin receptor-expressing neurons leads to decreased body weight and adiposity in mice. Endocrinology. 2012;153:4227–4237. doi: 10.1210/en.2012-1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dielen FM, van'T Veer C, Buurman WA, Greve JW. Leptin and soluble leptin receptor levels in obese and weight-losing individuals. J Clin Endocrinol Metab. 2002;87:1708–1716. doi: 10.1210/jcem.87.4.8381. [DOI] [PubMed] [Google Scholar]
- Wang X, Ge A, Cheng M, Guo F, Zhao M, Zhou X, Liu L, Yang N. Increased hypothalamic inflammation associated with the susceptibility to obesity in rats exposed to high-fat diet. Exp Diabetes Res. 2012;2012:847246. doi: 10.1155/2012/847246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauman J, Tavernier J. Leptin receptor signaling: pathways to leptin resistance. Front Biosci (Landmark Ed) 2011;17:2771–2793. doi: 10.2741/3885. [DOI] [PubMed] [Google Scholar]
- Wilsey J, Scarpace PJ. Caloric restriction reverses the deficits in leptin receptor protein and leptin signaling capacity associated with diet-induced obesity: role of leptin in the regulation of hypothalamic long-form leptin receptor expression. J Endocrinol. 2004;181:297–306. doi: 10.1677/joe.0.1810297. [DOI] [PubMed] [Google Scholar]
- Wintermantel TM, Campbell RE, Porteous R, Bock D, Gröne HJ, Todman MG, Korach KS, Greiner E, Pérez CA, Schütz G, Herbison AE. Definition of estrogen receptor pathway critical for estrogen positive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron. 2006;52:271–280. doi: 10.1016/j.neuron.2006.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Whiddon BB, Palmiter RD. Ablation of neurons expressing agouti-related protein, but not melanin concentrating hormone, in leptin-deficient mice restores metabolic functions and fertility. Proc Natl Acad Sci U S A. 2012;109:3155–3160. doi: 10.1073/pnas.1120501109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Yu Y, Szabo A, Han M, Huang XF. Central inflammation and leptin resistance are attenuated by ginsenoside Rb1 treatment in obese mice fed a high-fat diet. PLoS One. 2014;9:e92618. doi: 10.1371/journal.pone.0092618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuure WA, Roberts AL, Quennell JH, Anderson GM. Leptin signaling in GABA neurons, but not glutamate neurons, is required for reproductive function. J Neurosci. 2013;33:17874–17883. doi: 10.1523/JNEUROSCI.2278-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuure WA, Quennell JH, Anderson GM. Leptin responsive and GABAergic projections to the rostral preoptic area in mice. J Neuroendocrinol. 2016;2016:3. doi: 10.1111/jne.12357. [DOI] [PubMed] [Google Scholar]