

Abstract
It is generally thought that neurons in the thalamic reticular nucleus (TRN) form GABAergic synapses with other TRN neurons and that these interconnections are important for the function of the TRN. However, the existence of such intrinsic connections is controversial. We combine two complementary approaches to examine intrinsic GABAergic connections in the TRN of the mouse. We find that optogenetic stimulation of TRN neurons and their axons evokes GABAergic IPSCs in TRN neurons in mice younger than 2 weeks of age but fails to do so after that age. Blocking synaptic release from TRN neurons through conditional deletion of vesicular GABA transporter has no effect on spontaneous IPSCs recorded in TRN neurons aged 2 weeks or older while dramatically reducing GABAergic transmission in thalamic relay neurons. These results demonstrate that except for a short period after birth, the TRN of the mouse lacks intrinsic GABAergic connections.
SIGNIFICANCE STATEMENT The thalamic reticular nucleus has a critical role in modulating information transfer from the thalamus to the cortex. It has been proposed that neurons in the thalamic reticular nucleus are interconnected through GABAergic synapses and that these connections serve important functions. Our results show that except for the first 2 weeks after birth, the thalamic reticular nucleus of the mouse lacks intrinsic GABAergic connections.
Keywords: GABA, mouse, optogenetics, parvalbumin, reticular nucleus, thalamus
Introduction
The thalamic reticular nucleus (TRN) is a sheet of GABAergic neurons surrounding the dorsal and lateral portions of the thalamus. Situated in the path of axons connecting the thalamus and the cortex, the TRN receives excitatory inputs from collaterals of thalamocortical and corticothalamic axons and provides the major inhibitory input to thalamocortical neurons (Guillery and Harting, 2003; Pinault, 2004). Because of its location and connectivity, the TRN is often regarded as the gatekeeper of information transfer from the thalamus to the cortex and has been implicated in sensory detection, attention, and arousal (Crick, 1984; Steriade et al., 1993; McCormick and Bal, 1997; Zikopoulos and Barbas, 2006; McAlonan et al., 2008; Halassa et al., 2014; Lewis et al., 2015). Dysfunction of the TRN is associated with epilepsy (Huguenard and McCormick, 2007) and schizophrenia (Ferrarelli and Tononi, 2011).
It has been proposed that TRN neurons form chemical synapses between each other (Ahlsén and Lindström, 1982) and that these intrinsic GABAergic connections play an important role in selective attention by allowing cross-modal interactions in the thalamus (Crick, 1984; Ahrens et al., 2015). The TRN is implicated in generating synchronous oscillations in the thalamocortical circuit including spike-wave discharge that is characteristic of absence seizures (Steriade, 2005; Huguenard and McCormick, 2007). Intra-TRN GABAergic connections are thought to suppress spike-wave discharge by limiting the number and synchrony of burst discharges of TRN neurons (Huntsman et al., 1999; Sohal and Huguenard, 2003). However, the existence of intra-TRN GABAerigic projections is controversial. Although early studies reported that TRN neurons form axonal collaterals in the TRN (Cox et al., 1996), detailed analyses of the axonal projections of TRN neurons did not detect any intra-TRN projections (Pinault et al., 1997). Results from functional analyses were also inconclusive. Circuit-mapping studies using local glutamate application and glutamate uncaging provided support for intra-TRN projections (Sanchez-Vives et al., 1997; Shu and McCormick, 2002; Deleuze and Huguenard, 2006; Lam et al., 2006). However, extensive paired recordings of TRN neurons did not detect any chemical synapses formed between TRN neurons (Landisman et al., 2002). One possible explanation is that TRN neurons preferentially form long-range, cross-modal connections within the TRN, and such long-range connections would be difficult to detect with paired recording of neighboring cells.
In this study, we used two complementary approaches to examine the presence of intra-TRN synaptic connections. First, we expressed channelrhodopsin (ChR2) throughout the TRN and analyzed synaptic responses to stimulation of TRN neurons and their axons. Second, we genetically blocked GABAergic transmission from TRN neurons and analyzed spontaneous synaptic transmission. Our results demonstrate that except for a brief period after birth the TRN of the mouse lacks intrinsic GABAergic connections.
Materials and Methods
Mice.
Parvalbumin (PV, Pvalb)-cre (JAX stock #17320), Rosa26-tdTomato reporter (Ai14; JAX stock #7914), Rosa26-ChR2(H134R)-EYFP reporter (Ai32; JAX stock #12569), and vesicular GABA transporter (VGAT) conditional knock-out mice (JAX stock #12897) were housed in the Research Animal Facility at The Jackson Laboratory. Pvalb-cre, Ai14, and Ai32 mice were on a C57BL/6J (B6) background; the conditional VGAT strain, originally on a mixed 129;B6 background, was backcrossed to B6 for six generations before the experiments. Genotypes were determined by PCR using tail DNA. Information about PCR primers and reactions is available from The Jackson Laboratory website. Both male and female mice were used for the experiments. All procedures were approved by The Jackson Laboratory Animal Care and Use Committee.
Immunostaining and confocal microscopy.
Mice were anesthetized with tribromoethanol and perfused with 4% paraformaldehyde in 0.1 m PBS. Serial coronal sections 70 μm thick were cut on a vibratome. For each mouse, six [postnatal day 14 (P14)–P20] or seven (P42–P46) sections spaced 70 μm apart were used to cover the entire TRN along the rostral–caudal axis. The most rostral and caudal sections correspond to those of bregma −0.5 and −1.8 mm in adult mice, respectively (Paxinos and Franklin, 2001). Sections were incubated with antibodies of PV (1:3000, mouse; MAB1572, Millipore) and NeuN (1:500, rabbit; ABN78, Millipore), or GAD67 (1:1000; MAB5406, Millipore), for 48 h at 4°C. After wash, sections were incubated with Alexa Fluor–conjugated secondary antibodies (1:500, Invitrogen) for 2 h at room temperature. Sections were mounted in an antifade medium (Prolong Gold or Prolong Diamond, Invitrogen).
Confocal images were taken with a Leica SP5 microscope. For each mouse, six or seven coronal sections were analyzed. For each section, three or four nonoverlapping images (247 × 247 μm) were taken from the TRN along the dorsal–ventral axis with a 63× [1.4 numerical aperture (NA)] objective. These images were used for colocalization analyses. Cells expressing NeuN, PV, GAD67, or tdTomato were quantified manually using the Cell Counter plugin for ImageJ. In sections stained for NeuN and PV, the TRN was outlined using NeuN signal. In sections strained for GAD67, the TRN was outlined using GAD67 signal.
Slice electrophysiology.
Mice were anesthetized with tribromoethanol and decapitated. Horizontal or coronal sections 300 μm thick were prepared with a vibratome (Leica VT1200) and kept in artificial cerebral spinal fluid (ACSF) containing the following (in mm): 124 NaCl, 3.0 KCl, 1.5 CaCl2, 1.3 MgCl2, 1.0 NaH2PO4, 26 NaHCO3, and 20 glucose, saturated with 95% O2 and 5% CO2 at room temperature (21−23°C).
For patch-clamp recording, slices were transferred to a submersion type chamber where they were continuously perfused with ACSF saturated with 95% O2 and 5% CO2 at 32−33°C. Neurons in the TRN or ventral basal complex (VB) of the thalamus were viewed with a 40× objective and Nomarski optics. Whole-cell recordings were obtained from the soma with a Multiclamp 700B amplifier. For optogenetic experiments, the pipette solution contained the following (in mm): 120 Cs methylsulfate, 10 CsCl, 4 ATP-Mg, 0.3 GTP, 0.5 EGTA, 4 QX-314, and 10 HEPES, pH 7.2, 270–280 mOsm with sucrose. Junction potential was estimated to be 9 mV and corrected during recording. In some recordings of light-evoked IPSCs in VB neurons, the TRN was cut from the slice using the tip of a 25 gauge needle. For recordings of quantal GABAergic events, the pipette solution contained the following (in mm): 130 KCl, 4 ATP-Mg, 0.3 GTP-Na, 0.5 EGTA, and 10 HEPES, pH 7.2, 270–280 mOsm with sucrose. The series resistance, usually between 8 and 15 MΩ, was not compensated in voltage-clamp experiments. Loose patch recordings were obtained with pipettes filled ACSF. The seal resistance was between 40 and 200 MΩ, and the recording pipette was held near the zero-current potential.
ChR2-expressing neurons and their axons were activated by blue light (470 nm) delivered by a high-powered LED (M470L2, Thorlabs) coupled to the epifluorescence path of the microscope through a 40× objective (0.8 NA). The duration of light pulses was 3 ms for all experiments. The light intensity was controlled with the LED driver (LEDD1B, Thorlabs). Unless noted otherwise, full-field light stimulation was used, and the light spot on slices had a diameter of ∼550 μm. The power of light delivered to the slice was measured with a photodiode sensor (S120C, Thorlabs) placed under the objective. The maximum output power measured at the objective was 8.4 mW, and the maximum power density was 8.8 mW/mm2. Recordings were conducted with light power between 0.1 and 1.3 mW.
Experiments were conducted using AxoGraph X (AxoGraph Scientific). Data were filtered at 4 kHz and digitized at 20 kHz. Spontaneous synaptic events were analyzed using the event detection function of AxoGraph as described previously (Zhang et al., 2010).
Statistics.
Statistics were calculated using JMP (SAS). Data are given as means ± SEM.
Results
To select molecular markers for TRN neurons, we tested several markers of GABAergic interneurons using immunostaining on fixed brain sections from B6 mice at 2 and 6 weeks of age. We found that PV was strongly expressed in the TRN at 2 and 6 weeks of age (Fig. 1A). To quantify the percentage of TRN neurons expressing PV, we counted NeuN+ and PV+ cells across the entire TRN using six or seven coronal sections. At 2 weeks of age, 99% of NeuN+ somata in the TRN were also PV+, and the pattern remained the same at 6 weeks of age (Fig. 1A,D). We also examined PV expression genetically using the Pvalb-cre driver (Hippenmeyer et al., 2005) and the Rosa26-tdTomato reporter (Ai14; Madisen et al., 2010). Consistent with our immunostaining data in B6 mice, 98% of NeuN+ neurons in Pvalb-cre;Ai14 (PV-Ai14) mice expressed tdTomato (Fig. 1B,D). We also performed GAD67 immunostaining on sections from PV-Ai14 brains, and our results showed that 98% of GAD67+ neurons in the TRN also expressed tdTomato (Fig. 1C,D). Therefore, we conclude that at least 98% of TRN neurons in the mouse express PV.
Figure 1.

Over 98% of TRN neurons in the mouse express PV. A, Confocal images of brain sections immunostained for NeuN (red) and PV (green). The top and middle panels are from a P14 mouse; the bottom panel is from a mouse at P42. The TRN is encircled by a dashed line in the top panel; the middle and bottom panels are images of TRN neurons at higher magnification. B, Images of TRN neurons in a section from a PV-Ai14 mouse at P14 immunostained for NeuN (green). C, Images of TRN neurons in a section from a PV-Ai14 mouse at P19 immunostained for GAD67 (green). D, Quantification of TRN neurons expressing PV (blue bars) in B6 mice or Cre (red bars) in PV-Ai14 mice. In B6 mice, the percentage of TRN neurons expressing PV was 99.5 ± 0.2% at P14 (2041 cells from 3 mice) and 99.6 ± 0.1% at P42–P46 (2695 cells from 3 mice). In PV-Ai14 mice, the percentage of TRN neurons expressing Cre was 98.5 ± 0.5% at P14 (2554 cells from 3 mice immunostained for NeuN) and 98.3 ± 0.3% at P19–P20 (3067 cells from 3 mice immunostained for GAD67). Scale bars: A, top, 250 μm; middle, bottom, 40 μm; B, 40 μm; C, 50 μm. D, Dorsal; M, medial; L, lateral; V, ventral.
We used the Pvalb-cre driver and the Rosa26-ChR2(H134R)-EYFP reporter (Ai32; Madisen et al., 2012) to express ChR2 in TRN neurons. Patch-clamp recordings were performed in acute slices obtained from Pvalb-cre;Ai32 (PV-ChR2) mice at age P15–P20. To ensure optimal activation of neurons and axons, mice homozygous for Ai32 were used. We first recorded activity of TRN neurons in loose patch mode with ACSF in the recording pipette. Full-field application of blue light pulses (470 nm, 3 ms in duration) consistently evoked action potentials in all TRN neurons that we recorded (Fig. 2A, top trace; 29 of 29 cells from 4 mice). A single light pulse of 0.91 mW produced 3.9 ± 0.3 action potentials (n = 29 cells), and the time between the onset of light pulse and that of the first action potential was 4.7 ± 0.5 ms (n = 29 cells). This result suggests that activation of ChR2 was sufficient to trigger action potential in TRN neurons of PV-ChR2 mice. We next recorded ChR2 currents from TRN neurons using whole-cell recording. Cells were voltage clamped at −65 mV, the reversal potential of Cl−. All TRN neurons showed light-evoked ChR2 currents (Fig. 2B). The peak amplitude of ChR2 currents, obtained at the light power of 0.91 mW, was 669 ± 38 pA (n = 49 cells from 7 mice). These results indicate that ChR2 was highly expressed in TRN neurons of PV-ChR2 mice.
Figure 2.
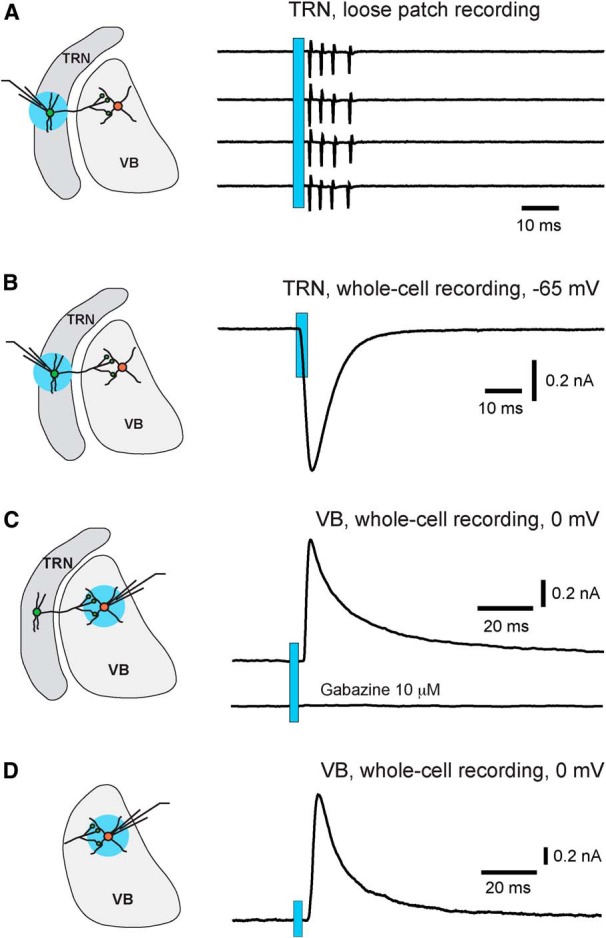
Light stimulation reliably activates TRN neurons and their axons in PV-ChR2 mice. All results were obtained from PV-ChR2 mice at age P15–P20. A, Light-evoked action potentials in a TRN neuron recorded in loose patch mode. The blue circle in the diagram on the left indicates the location of light stimulation. The light spot was 0.55 mm in diameter, and the power applied to the slice was 0.63 mW. The traces on the right are four consecutive trials with the blue bar indicating the application of light pulses. B, Light-evoked ChR2 currents recorded from a TRN neuron in whole-cell mode. The cell was voltage clamped at −65 mV, the reversal potential for Cl− in this experiment. The light power was 0.91 mW. C, Light-evoked IPSC in a VB neuron (top trace) and the blockade by the GABAA antagonist gabazine (bottom trace). VB neurons were recorded in whole-cell mode, and the reversal potential for Cl− was at −65 mV. The light spot was centered on the recorded VB neuron, and the light power was 0.63 mW. D, Light-evoked IPSCs in a VB neuron from a slice in which the TRN had been removed. At light power of 0.63 mW, the mean peak amplitude of light-evoked IPSCs recorded in slices without the TRN was 1294 ± 165 pA (n = 12 cells from 2 slices).
To determine whether the light pulses that we used are sufficient to activate synaptic terminals of TRN neurons, we performed whole-cell recording from VB neurons, which are targets of TRN projections. Light pulses were applied to the VB. The reversal potential of Cl− was at −65 mV, and IPSCs were recorded at 0 mV, the reversal potential of ChR2 currents. All VB neurons showed light-evoked IPSCs that were blocked by the GABAA antagonist gabazine (Fig. 2C). At light power of 0.63 mW, the peak amplitude of light-evoked IPSCs was 972 ± 110 pA (n = 14 cells from 4 mice). Reducing the size of the light spot from 550 to ∼250 μm in diameter had no effect on light-evoked IPSCs (data not shown). Removing the TRN from the slice also did not affect light-evoked IPSCs in VB neurons (Fig. 2D). These results indicate that light pulses used in these experiments reliably activated axons and axonal terminals of TRN neurons.
We next examined light-evoked IPSCs in TRN neurons. If TRN neurons were interconnected through GABAergic synapses, we would expect that a significant number of recorded neurons would show IPSCs in response to light pulses applied to the TRN. Instead, even with light powers twice of that used to evoke IPSCs in VB neurons, 98% of the TRN neurons did not show any light-evoked IPSCs (Fig. 3A; 48 of 49 cells from 7 mice). The same results were obtained in horizontal (n = 24 cells from 4 mice) and coronal slices (n = 25 cells from 3 mice). Only one of the 49 cells recorded showed small light-evoked IPSCs (data not shown). These findings demonstrated that activation of TRN neurons or their axons does not produce IPSCs in the vast majority of TRN neurons at P15–P20.
Figure 3.

Lack of light-evoked IPSCs in TRN neurons at age P15 or older. All results were obtained from PV-ChR2 mice. Whole-cell recordings were performed, and the holding potential was at 0 mV, the reversal potential of ChR2 currents. The light power was 1.3 mW for all the experiments presented here. A, Lack of light-evoked IPSCs in a TRN neuron at P16. Light pulses were applied either centered at (circle 2, left) or 0.4 mm away (circles 1 and 3) from the recorded cell. Results of five consecutive trials at each spot are shown on the right. B, Lack of light-evoked IPSCs in a TRN neuron at P62. C, Light-evoked IPSCs and the blockade by gabazine in a TRN neuron at P12. The average peak amplitude of light-evoked IPSCs at P11–P13 was 77 ± 17 pA (n = 9 cells).
We also examined light-evoked responses in PV-Ai32 mice between 5 and 11 weeks of age. All TRN neurons at these ages showed large ChR2 currents; at light power of 0.91 mW, the mean peak amplitude of ChR2 currents was 1742 ± 101 pA (n = 22 cells). However, we did not detect any light-evoked IPSCs in these TRN neurons (Fig. 3B; n = 5 cells at P34, n = 9 cells at P62, n = 8 cells at P79). These results suggest a lack of intrinsic GABAergic connections in the TRN of young adult mice.
Previous studies conducted at younger ages have reported that some TRN neurons receive GABAergic inputs from other cells in the TRN (Zhang and Jones, 2004; Deleuze and Huguenard, 2006; Lam et al., 2006). To determine whether intra-TRN GABAergic connections are more common in immature TRN, we performed additional experiments in slices from PV-ChR2 mice at age P11–P13. We found that 32% of TRN neurons showed light-evoked IPSCs (Fig. 3C; 9 of 28 cells from 5 mice; p < 0.001 vs P15–P20, Fisher's exact test). This result is consistent with previous studies at these ages and suggests that chemical synapses are formed between TRN neurons in newborns and that these connections are subsequently downregulated during early life.
An alternative explanation for the lack of evoked IPSCs in TRN neurons of PV-ChR2 mice is that local axon collaterals in the TRN may express low levels of ChR2, and thus most of the cut axon collaterals cannot be activated by light stimulation. To examine this possibility, we deleted the vesicular GABA transporter from TRN neurons using the Pvalb-cre driver and the conditional Vgat allele (Vgat F/F; Tong et al., 2008), and analyzed GABAergic transmission in slices. Miniature IPSCs (mIPSCs) were dramatically reduced in VB neurons of Pvalb-cre;Vgat F/F (PV-Vgat−/−) mice (Fig. 4A–C), thus confirming the deletion of Vgat from TRN neurons. However, deletion of Vgat from TRN neurons had no effect on mIPSCs in TRN neurons (Fig. 4D–F). There was no difference between mutant and control mice in the frequency or amplitude of mIPSCs recorded from TRN neurons. These results further suggest that GABAergic inputs to the TRN come from external rather than intra-TRN sources.
Figure 4.

Conditional deletion of VGAT from TRN neurons reduces GABAergic transmission in VB neurons but not TRN neurons. A, Quantal IPSCs (mIPSCs) were recorded from VB neurons of WT (top trace) and PV-Vgat−/− (bottom trace) mice. The reversal potential of Cl− was at 0 mV, and mIPSCs were recorded at −70 mV in the presence of tetrodotoxin (0.3 μm), DNQX (10 μm), and kynurenic acid (1 mm). B, C, Frequency and peak amplitude of mIPSCs of VB neurons from WT (filled circles) and PV-Vgat−/− (open circles) mice at age P15–P18. The average frequency of mIPSCs of VB neurons was 5.5 ± 0.6 Hz (n = 15 cells from 3 mice) for WT and 1.7 ± 0.2 Hz (n = 18 cells from 3 mice; ***p < 0.0001 vs WT, Wilcoxon test) for mutant mice; the average peak amplitude of mIPSCs of VB neurons was 28.7 ± 2.3 pA for WT and 16.5 ± 1.7 pA for mutant mice (***p = 0.0002 vs WT, Wilcoxon test). D–F, Results obtained from TRN neurons of WT and PV-Vgat−/− mice aged P15–P18. The average frequency of mIPSCs of TRN neurons was 3.4 ± 0.3 Hz (n = 20 cells from 3 mice) for WT mice and 3.1 ± 0.2 Hz (n = 23 cells from 3 mice; p = 0.53, Wilcoxon test) for mutant mice; the average peak amplitude of mIPSCs of TRN neurons was 35.9 ± 1.6 pA for WT mice and 38.9 ± 2.0 pA for mutant mice (p = 0.47, Wilcoxon test).
Discussion
Intrinsic GABAergic connections in the TRN
Despite numerous studies on GABAergic transmission in the TRN, the evidence for intra-TRN connections remained controversial. Early in vivo studies showed that stimulation of the visual cortex of cats evoked IPSPs in neurons in the perigeniculate nucleus (PGN), and it was proposed that this inhibition was caused by GABAergic connections between PGN neurons (Ahlsén and Lindström, 1982). Consistent with this idea, local applications of glutamate in the PGNs of ferrets in acute slices evoked GABAergic IPSPs in nearby PGN neurons (Sanchez-Vives et al., 1997; Shu and McCormick, 2002). Similar results were obtained in the rat TRN with laser-guided glutamate uncaging (Deleuze and Huguenard, 2006; Lam et al., 2006) and in the mouse TRN with focal electrical stimulation of cortical layer 6 (Zhang and Jones, 2004). However, extensive paired recording from TRN neurons in rats and mice revealed electric coupling but rarely chemical synapses between TRN neurons (Landisman et al., 2002; Long et al., 2004; Parker et al., 2009). Furthermore, optogenetic stimulation of layer 6 corticothalamic neurons in the mouse failed to evoke IPSCs in TRN neurons (Cruikshank et al., 2010; Crandall et al., 2015). Besides the differences in species, the discrepancy observed in rodents may have a developmental cause. Studies supporting the presence of intra-TRN GABAergic connections were conducted in rodents younger than 2 weeks of age (Zhang and Jones, 2004; Deleuze and Huguenard, 2006; Lam et al., 2006), whereas those against were mostly conducted after 2 weeks of age (Landisman et al., 2002; Cruikshank et al., 2010). In one study where paired recordings were made from mice aged between P1 and P14, the incidence of GABAergic connections between TRN neurons was found to be 3% (Parker et al., 2009). This is likely an underestimation, as these paired recordings were made from neighboring cells.
In this study, we used the Pvalb-cre driver and the Ai32 reporter to express ChR2 in about 98% of TRN neurons. We also used whole-field light stimulation to simultaneously activate a large number of TRN axons. This is a major advantage over previous paired recording and glutamate uncaging studies, in which only one or a few TRN neurons were activated at a time. We found that activation of TRN neurons and their axons failed to evoke IPSCs in TRN neurons from mice older than P15. To complement the optogenetic approach, we genetically blocked GABAergic transmission from TRN neurons using conditional Vgat deletion. We found that Vgat deletion from TRN neurons had no effect on mIPSCs in TRN neurons. Together, our results demonstrate a lack of intra-TRN GABAergic connections in mice older than P15.
We found that between P11 and P13, 30% of TRN neurons in PV-ChR2 mice showed light-evoked GABAergic IPSCs. This is consistent with previous studies conducted at these ages (Zhang and Jones, 2004; Deleuze and Huguenard, 2006; Lam et al., 2006). Thus, it is likely that intra-TRN GABAergic connections are present in newborn mice but they are eliminated after 2 weeks of age. However, we cannot exclude the possibility that at least some of these light-evoked IPSCs are due to activation of external GABAergic inputs.
Our results suggest that few if any TRN neurons form axodendritic synapses with other TRN neurons in adult mice. This is consistent with anatomical evidence. Single-cell labeling in vivo showed that only 10% of TRN neurons in the rat have local axonal collaterals, and none of the axonal collaterals appear to form synapses with other TRN neurons (Pinault et al., 1997). On the other hand, electron microscopy studies revealed that dendrites of TRN neurons in cats and rats form symmetrical synapses (Deschênes et al., 1985; Pinault et al., 1997). The function of these dendrodendritic synapses is unknown. Several studies showed that action potentials generated at the soma of TRN neurons trigger large calcium increases in the distal dendrites (Crandall et al., 2010; Chausson et al., 2013; Connelly et al., 2015), which should be sufficient to trigger synaptic release at dendrodendritic synapses. However, due to cable properties of dendrites, small synaptic responses generated at distal dendrites may not be detectable with somatic recordings. Thus, we cannot completely exclude the presence of dendrodendritic GABAergic synapses in the TRN.
External GABAergic inputs to the TRN
Our results suggest that in the mouse, the vast majority, if not all, of GABAergic inputs to the TRN come from other brain regions. Previous studies have identified a number of external GABAergic inputs to the TRN. These include the basal forebrain in rats (Asanuma, 1989; Asanuma and Porter, 1990) and cats (Bickford et al., 1994), the substantia nigra pars reticulata in cats (Pare et al., 1990), and the globus pallidus in monkeys (Hazrati and Parent, 1991; Asanuma, 1994) and rats (Govindaiah et al., 2010). Whether similar GABAergic inputs are present in the mouse is unknown. A recent optogenetic study in the mouse showed that GABAergic neurons in the lateral hypothalamus innervate the TRN and this projection has an important role in regulating arousal (Herrera et al., 2016). GABAergic neurons are highly diverse, and it is unknown whether specific subtypes of GABAergic neurons project to the TRN. The lack of optogenetically evoked IPSCs in the TRNs of young adult PV-ChR2 mice suggests that external GABAergic inputs to the TRN primarily come from PV-negative neurons.
GABAergic synaptic transmission in the TRN has critical roles in thalamocortical function and in the pathophysiology of epilepsy. Our results help clarify the issue of intrinsic GABAergic connections. Further studies are needed to identify and ascertain GABAergic projections to the TRN and to elucidate the physiological role of these inputs.
Footnotes
This work was supported by National Institutes of Health Grants NS064013 and NS082658. We thank Martin Deschênes for comments on a previous version of this manuscript and Bradley Clemens and Hong Liu for technical assistance.
References
- Ahlsén G, Lindström S. Mutual inhibition between perigeniculate neurones. Brain Res. 1982;236:482–486. doi: 10.1016/0006-8993(82)90731-4. [DOI] [PubMed] [Google Scholar]
- Ahrens S, Jaramillo S, Yu K, Ghosh S, Hwang GR, Paik R, Lai C, He M, Huang ZJ, Li B. ErbB4 regulation of a thalamic reticular nucleus circuit for sensory selection. Nat Neurosci. 2015;18:104–111. doi: 10.1038/nn.3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asanuma C. Axonal arborizations of a magnocellular basal nucleus input and their relation to the neurons in the thalamic reticular nucleus of rats. Proc Natl Acad Sci U S A. 1989;86:4746–4750. doi: 10.1073/pnas.86.12.4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asanuma C. GABAergic and pallidal terminals in the thalamic reticular nucleus of squirrel monkeys. Exp Brain Res. 1994;101:439–451. doi: 10.1007/BF00227337. [DOI] [PubMed] [Google Scholar]
- Asanuma C, Porter LL. Light and electron microscopic evidence for a GABAergic projection from the caudal basal forebrain to the thalamic reticular nucleus in rats. J Comp Neurol. 1990;302:159–172. doi: 10.1002/cne.903020112. [DOI] [PubMed] [Google Scholar]
- Bickford ME, Günlük AE, Van Horn SC, Sherman SM. GABAergic projection from the basal forebrain to the visual sector of the thalamic reticular nucleus in the cat. J Comp Neurol. 1994;348:481–510. doi: 10.1002/cne.903480402. [DOI] [PubMed] [Google Scholar]
- Chausson P, Leresche N, Lambert RC. Dynamics of intrinsic dendritic calcium signaling during tonic firing of thalamic reticular neurons. PLoS One. 2013;8:e72275. doi: 10.1371/journal.pone.0072275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly WM, Crunelli V, Errington AC. The global spike: conserved dendritic properties enable unique Ca2+ spike generation in low-threshold spiking neurons. J Neurosci. 2015;35:15505–15522. doi: 10.1523/JNEUROSCI.2740-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CL, Huguenard JR, Prince DA. Heterogeneous axonal arborizations of rat thalamic reticular neurons in the ventrobasal nucleus. J Comp Neurol. 1996;366:416–430. doi: 10.1002/(SICI)1096-9861(19960311)366:3<416::AID-CNE4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Crandall SR, Govindaiah G, Cox CL. Low-threshold Ca2+ current amplifies distal dendritic signaling in thalamic reticular neurons. J Neurosci. 2010;30:15419–15429. doi: 10.1523/JNEUROSCI.3636-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crandall SR, Cruikshank SJ, Connors BW. A corticothalamic switch: controlling the thalamus with dynamic synapses. Neuron. 2015;86:768–782. doi: 10.1016/j.neuron.2015.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. Function of the thalamic reticular complex: the searchlight hypothesis. Proc Natl Acad Sci U S A. 1984;81:4586–4590. doi: 10.1073/pnas.81.14.4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruikshank SJ, Urabe H, Nurmikko AV, Connors BW. Pathway-specific feedforward circuits between thalamus and neocortex revealed by selective optical stimulation of axons. Neuron. 2010;65:230–245. doi: 10.1016/j.neuron.2009.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleuze C, Huguenard JR. Distinct electrical and chemical connectivity maps in the thalamic reticular nucleus: potential roles in synchronization and sensation. J Neurosci. 2006;26:8633–8645. doi: 10.1523/JNEUROSCI.2333-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschênes M, Madariaga-Domich A, Steriade M. Dendrodendritic synapses in the cat reticularis thalami nucleus: a structural basis for thalamic spindle synchronization. Brain Res. 1985;334:165–168. doi: 10.1016/0006-8993(85)90580-3. [DOI] [PubMed] [Google Scholar]
- Ferrarelli F, Tononi G. The thalamic reticular nucleus and schizophrenia. Schizophr Bull. 2011;37:306–315. doi: 10.1093/schbul/sbq142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindaiah G, Wang T, Gillette MU, Crandall SR, Cox CL. Regulation of inhibitory synapses by presynaptic D(4) dopamine receptors in thalamus. J Neurophysiol. 2010;104:2757–2765. doi: 10.1152/jn.00361.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillery RW, Harting JK. Structure and connections of the thalamic reticular nucleus: advancing views over half a century. J Comp Neurol. 2003;463:360–371. doi: 10.1002/cne.10738. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Chen Z, Wimmer RD, Brunetti PM, Zhao S, Zikopoulos B, Wang F, Brown EN, Wilson MA. State-dependent architecture of thalamic reticular subnetworks. Cell. 2014;158:808–821. doi: 10.1016/j.cell.2014.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazrati LN, Parent A. Projection from the external pallidum to the reticular thalamic nucleus in the squirrel monkey. Brain Res. 1991;550:142–146. doi: 10.1016/0006-8993(91)90418-U. [DOI] [PubMed] [Google Scholar]
- Herrera CG, Cadavieco MC, Jego S, Ponomarenko A, Korotkova T, Adamantidis A. Hypothalamic feedforward inhibition of thalamocortical network controls arousal and consciousness. Nat Neurosci. 2016;19:290–298. doi: 10.1038/nn.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, Arber S. A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol. 2005;3:e159. doi: 10.1371/journal.pbio.0030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR, McCormick DA. Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends Neurosci. 2007;30:350–356. doi: 10.1016/j.tins.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Huntsman MM, Porcello DM, Homanics GE, DeLorey TM, Huguenard JR. Reciprocal inhibitory connections and network synchrony in the mammalian thalamus. Science. 1999;283:541–543. doi: 10.1126/science.283.5401.541. [DOI] [PubMed] [Google Scholar]
- Lam YW, Nelson CS, Sherman SM. Mapping of the functional interconnections between thalamic reticular neurons using photostimulation. J Neurophysiol. 2006;96:2593–2600. doi: 10.1152/jn.00555.2006. [DOI] [PubMed] [Google Scholar]
- Landisman CE, Long MA, Beierlein M, Deans MR, Paul DL, Connors BW. Electrical synapses in the thalamic reticular nucleus. J Neurosci. 2002;22:1002–1009. doi: 10.1523/JNEUROSCI.22-03-01002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis LD, Voigts J, Flores FJ, Schmitt LI, Wilson MA, Halassa MM, Brown EN. Thalamic reticular nucleus induces fast and local modulation of arousal state. eLife. 2015:4. doi: 10.7554/eLife.08760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long MA, Landisman CE, Connors BW. Small clusters of electrically coupled neurons generate synchronous rhythms in the thalamic reticular nucleus. J Neurosci. 2004;24:341–349. doi: 10.1523/JNEUROSCI.3358-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Mao T, Koch H, Zhuo JM, Berenyi A, Fujisawa S, Hsu YW, Garcia AJ, 3rd, Gu X, Zanella S, Kidney J, Gu H, Mao Y, Hooks BM, Boyden ES, Buzsáki G, Ramirez JM, Jones AR, Svoboda K, Han X, et al. A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat Neurosci. 2012;15:793–802. doi: 10.1038/nn.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlonan K, Cavanaugh J, Wurtz RH. Guarding the gateway to cortex with attention in visual thalamus. Nature. 2008;456:391–394. doi: 10.1038/nature07382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Bal T. Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci. 1997;20:185–215. doi: 10.1146/annurev.neuro.20.1.185. [DOI] [PubMed] [Google Scholar]
- Paré D, Hazrati LN, Parent A, Steriade M. Substantia nigra pars reticulata projects to the reticular thalamic nucleus of the cat: a morphological and electrophysiological study. Brain Res. 1990;535:139–146. doi: 10.1016/0006-8993(90)91832-2. [DOI] [PubMed] [Google Scholar]
- Parker PR, Cruikshank SJ, Connors BW. Stability of electrical coupling despite massive developmental changes of intrinsic neuronal physiology. J Neurosci. 2009;29:9761–9770. doi: 10.1523/JNEUROSCI.4568-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2001. [Google Scholar]
- Pinault D. The thalamic reticular nucleus: structure, function and concept. Brain Res Brain Res Rev. 2004;46:1–31. doi: 10.1016/j.brainresrev.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Pinault D, Smith Y, Deschênes M. Dendrodendritic and axoaxonic synapses in the thalamic reticular nucleus of the adult rat. J Neurosci. 1997;17:3215–3233. doi: 10.1523/JNEUROSCI.17-09-03215.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Vives MV, Bal T, McCormick DA. Inhibitory interactions between perigeniculate GABAergic neurons. J Neurosci. 1997;17:8894–8908. doi: 10.1523/JNEUROSCI.17-22-08894.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, McCormick DA. Inhibitory interactions between ferret thalamic reticular neurons. J Neurophysiol. 2002;87:2571–2576. doi: 10.1152/jn.00850.2001. [DOI] [PubMed] [Google Scholar]
- Sohal VS, Huguenard JR. Inhibitory interconnections control burst pattern and emergent network synchrony in reticular thalamus. J Neurosci. 2003;23:8978–8988. doi: 10.1523/JNEUROSCI.23-26-08978.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M. Sleep, epilepsy and thalamic reticular inhibitory neurons. Trends Neurosci. 2005;28:317–324. doi: 10.1016/j.tins.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- Tong Q, Ye CP, Jones JE, Elmquist JK, Lowell BB. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci. 2008;11:998–1000. doi: 10.1038/nn.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Jones EG. Corticothalamic inhibition in the thalamic reticular nucleus. J Neurophysiol. 2004;91:759–766. doi: 10.1152/jn.00624.2003. [DOI] [PubMed] [Google Scholar]
- Zhang ZW, Zak JD, Liu H. MeCP2 is required for normal development of GABAergic circuits in the thalamus. J Neurophysiol. 2010;103:2470–2481. doi: 10.1152/jn.00601.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zikopoulos B, Barbas H. Prefrontal projections to the thalamic reticular nucleus form a unique circuit for attentional mechanisms. J Neurosci. 2006;26:7348–7361. doi: 10.1523/JNEUROSCI.5511-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]