

Abstract
IMPORTANCE
Repeats of CAG in the ataxin 2 gene (ATXN2) in the long-normal range (sometimes referred to as intermediate) have been identified as modifiers of amyotrophic lateral sclerosis (ALS) risk. Prior studies have used thresholding considering various cutoffs for ATXN2 repeat length.
OBJECTIVE
To calculate association between ATXN2 CAG repeat alleles and increased risk of ALS across multiple ethnic groups.
DATA SOURCES
The MEDLINE database was searched for studies published by December 29, 2013, reporting ATXN2 CAG repeat length in patients with ALS and controls.
STUDY SELECTION
Studies were included if they reported original data on relative risks or odds ratios (ORs) from ALS and control populations for individual ATXN2 alleles. Review articles that reported no new data were not included in the analysis.
DATA EXTRACTION AND SYNTHESIS
Analysis of allele distribution was performed to ensure that all studies followed identical allele sizing. The ORs, 95% confidence intervals, and population attributable risk percentages were calculated according to standard procedures.
MAIN OUTCOMES AND MEASURES
Occurrence of ALS associated with ATXN2 repeat alleles, expressedas ORs.
RESULTS
Nine studies were analyzed, including 7505 controls and 6151 sporadic ALS cases. The ALS and control cohorts were recruited from different geographical and ethnic regions including the United States, French Canada/Canada, Belgium and the Netherlands, Germany, Italy, mainland China, Turkey, and Flanders-Belgium. The ATXN2 CAG repeat lengths ranged from 13 to 39 in patients with ALS and from 13 to 34 in controls. The ORs were less than 1.00 for alleles with 25 to 28 repeats. The OR was 1.55 for 30 repeats, but this elevation was not statistically significant (95% CI, 0.88–2.73). The ORs were 2.70 (95% CI, 1.47–4.93) for 31 CAG repeats, 11.09 (95% CI, 4.16–29.57) for 32 repeats, and 5.76 (95% CI, 1.79–18.57) for 33 repeats.
CONCLUSIONS AND RELEVANCE
In contrast to prior studies with smaller numbers, risk for ALS associated with long-normal alleles is complex. Alleles with 27 and 28 repeats lower ALS risk slightly. The risk for ALS increases beginning with 29 repeats and reaches a maximum at 32 and 33 repeats. Of note, alleles with repeats of these lengths are known to be predisposed to meiotic expansion to full-penetrance mutant alleles. In patients with ALS, alleles with 31 to 33 repeats may have undergone preferential expansion in motor neurons during mitosis or DNA repair. Our meta-analysis provides a framework for counseling individuals with long-normal ATXN2 repeats.
Spinocerebellar ataxia type 2 (SCA2) is a rare autosomal dominant neurodegenerative disorder caused by expanded glutamine repeats located in the N-terminal region of the ataxin 2 gene (ATXN2; GenBank BC114546). The ATXN2 polyglutamine allele length, although variable, is most commonly 22 repeats with rarer nonpathological alleles ranging from 14 to 31 repeats. Disease alleles causing ataxia contain 33 or more repeats.1,2 Longer pathogenic polyglutamine repeat lengths are inversely correlated with the age at onset.3 Pathology of SCA2 is characterized by considerable neuronal loss in the cerebellar Purkinje cell layer as well as all 4 deep cerebellar nuclei.4 In addition to cerebellar ataxia, patients with SCA2 may show other clinical signs such as saccade slowing, early hyporeflexia, severe tremors, and myoclonus.5
A recent study by Elden et al6 showed that ATXN2 was a potent modifier of transactive response DNA-binding protein 43 kDa (TDP-43) through an RNA-dependent mechanism. TDP-43 has been found to be a major component of ubiquitinated cytoplasmic inclusions in neurons of patients with amyotrophic lateral sclerosis (ALS).7,8 Interestingly, Elden and colleagues went on to show that ATXN2 alleles with 27 to 33 repeats, a class of alleles they designated as intermediate, were significantly associated with ALS. Since then, many studies9–16 have shown that long-normal ATXN2 alleles contribute to an increased risk of ALS in a variety of ethnic backgrounds. Unfortunately, there seems to be a lack of consistency when defining the boundaries of intermediate-length repeats among the studies.
The purpose of our study was to perform a meta-analysis of published data to determine allele-specific risk for ALS for alleles ranging from 24 to 33 repeats.
Methods
Search Strategy
We undertook a MEDLINE database search on PubMed from July 2012 to December 2013 for studies that tracked ATXN2 CAG repeat length in patients with ALS. The Medical Subject Heading terms used were SCA2 OR ataxin-2 OR ATXN2 AND ALS OR amyotrophic lateral sclerosis. The analysis was restricted to articles published in English. The final search was conducted on December 29, 2013. Studies were included if they reported original data, either directly in the publication or provided on request, on relative risks or odds ratios (ORs) from case and control populations, with cases defined as patients with ALS and control samples representative of the general population or controls who were matched for age and/or geographical region to the cases. We excluded review articles that reported no new data and studies that reported on interrupted ATXN2 repeat lengths.
Statistical Analysis
Data were analyzed for genetic association between ATXN2 and ALS using Mantel-Haenszel methods in Stata version IC/12.1 statistical software (StataCorp LP). The ORs were determined for allele counts greater than 23, taking into account the weight of each study. The ORs for alleles with more than 34 repeats couldnot be computed as these alleles were not found in controls. A repeat of 0.5 was added to the control group so that ORs could be computed in instances in which controls were not reported for the respective repeat length. The addition of the small constant makes the estimate of Mantel-Haenszel pool ORs close to the maximum likelihood estimate.17,18 Population attributable risk percentages were calculated using standard methods.19
Study Populations
Most patients with ALS in previously published studies6,9–16 were diagnosed according to the El Escorial revised criteria.20 In all examined studies, control samples were regionally matched to unrelated participants with no reported history of neurological disease.
Results
From the MEDLINE database search, PubMed yielded 28 articles, of which 14 fit the inclusion criteria for the meta-analysis. To be included within the meta-analysis, specific data for individual ATXN2 allele frequencies greater than 23 for both the ALS and control cohorts were required. When sufficient data were found in the publication, the data were extracted from the published information. When adequate data were not published in the article, first and senior authors were contacted via e-mail to request additional information. Of the 14 eligible publications, 9 provided adequate data for analysis. Five of the 14 eligible studies did not respond to e-mails requesting additional data.
From the 9 studies, 7505 controls and 6151 sporadic ALS cases were analyzed. The ALS and control cohorts were recruited from different geographical and ethnic regions including the United States,6 French Canada/Canada,10 Belgium and the Netherlands,12 Germany,11 Italy,9,16 mainland China,15 Turkey,13 and Flanders-Belgium.14 The ATXN2 CAG repeat lengths ranged from 13 to 39 in patients with ALS and from 13 to 34 in controls. Overall ATXN2 CAG repeat length distribution is shown in Figure 1 and in the eTable in the Supplement. Repeat lengths 23 or less represented 97.19% of alleles in the control population and 96.64% of alleles in patients with ALS. This is congruent with prior studies.1,21,22 As previously reported,22,23 the third most common repeat allele in controls and patients contained 27 repeats, suggesting that all studies used similar sizing standards, allowing comparisons across studies.
Figure 1. Distribution of Ataxin 2 Gene (ATXN2) Polyglutamine Repeat Lengthsin Both Amyotrophic Lateral Sclerosis and Control Cohorts.
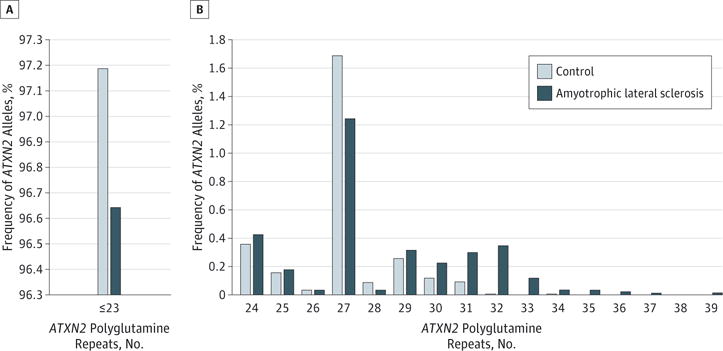
Distributions of ATXN2 polyglutamine repeat lengths are shown for 23 or fewer CAG repeats (A) and for at least 24 CAG repeats (B).
In contrast to prior studies that used a threshold for normal and predisposing alleles, which was often set post hoc, we had a sufficiently large number of alleles for each repeat size that allowed us to determine ORs specific for individual alleles. This meta-analysis revealed ORs of 1.00 or lower for alleles with 25 to 28 repeats. The first study examining ALS risk for CAG repeats between 27 and 33 reported a significantly increased risk for this class of alleles.6 The Table lists the ORs, 95% confidence intervals, and population attributable risk percentages for each allele summed across all studies. Contrary to previous studies, our analysis showed that ALS risk was decreased with CAG repeat lengths of 27 (OR = 0.77; 95% CI, 0.63–0.95) and 28 (OR = 0.44; 95% CI, 0.18–1.04). The OR was 1.55 for 30 repeats, but this higher OR value did not indicate a statistically significant risk increase for ALS (95% CI, 0.88–2.73). Beginning with 31 repeats, ORs were significantly elevated (Table). The population attributable risk percentage for alleles with more than 30 repeats, however, was relatively low in keeping with the overall rare occurrence of these alleles in the general population (Table).
Table.
Summary of Odds Ratios, 95% Confidence Intervals, and Population Attributable Risk Percentages for Ataxin 2 Gene Alleles With 24 to 33 CAG Repeats
| CAG Repeats, No. | Odds Ratio (95% CI) | Population Attributable Risk % |
|---|---|---|
| 24 | 1.10 (0.74–1.64) | 0.0415 |
| 25 | 0.70 (0.39–1.25) | 0.0113 |
| 26 | 0.80 (0.23–2.75) | −0.0003 |
| 27 | 0.77 (0.63–0.95) | −0.2434 |
| 28 | 0.44 (0.18–1.04) | −0.0306 |
| 29 | 1.15 (0.73–1.82) | 0.0333 |
| 30 | 1.55 (0.88–2.73) | 0.0616 |
| 31 | 2.70 (1.47–4.93) | 0.1182 |
| 32 | 11.09 (4.16–29.57) | 0.1948 |
| 33 | 5.76 (1.79–18.57) | 0.0694 |
The ORs of each study for alleles of 30, 31, 32, and 33 repeats are shown in Figure 2. The ORs were 2.70 (95% CI, 1.47–4.93) for 31 CAG repeats, 11.09 (95% CI, 4.16–29.57) for 32 repeats, and 5.76 (95% CI, 1.79–18.57) for 33 repeats. Not all studies were informative for each allele class, when the respective allele was not present in cases and in controls. This is particularly true for alleles of more than 34 repeats that were found in patients with ALS but not in controls (Figure 1).
Figure 2. Odds Ratios (ORs) for Individual Ataxin 2 Gene (ATXN2) Alleles With30 to 33 CAG Repeats Compared With Alleles With 23or Fewer CAG Repeats.
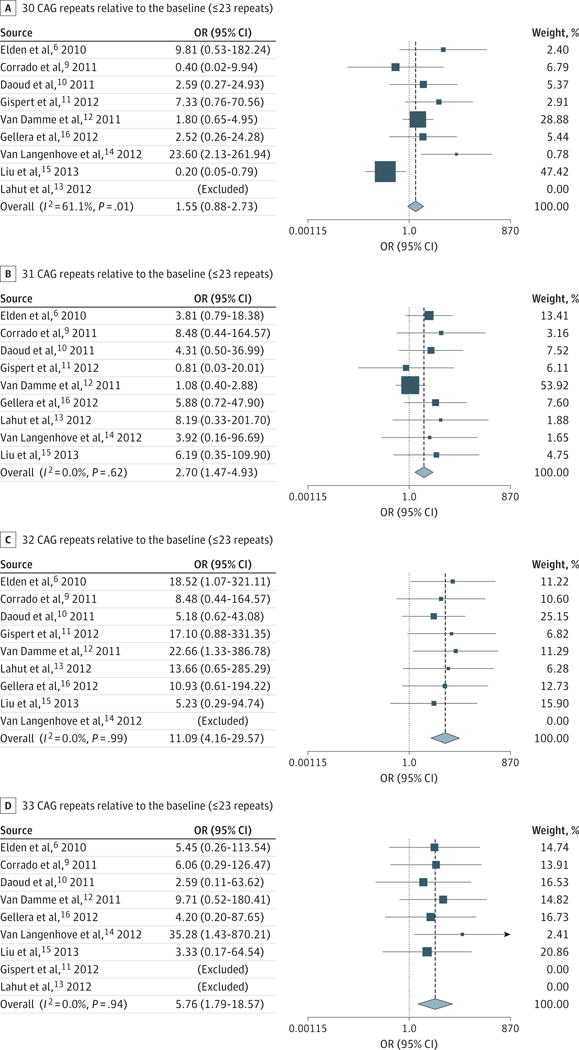
Data are shown for 30 (A), 31 (B), 32 (C), and 33 (D) CAG repeats relative to the baseline of 23 or fewer CAG repeats. Horizontal lines indicate 95% CI; arrow (D) indicates an off-scale 95% CI. Dashed line indicates the pooled OR, with the width of the diamond representing the 95% CI. Square size is representative of the size or weight of the individual study.
Discussion
Our analysis used 9 studies from the previously reported literature. Our meta-analysis supports previous observations of an increased risk of ALS with certain expanded ATXN2 alleles. This meta-analysis, however, provides a differentiated picture with some of the intermediate alleles associated with reduced risk and others with increased risk.
Elden et al6 originally defined the SCA2 repeat length for increased ALS risk to be 27 or more repeats. Since that 2010 publication, the clinically relevant range has been defined as widely as 29 or more repeats12 or more restricted to 31 to 34 repeats.11 Our meta-analysis has further defined the clinically relevant range of expanded ATXN2 polyglutamine regions that increase ALS risk as 31 or more CAG repeats. While there are inconsistencies in defining at which point polyglutamine ATXN2 expansions increase ALS risk in previously published studies, these discrepancies may be due to the diverse ethnic backgrounds investigated in existing research and to unrecognized population substructures in cases and controls.
Sizing of CAG repeats can be challenging. Because none of the studies reported a common sizing standard, we assessed the quality of sizing by examining the frequency distribution of alleles. All studies showed the expected distribution with 22 repeats as the most common allele, followed by alleles with 23 and 27 repeats.1,21,22 The frequency of the 27-repeat allele in individual studies, however, showed significant variability ranging from0.2%13 to 3.6%.12 Similar observations hold true for the 28- and 29-repeat alleles. The observed variation in frequency likely represents unrecognized population substructure and can, in retrospect, explain the spurious association of increased ALS risk for these alleles. It is also possible that these alleles have an increased risk in specific populations only.
Although the stated objectives of the studies included in our meta-analysis were similar, there were some differences in the definition and composition of ALS and control populations. The ALS and control cohorts were recruited from different geographical and ethnic regions including the United States,6 French Canada/Canada,10 Belgium and the Netherlands,12 Germany,11 Italy,9,16 mainland China,15 Turkey,13 and Flanders-Belgium.14 Additionally, the source and type of controls differed from one study to the next; controls were defined as neurologically normal,6,11,12 neurologically normal without any known history of neurological disorders,13,15 neurologically normal matched for age,10 neurologically normal matched for age and ethnicity,9,10,15 and neurologically normal with exclusion of incipient Parkinson disease through midbrain sonography.11
As shown in both Figure 1 and the eTable in the Supplement, several studies identified pathogenic SCA2 alleles with 34 or more repeats in patients with ALS. In total, 15 patients with ALS and full-length pathological SCA2 repeats (≥34) presented with a motor neuron syndrome as opposed to a cerebellar syndrome. A recent study described a family with coexistingSCA2 andALS.24 The proband with ATXN2 CAG repeats of 40 and 22 presented with degenerative ataxia, while her uncle had an upper and lower motor neuron syndrome with CAG repeats of 39 and 22. Familial ALS, caused by mutant ATXN2 alleles, was also observed in a Cuban population.25 This study, in addition to our findings, further supports the notion that expanded ATXN2 CAG repeats can give rise to several neurological disorders with a wide range of phenotypes.
It is not known what causes the shift from a typical cerebellar phenotype to a rare motor phenotype in SCA2. In principle, genetic, environmental, and stochastic factors could play a role. The occurrence of individuals with the typical cerebellar phenotype and the ALS phenotype in the same pedigree argues against importance of cis-acting factors. Nonallelic modifiers (genetic background) cannot be excluded, but the presence of the SCA2 ALS phenotype in different ethnic groups may make this mechanism less-likely. The relative rarity of the ALS phenotype suggests to us that stochastic events such as expansion of long-normal ATXN2 alleles in motor neurons during mitosis or DNA repair, both of which have genetic, epigenetic, or environmental underpinnings, may explain the exclusive motor neuron phenotype in some individuals. The possibility that only ataxin 2 molecules with a specific and narrow window of polyglutamine repeat lengths have an abnormal interaction with TDP-43 seems less likely.6 At this point, however, experimental data are lacking both in model systems and in human patients.
The ATXN2 alleles with 32 and 33 repeats are considered extremely rare and have been associated with very late-onset cerebellar ataxia for 32 repeats in the homozygous state in one publication26 (and by personal observation by one of us [S.M.P.]) and 33 repeats in the heterozygous state in another.2 Consistent with that notion, only 1 allele with 32 repeats and no alleles with 33 repeats were found among approximately 15 000 control alleles. Among approximately 12 000 ALS alleles, there were 43 alleles with 32 repeats and 15 alleles with 33 repeats.
In ALS, it is highly probable that both known and unknown susceptibility genes interact with environmental exposures to modulate disease risk.27 In addition to long-normal ATXN2 repeats, several other genes and environmental factors have been shown to have an association with ALS. Careers such as farming (OR = 2.0; 95% CI, 1.1–3.5)28 and some electrical occupations (OR = 2.3; 95% CI, 1.29–4.09)29 may be related to an increased risk of ALS.
Additionally, having ever smoked a cigarette (OR = 2.0; 95% CI, 1.3–3.2)30 or having a high intake of dietary fat (OR = 2.78; 95% CI, 0.9–8.0)31 have shown association with ALS; however, dietary fiber (OR = 0.3; 95% CI, 0.1–0.7)31 may have a preventive effect. Thus, ORs of 2.70, 11.09, and 5.76 for 31, 32, and 33 ATXN2 repeats, respectively, represent significant risk factors for ALS.
Conclusions
This meta-analysis provides a new framework for interpretation and counseling of patients with regard to ATXN2 alleles. Alleles with up to 30 CAG repeats can be considered normal with no apparent increased risk for neurodegenerative disease. In fact, alleles of 27 and 28 repeats had a slight but significantly reduced risk of ALS. Our analysis confirms that there is an increased risk for ALS with alleles of 31, 32, and 33 repeats. It is not known whether repeat interruptions modify this risk. Counseling also needs to include information that long-normal alleles show increased meiotic instability and can expand to full-mutation alleles in offspring. For alleles of 34 or more repeats, most individuals will develop a cerebellar degeneration but some may present with motor neuron disease (Figure 1). Of note, many patients with SCA2 presenting with cerebellar dysfunction will develop symptoms and signs of motor neuron disease late in the course of their illness.
Supplementary Material
Acknowledgments
Funding/Support: This work was supported in part by grants RC4NS073009, R21NS079852, R21NS081182, and R01NS33123 from the National Institute of Health (Dr Pulst) and by the University of Utah’s ACCESS and Bioscience Undergraduate Research Programs.
Role of the Funder/Sponsor: The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Author Contributions: Dr Pulst had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Pulst.
Acquisition, analysis, or interpretation of data: All authors.
Drafting of the manuscript: Neuenschwander, Thai, Pulst.
Critical revision of the manuscript for important intellectual content: Figueroa, Pulst.
Statistical analysis: Neuenschwander, Thai.
Obtained funding: Pulst.
Administrative, technical, or material support: Figueroa.
Study supervision: Pulst.
Conflict of Interest Disclosures: Dr Pulst has received royalties from Cedars-Sinai Medical Center and is a consultant for Progenitor Lifesciences. No other disclosures were reported.
Additional Information: After the manuscript was accepted for publication, Lattante et al described the contribution of ATXN2 intermediary polyglutamine expansions in a spectrum of neurodegenerative disorders (Lattante S, Millecamps S, Stevanin G, et al; French Research Network on FTD and FTD-ALS. Contribution of ATXN2 intermediary polyQ expansions in a spectrum of neurodegenerative disorders. Neurology. 2014;83[11]:990–995).
Additional Contributions: We are grateful to the patients with ALS and their spouses. We thank the investigators who sent detailed data for use in the meta-analysis. They received no compensation for these contributions.
References
- 1.Pulst SM, Nechiporuk A, Nechiporuk T, et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet. 1996;14(3):269–276. doi: 10.1038/ng1196-269. [DOI] [PubMed] [Google Scholar]
- 2.Fernandez M, McClain ME, Martinez RA, et al. Late-onset SCA2: 33 CAG repeats are sufficient to cause disease. Neurology. 2000;55(4):569–572. doi: 10.1212/wnl.55.4.569. [DOI] [PubMed] [Google Scholar]
- 3.Pulst SM, Santos N, Wang D, et al. Spinocerebellar ataxia type 2: polyQ repeat variation in the CACNA1A calcium channel modifies age of onset. Brain. 2005;128(pt 10):2297–2303. doi: 10.1093/brain/awh586. [DOI] [PubMed] [Google Scholar]
- 4.Scherzed W, Brunt ER, Heinsen H, et al. Pathoanatomy of cerebellar degeneration in spinocerebellar ataxia type 2 (SCA2) and type 3 (SCA3) Cerebellum. 2012;11(3):749–760. doi: 10.1007/s12311-011-0340-8. [DOI] [PubMed] [Google Scholar]
- 5.Lastres-Becker I, Rüb U, Auburger G. Spinocerebellar ataxia 2 (SCA2) Cerebellum. 2008;7(2):115–124. doi: 10.1007/s12311-008-0019-y. [DOI] [PubMed] [Google Scholar]
- 6.Elden AC, Kim HJ, Hart MP, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466(7310):1069–1075. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136(6):1001–1004. doi: 10.1016/j.cell.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pesiridis GS, Lee VM, Trojanowski JQ. Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum Mol Genet. 2009;18(R2):R156–R162. doi: 10.1093/hmg/ddp303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corrado L, Mazzini L, Oggioni GD, et al. ATXN-2 CAG repeat expansions are interrupted in ALS patients. Hum Genet. 2011;130(4):575–580. doi: 10.1007/s00439-011-1000-2. [DOI] [PubMed] [Google Scholar]
- 10.Daoud H, Belzil V, Martins S, et al. Association of long ATXN2 CAG repeat sizes with increased risk of amyotrophic lateral sclerosis. Arch Neurol. 2011;68(6):739–742. doi: 10.1001/archneurol.2011.111. [DOI] [PubMed] [Google Scholar]
- 11.Gispert S, Kurz A, Waibel S, et al. The modulation of amyotrophic lateral sclerosis risk by ataxin-2 intermediate polyglutamine expansions is a specific effect. Neurobiol Dis. 2012;45(1):356–361. doi: 10.1016/j.nbd.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 12.Van Damme P, Veldink JH, van Blitterswijk M, et al. Expanded ATXN2 CAG repeat size in ALS identifies genetic overlap between ALS and SCA2. Neurology. 2011;76(24):2066–2072. doi: 10.1212/WNL.0b013e31821f445b. [DOI] [PubMed] [Google Scholar]
- 13.Lahut S, Ömür Ö, Uyan Ö, et al. ATXN2 and its neighbouring gene SH2B3 are associated with increased ALS risk in the Turkish population. PLoS One. 2012;7(8):e42956. doi: 10.1371/journal.pone.0042956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Langenhove T, van der Zee J, Engelborghs S, et al. Ataxin-2 polyQ expansions in FTLD-ALS spectrum disorders in Flanders-Belgian cohorts. Neurobiol Aging. 2012;33(5):e17–e20. doi: 10.1016/j.neurobiolaging.2011.09.025. [DOI] [PubMed] [Google Scholar]
- 15.Liu X, Lu M, Tang L, Zhang N, Chui D, Fan D. ATXN2 CAG repeat expansions increase the risk for Chinese patients with amyotrophic lateral sclerosis. Neurobiol Aging. 2013;34(9):e5–e8. doi: 10.1016/j.neurobiolaging.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 16.Gellera C, Ticozzi N, Pensato V, et al. ATAXIN2 CAG-repeat length in Italian patients with amyotrophic lateral sclerosis: risk factor or variant phenotype? implication for genetic testing and counseling. Neurobiol Aging. 2012;33(8):e15–e21. doi: 10.1016/j.neurobiolaging.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 17.Lane PW. Meta-analysis of incidence of rare events. Stat Methods Med Res. 2013;22(2):117–132. doi: 10.1177/0962280211432218. [DOI] [PubMed] [Google Scholar]
- 18.Friedrich JO, Adhikari NK, Beyene J. Inclusion of zero total event trials in meta-analyses maintains analytic consistency and incorporates all available data. BMC Med Res Methodol. 2007;7:5. doi: 10.1186/1471-2288-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaelin MA, Bayona M. Attributable risk applications in epidemiology. http://www.collegeboard.com/prod_downloads/yes/4297_MODULE_17.pdf. Accessed March 28,2014.
- 20.Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 21.Sanpei K, Takano H, Igarashi S, et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet. 1996;14(3):277–284. doi: 10.1038/ng1196-277. [DOI] [PubMed] [Google Scholar]
- 22.Imbert G, Saudou F, Yvert G, et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet. 1996;14(3):285–291. doi: 10.1038/ng1196-285. [DOI] [PubMed] [Google Scholar]
- 23.Figueroa KP, Farooqi S, Harrup K, Frank J, O’Rahilly S, Pulst SM. Genetic variance in the spinocerebellar ataxia type 2 (ATXN2) gene in children with severe early onset obesity. PLoS One. 2009;4(12):e8280. doi: 10.1371/journal.pone.0008280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tazen S, Figueroa K, Kwan JY, et al. Amyotrophic lateral sclerosis and spinocerebellar ataxia type 2 in a family with full CAG repeat expansions of ATXN2. JAMA Neurol. 2013;70(10):1302–1304. doi: 10.1001/jamaneurol.2013.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laffita-Mesa JM, Rodríguez Pupo JM, Moreno Sera R, et al. De novo mutations in ataxin-2 gene and ALS risk. PLoS One. 2013;8(8):e70560. doi: 10.1371/journal.pone.0070560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Almaguer-Mederos LE, Falcón NS, Almira YR, et al. Estimation of the age at onset in spinocerebellar ataxia type 2 Cuban patients by survival analysis. Clin Genet. 2010;78(2):169–174. doi: 10.1111/j.1399-0004.2009.01358.x. [DOI] [PubMed] [Google Scholar]
- 27.Al-Chalabi A, Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol. 2013;9(11):617–628. doi: 10.1038/nrneurol.2013.203. [DOI] [PubMed] [Google Scholar]
- 28.McGuire V, Longstreth WT, Jr, Nelson LM, et al. Occupational exposures and amyotrophic lateral sclerosis: a population-based case-control study. Am J Epidemiol. 1997;145(12):1076–1088. doi: 10.1093/oxfordjournals.aje.a009070. [DOI] [PubMed] [Google Scholar]
- 29.Noonan CW, Reif JS, Yost M, Touchstone J. Occupational exposure to magnetic fields in case-referent studies of neurodegenerative diseases. Scand J Work Environ Health. 2002;28(1):42–48. doi: 10.5271/sjweh.645. [DOI] [PubMed] [Google Scholar]
- 30.Nelson LM, McGuire V, Longstreth WT, Jr, Matkin C. Population-based case-control study of amyotrophic lateral sclerosis in western Washington State, I: cigarette smoking and alcohol consumption. Am J Epidemiol. 2000;151(2):156–163. doi: 10.1093/oxfordjournals.aje.a010183. [DOI] [PubMed] [Google Scholar]
- 31.Nelson LM, Matkin C, Longstreth WT, Jr, McGuire V. Population-based case-control study of amyotrophic lateral sclerosis in western Washington State, II: diet. Am J Epidemiol. 2000;151(2):164–173. doi: 10.1093/oxfordjournals.aje.a010184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.