

Abstract
Oral cancer kills about 1 person every hour, each day in the United States and is the 6th most prevalent cancer worldwide. The pro-inflammatory cytokine ‘macrophage migration inhibitory factor’ (MIF) has been shown to be expressed in oral cancer patients, yet its precise role in oral carcinogenesis is not clear. In this study, we examined the impact of global Mif deletion on the cellular and molecular process occurring during oral carcinogenesis using a well-established mouse model of oral cancer with the carcinogen 4-nitroquinoline-1-oxide (4NQO). C57BL/6 Wild-type (WT) and Mif knock-out mice were administered with 4NQO in drinking water for 16 weeks, then regular drinking water for 8 weeks. Mif knock-out mice displayed fewer oral tumor incidence and multiplicity, accompanied by a significant reduction in the expression of pro-inflammatory cytokines Il-1β, Tnf-α, chemokines Cxcl1, Cxcl6 and Ccl3 and other molecular biomarkers of oral carcinogenesis Mmp1 and Ptgs2. Further, systemic accumulation of myeloid-derived tumor promoting immune cells was inhibited in Mif knock-out mice. Our results demonstrate that genetic Mif deletion reduces the incidence and severity of oral carcinogenesis, by inhibiting the expression of chronic pro-inflammatory immune mediators. Thus, targeting MIF is a promising strategy for the prevention or therapy of oral cancer.
Keywords: Oral, Cancer, Inflammation, Cytokine, Myeloid
INTRODUCTION
Oral cancer kills about 1 person every hour, each day in the United States and is the 6th most prevalent cancer worldwide, with about 280,000 cases reported annually1, 2. Overall survival rates for oral cancers (61% at 5 years, 51% at 10 years) have not significantly changed over the past 3 decades3. Major risk factors for oral cancer include tobacco smoking, excessive alcohol consumption and exposure to human papilloma virus (HPV), which create a chronic inflammatory micro-environment that favors oral carcinogenesis4, 5. Chronic inflammation, characterized by the sustained production of pro-inflammatory cytokines and the recruitment of immune cells, is recognized as a major player in the development of numerous cancers including cancers of the head and neck6.
The cytokine Macrophage Migration Inhibitory Factor (MIF) is a pleiotropic cytokine produced by immune and non-immune cells, which mediates chronic inflammatory immune responses7, 8. Although one of the first cytokines to be discovered in the mid 1960’s, the role of MIF as a biomarker and mediator of carcinogenesis has only become recognized by researchers within the past decade. MIF has been shown to constitute an important link between chronic inflammation and cancer7, and it is implicated in cancers of the breast9, colon10, prostate11, liver12, lung13 and skin14. In vitro and in vivo studies reveal that MIF binds to, and inhibits the activity of, the tumor suppressor gene p53, known to play a major role in cell cycle arrest and apoptosis in response to DNA damage15, 16. MIF signaling through the receptor CD74 leads to sustained ERK1/2 activation, resulting in the activation of genes involved in cell cycle progression. Interestingly, deregulation of the ERK1/2 MAP kinase pathway has been implicated in a third of all human cancers17. Further, MIF promotes angiogenesis, and under hypoxic conditions characteristic of tumor microenvironments, MIF has been shown to support tumor growth18, 19.
Clinical studies in oral cancer patients have demonstrated a correlation between MIF expression and oral tumor progression. One such study conducted by Franca et al. (2012) observed expression of MIF in oral tumor stromal cells as well as in infiltrating immune cells of patients with intra oral and lip tumors20. Further studies by Kindt et al.21, revealed a positive association between levels of MIF in tissue and tumor progression in oral cavity carcinomas. In another recent study, high MIF protein was observed in the saliva and serum of oral squamous cell carcinoma (OSCC) patients, and serologic MIF concentrations were predictive of tumor recurrence in post-surgical patients22. These studies suggest that expression of MIF in oral tumors could either be a consequence of oral carcinogenesis, or it could be that MIF actively plays a causative role in oral tumor formation. A recent study using MIF small interfering RNA (siRNA) knock-down technology demonstrated a reduction in cell proliferation, cell migration and colony formation in OSCC cells in vitro23, which suggests a causative role for MIF during oral carcinogenesis. However, there still remains a major gap in our understanding of the precise role MIF plays during oral carcinogenesis in vivo. In this study, we examined the impact of global MIF deletion on the cellular and molecular process occurring during oral carcinogenesis using a well-established mouse model of oral cancer. This model, which uses the carcinogen 4-nitroquinoline-1-oxide (4NQO) to induce oral carcinogenesis in C57BL/6 mice, has been shown to mimic the pathology of human oral cancer24, 25. Our results demonstrate that MIF plays an active role during oral carcinogenesis. We propose that targeting MIF is a potential strategy for prevention or therapy of oral cancers.
MATERIAL AND METHODS
Mice
C57BL/6 wild type (WT) and MIF knock out Mif−/− mice were maintained at the Ohio State University animal facility according to animal protocols and University Laboratory Animal Resources (ULAR) regulations. All experiments were approved by the Institutional Animal Care and Use Committee (Protocol # 2010A00000085) and Institutional Biosafety Committee (IBC) of the Ohio State University.
Chemicals
4-nitroquinoline-1-oxide (4NQO) was obtained from Sigma-Aldrich (St. Louis, MO) and stored in foil-wrapped containers at −20°C. Working solutions of 100μg/mL were prepared weekly in amber colored drinking water bottles and administered to mice for 16 weeks. This dose regimen has been shown to be effective at inducing tongue cancer in mice with minimal adverse effects.
OSCC Patient Samples
Oral tissue samples for total RNA isolation were obtained from OSCC patients (N = 38) enrolled in a Phase 0 clinical trial study (ClinicalTrials.gov ID NCT01465776) approved by the Internal Review Board at the Ohio State University Wexner Medical Center/The Arthur G. James and Richard J. Solove Research Institute.
Mouse oral carcinogenesis protocol
15 WT and 15 Mif−/− (6 – 8 week old) C57BL/6 mice were placed on AIN-76A diet and used to determine sensitivity to oral carcinogen, 4NQO. Both groups received 4NQO (100 μg/mL) in their drinking water for 16 weeks. Following 4NQO treatment, mice were placed on drinking water for 8 additional weeks. The use of 100 μg/mL for the induction of tongue cancer in mice has been previously reported by Ohkoshi et al.25, with minimal mortality (<6%). Mice were sacrificed at 24 weeks after the onset of treatment, the tongues harvested, and tumors/lesions evaluated macroscopically and microscopically. Tumor numbers and sizes were measured, and whole tongues were bisected, with one part of the tissue placed in buffered formalin for subsequent histological analysis, while the other part of the tongue tissue was stored in RNAlater for subsequent RNA isolation and gene expression analysis.
Microarray analysis
Total RNA was isolated from tumor and distant, non-involved phenotypically “normal” tissues of current OSCC patients (N = 6 per group) using an RNeasy Fibrous Tissue Kit (Qiagen, Valencia, CA, USA). Biopsies were obtained prior to treatment. RNA quantity, quality and integrity were confirmed by Nanodrop and Agilent Bioanalyzer before inclusion in the array. Microarray processing was performed at the Micro Array Shared Resource, The Ohio State University. RNA amplification, fragmentation and labelling were carried out according to manufacturer’s protocols (Affymetrix, Santa Clara, CA, USA). The arrays (Affymetrix GeneChip) were hybridized for 16 hours at 45°C and 60rpm. Washing and staining of arrays was performed at the fluidics station 450 according to manufacturer’s protocol (Affymetrix, Santa Clara, CA, USA). The microarrays were scanned using Affymetrix GeneChip Scanner 3000 7G using the Affymetrix GeneChip Command Console (AGCC) software (Affymetrix, Santa Clara, CA, USA). Background correction and quantile normalization was performed to adjust technical bias, and expression levels were summarized over probeset using RMA method. A filtering method based on percentage of arrays above noise cutoff was applied to filter out low expression genes. Affymetrix software Expression Console and statistical package R was used for the analysis. Microarray expression data have been submitted to the Gene Expression Omnibus (GSE accession # 74530).
Ingenuity Pathway Analysis of gene expression arrays
Molecular interactions among differentially regulated genes between tumor and adjacent non-tumor tissue in OSCC patients were explored using Ingenuity Pathway Analysis (IPA) (Qiagen Valencia, CA, USA). Each gene identifier was mapped to its corresponding gene in the Ingenuity Pathway Knowledge Base (IPKB). Families of genes which were up- or down-regulated in oral tumor tissue compared to adjacent non-tumor tissue were integrated into predictive network models based on gene interactions within a biological pathway defined in literature as contained in the IPKB.
Real Time PCR
Total RNA was extracted from OSCC tissues and adjacent non-tumor tissues of OSCC patients using the RNeasy Fibrous Tissue Kit (Qiagen, Valencia, CA, USA). Total RNA from tongue tissues of carcinogen treated and untreated WT and Mif−/− mice were extracted using the RNeasy mini kit (Qiagen Valencia, CA, USA). RNA was reverse transcribed to cDNA using the High capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Human and mouse primer sequences and cycling conditions for real-time PCR were obtained using the PRIMER BANK website (http://pga.mgh.harvard.edu/primerbank/index.html), Harvard Medical School. PCR amplification was performed in a 7900HT Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using SYBR Green (Qiagen Valencia, CA, USA) for detection. Data were normalized to the housekeeping gene GAPDH.
RT2 Profiler PCR Arrays
Total RNA from tongue tissues of WT and Mif−/− mice (N = 3 per group) was isolated using the RNeasy mini kit (Qiagen Valencia, CA, USA). RNA was reverse transcribed to cDNA using the RT2 HT First Strand kit (Qiagen Valencia, CA, USA). cDNA was applied to a RT2 profiler PCR Array plate containing a panel of mouse inflammatory response genes (Qiagen Valencia, CA, USA). Plates were run on a 7900HT Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using SYBR green for detection. Data were analyzed using the web-based RT2 profiler PCR Array data analysis software (www.SABiosciences.com/pcrarraydataanalysis.php).
Flow cytometry
Single cell suspensions were prepared from spleens and draining lymph nodes of WT and Mif−/− mice, washed in PBS and blocked with normal mouse serum. Cells were incubated with fluorochrome conjugated antibodies against the cell surface markers: CD4, CD8, CD3, NK1.1, F4/80, CD11b, Ly6C and Ly6G (Biolegend, SanDiego, CA). For FoxP3 intracellular staining, surface-stained cells were fixed and permeabilized with FoxP3 Fix/Perm solution (Biolegend, SanDiego, CA), then stained with PE-conjugated anti-FoxP3 antibodies (Biolegend, SanDiego, CA). Cells were acquired on a BD FACS Calibur or BD FACS Aria (BD Biosciences, San Jose, CA) and analysis was performed using the FlowJo software (Tree Star, Inc., Ashland, OR).
Histological grading of microscopic lesions
Formalin fixed tongue tissues of carcinogen induced WT and Mif−/− mice were paraffin embedded and serial sections were cut and mounted on slides. Slides were stained with hematoxylin and eosin and scanned at 25× and 100× magnifications. Each view in the field was categorized into 1 of 5 histologic categories as described previously26, namely normal epithelium, low-grade dysplasia, moderate dysplasia, high-grade dysplasia, carcinoma in situ, or squamous cell carcinoma. Low-grade (mild) dysplasia was characterized by changes in the epithelium such as basilar crowding and hyperplasia, cellular disorganization, and maturational disturbances not extending more than one third of the epithelial thickness with little interruption of the keratin layer. High-grade (severe) dysplasia included the preceding parameters extending beyond one half of the epithelial thickness but not affecting the entirety of the epithelium. Additional features included frequent mitotic figures, cellular pleomorphism, nuclear atypia, and some early disturbance of the keratin layer. Invasive carcinoma appeared as a full-thickness epithelial change with the previously stated features, with an expansion of multiple layers of cells into the suprabasal and intermediate layers, and with disturbance of the keratin layer and invasion through the basement membrane. Histological scoring was performed by an oral and maxillofacial pathologist (H.I).
Statistical Analysis
Data are presented as mean ± SEM. Gene expression values were normalized to the reference gene GAPDH and tested for significant changes between tumor tissue and matched adjacent non-tumor tissue for clinical patients, or carcinogen treated and untreated mice as well as WT and Mif−/− mice in our animal studies. Statistical analyses were done using Prism 5 (GraphPad Software, San Diego, CA). Student’s t test was employed to determine statistical significance of values obtained. The statistical package R was used for the analysis of microarray data. Comparative analysis of MIF expression in relation to clinicopathological parameters were performed using ANOVA. Significant differences were discriminated using Tukey’s post hoc test. P values <0.05 were considered statistically significant.
RESULTS
MIF is overexpressed in human oral cancers and in experimentally induced oral cancers
Previous studies have shown that MIF protein is expressed in oral tumors, and its expression correlates with oral tumor progression and is predictive of oral tumor recurrence in patients20–22. However, constitutive MIF protein expression occurs in various tissues. Therefore, we examined whether transcriptional MIF expression is upregulated during oral carcinogenesis. To do this, we examined mRNA expression of MIF and associated signaling mediators in oral cancer patients enrolled in a phase 1 clinical trial (ClinicalTrials.gov ID NCT01465776). Real time PCR analysis of oral tumors and adjacent non-involved oral tissue showed a significant upregulation of MIF transcripts in oral tumor samples of patients compared to normal non-involved tissue from the same patient in about 37.5% of samples (Figure 1A). MIF expression was compared against patient gender, tumor location, tumor stage, smoking status and alcohol use. Our results show no significant association between MIF and these clinical parameters, except for gender, were we observed a slight (p = 0.048) association (Supplemental Table 1), although this was seen in a smaller cohort than what was used in other studies20, 21.
Figure 1. Expression of MIF and associated pro-inflammatory mediators during oral carcinogenesis.
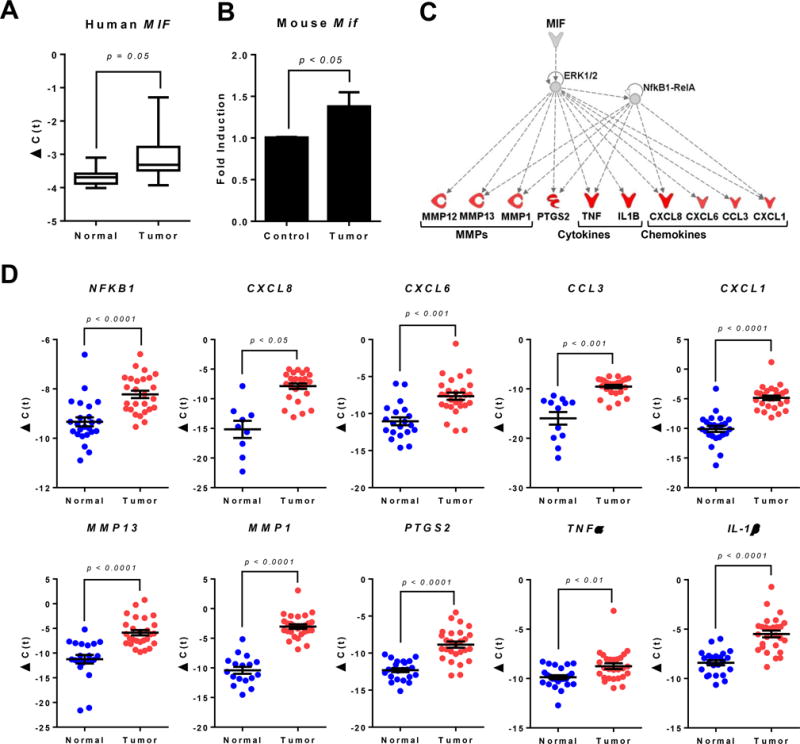
(A) Gene expression of MIF in tumors of oral cancer patients compared to adjacent non-involved tissue. Data are expressed as delta C(t) using β-actin as housekeeping gene. (B) Gene expression of Mif in oral tissues of 4NQO-induced C57BL/6 mice compared to non-carcinogen induced control C57BL/6 mice. Data are expressed as fold induction over control using β-actin as housekeeping gene. (C) Ingenuity Pathway Analysis (IPA) network model of microarray data from clinical samples of oral cancer patients showing upregulated genes potentially regulated by MIF during oral carcinogenesis. (D) Gene expression of pro-inflammatory mediators in tumors of oral cancer patients compared to adjacent non-involved tissue obtained from the same patient. Data presented are biological replicates and are expressed as delta C(t) using β-actin as housekeeping gene. P values were obtained after a paired t-test.
Ingenuity Pathway Analysis (IPA) of microarray data from oral cancer patients showed that transcriptional targets of the MIF-CD74-ERK1/2 MAP kinase signaling pathway (pro-inflammatory cytokines IL-1β, TNF-α, chemokines CXCL1, CXCL6, CXCL8 and CCL3 as well as matrix metalloproteinases MMP1, MMP12 and MMP13) are overexpressed in malignant oral lesions (Figure 1C). These results were further validated by real time PCR analysis (Figure 1D). Similarly, in 4NQO-induced oral tumors of C57BL/6 mice, Mif mRNA is significantly increased compared to control C57BL/6 mice (Figure 1B). Taken together, our results indicate that upregulation of MIF mRNA is associated with oral carcinogenesis in humans and mice.
Mice genetically deficient in Mif display fewer oral lesions during 4NQO-induced oral carcinogenesis
Although we and others show a correlation between MIF expression and increased oral carcinogenesis, it is not known whether MIF expression is merely a consequence of oral carcinogenesis, or MIF actively plays a causative role in oral tumor formation in vivo. To examine this, we used mice genetically deficient in the Mif gene (Mif−/−) combined with an in vivo model of oral carcinogenesis using the oral carcinogen 4NQO. The 4NQO mouse model of oral carcinogenesis is well-established and mimics the pathology of human oral cancer24, 27, 28. Following 16 weeks of oral 4NQO administration, then 8 weeks of regular drinking water (Figure 2A), we observed reduced oral tumor incidence and multiplicity in Mif−/− mice compared to WT mice treated with 4NQO (Figure 2B). These studies demonstrate that genetic deletion of Mif in mice reduces oral carcinogenesis experimentally induced by the oral carcinogen 4NQO, and suggests that MIF is a potential target for oral cancer prevention and treatment strategies.
Figure 2. Mif deficient mice show fewer oral lesions during 4NQO-induced oral carcinogenesis.
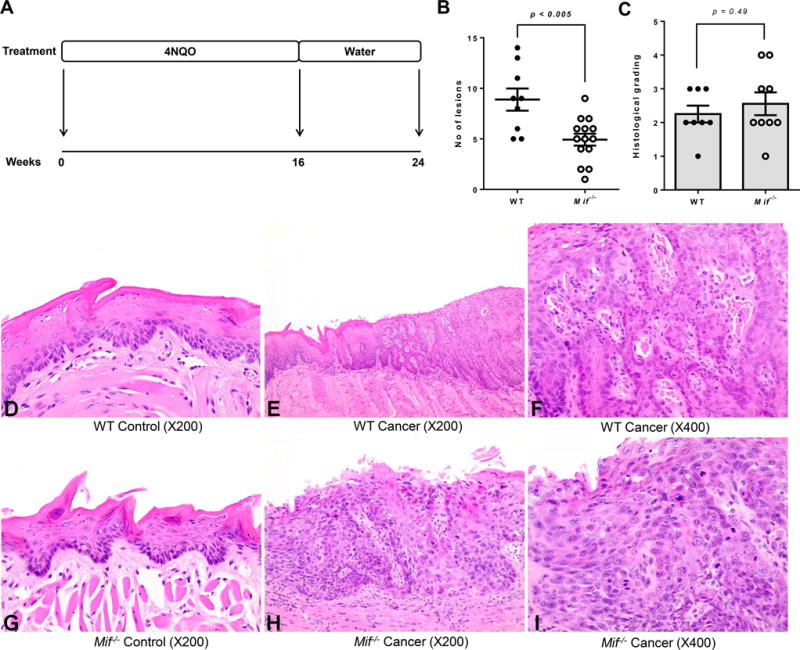
(A) Experimental oral carcinogenesis model. C57BL/6 WT and Mif−/− mice (N = 15) were administered 4NQO (100μg/mL) in their drinking water for 16 weeks, then placed on regular drinking water for an additional 8 weeks. Mice were sacrificed 24 weeks post-4NQO administration. (B) Tumor lesion numbers of C57BL/6 WT and Mif−/− mice exposed to the oral carcinogen 4NQO. (C) Histological grading of carcinogen induced WT and Mif−/− mice as determined by an oral and maxillofacial pathologist [H.I] using established criteria: 0 – Normal epithelium; 1 – mild dysplasia; 2 – moderate dysplasia; 3 – severe dysplasia; 4 – invasive carcinoma. (D–I) Representative Hematoxylin and eosin (H&E) stained images of tongue tissues from 4NQO treated and untreated WT and Mif−/− mice. (D) Untreated WT mice (Mag. ×200) Normal Squamous Mucosa (E) Carcinogen induced WT mice (Mag. ×200) Section showing abrupt transition from normal squamous mucosa to full thickness epithelial dysplasia, consistent with severe squamous dysplasia. (F) Carcinogen induced WT mice (Mag. ×400) High power view of epithelial dysplasia (in Figure 2E) showing full thickness dysmaturation. (G) Untreated Mif−/− mice (Mag. ×200) Normal Squamous Mucosa. (H) Carcinogen induced Mif−/− mice (Mag. X200) Section showing full thickness epithelial dysplasia, prototypic for severe squamous dysplasia. (I) Carcinogen induced Mif−/− mice (Mag. ×400) High power view of the full thickness epithelial dysplasia (in Figure 2H). Brisk suprabasilar mitosis is easily discernible.
Next, we performed histological examination of tongue tissues from carcinogen induced WT and Mif−/− mice. Although tongue lesion numbers were lower in Mif−/− compared to WT carcinogen-induced mice, histological grading showed no significant differences in the tumor stages of lesions that had progressed beyond dysplasia (Figure 2C–I), which suggests that MIF-mediated effects occur primarily during the early stages of oral carcinogenesis.
Expression of pro-inflammatory immune mediators is attenuated in Mif−/− mice
Since Mif−/− mice displayed fewer premalignant and malignant lesions compared to WT mice, we next investigated potential mechanisms by which the absence of MIF inhibited oral carcinogenesis. Chronic inflammation has become recognized as an enabling characteristic during the multistep process of carcinogenesis6. Furthermore, numerous studies have shown that MIF provides a link between chronic inflammation and cancer7. We therefore examined gene expression levels of pro-inflammatory immune mediators in the tongues of WT and Mif−/− mice induced with oral carcinogen. RT2 profiler arrays of mouse inflammatory genes revealed that the expression of pro-inflammatory cytokines Il-1β, Tnf-α, chemokines Cxcl1, Cxcl6 and Ccl3 were significantly reduced in oral carcinogen treated Mif−/− mice (Figure 3A) These results were confirmed by RT-qPCR (Figure 3B). These pro-inflammatory mediators contribute to infiltration of tumor promoting myeloid cell populations, which contribute to tumor progression. Further, as shown by our pathway analysis of microarray data from oral cancer patients enrolled in our clinical trial (Figure 1B), these mediators are associated with oral carcinogenesis. Taken together, these data suggests that MIF contributes to oral carcinogenesis by promoting a chronic pro-inflammatory microenvironment characterized by the production pro-inflammatory cytokines and chemokines, which contributes to activation and infiltration of tumor promoting myeloid cells, thereby favoring tumor growth.
Figure 3. Expression of pro-inflammatory immune mediators is attenuated in Mif−/− mice.
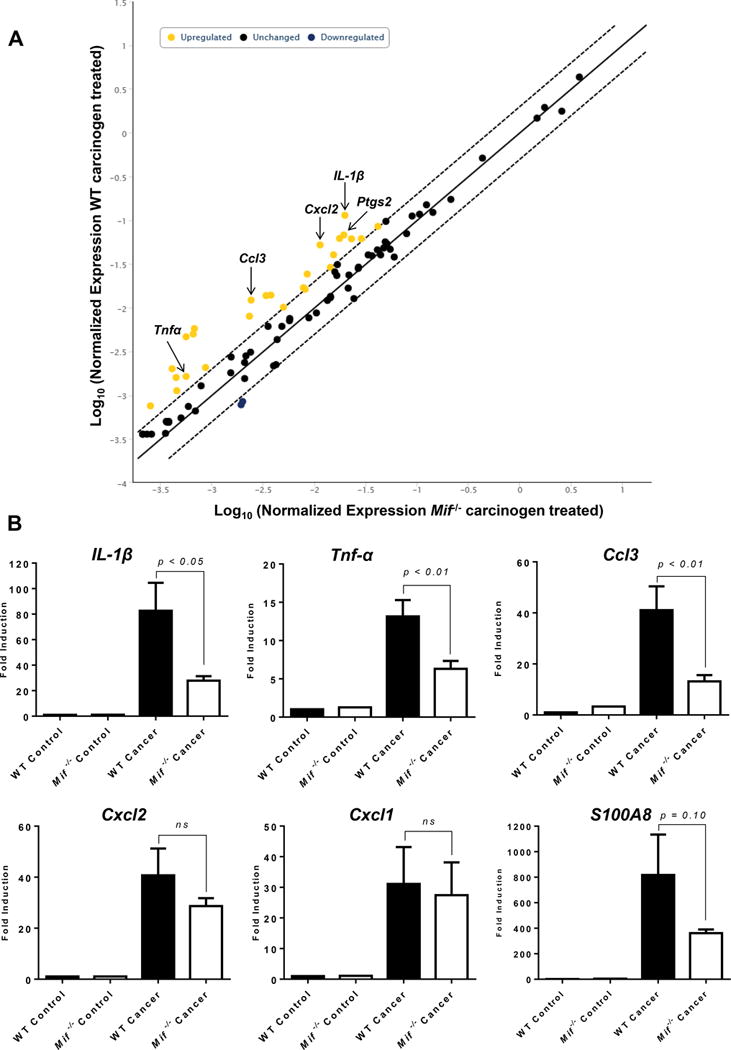
(A) RT2 profiler PCR array analysis of mouse inflammatory response genes in C57BL/6 WT and Mif−/− mice treated with the oral carcinogen 4NQO. Yellow dots represent genes significantly upregulated in carcinogen induced WT mice, compared to Mif−/− mice. Three representative WT and Mif−/− cancer induced mice, and a WT and a Mif−/− mouse controls were used for the array. (B) Real time PCR analysis of pro-inflammatory genes IL-1β, TNF-α, Ccl3, Cxcl2, Cxcl1 and S100A8 in C57BL/6 WT and Mif−/− mice treated with the oral carcinogen 4NQO. Data are expressed as fold induction over untreated control mice using β-actin as housekeeping gene.
Accumulation of myeloid cell populations is impaired in oral carcinogen induced Mif−/− mice
Next, we examined the impact of Mif deletion on systemic accumulation of myeloid cell populations in the spleens of tumor bearing mice. Immature myeloid cells (MDSCs) accumulate in the spleens of oral tumor bearing mice and they contribute to suppression of antitumor immune responses. Flow cytometric analysis of spleens of carcinogen treated mice revealed a significantly reduced accumulation of CD11b+ Gr-1+ cells in Mif−/− compared to WT mice (Figure 4A). Interestingly, unlike tumor bearing WT mice, CD11b+ Gr-1low Ly6Chi cells (monocytic MDSCs) do not accumulate in tumor bearing Mif−/− spleens (Figure 4B). In draining lymph nodes, although myeloid populations were elevated in both groups of mice, the proportion of CD11b+ Gr-1+ cells were lower in Mif−/− mice compared to WT mice while CD11b+ Ly6Chi cell populations were comparable between both mouse groups (Figure 4C). Accumulation of MDSCs has been associated with disease progression and poor prognosis in many cancers including head and neck cancers29–33. These results show that MIF plays a crucial role in creating and maintaining an inflammatory microenvironment, characterized by the accumulation of pro-tumorigenic CD11b+ Gr-1+ cells, which promotes oral carcinogenesis.
Figure 4. Reduced accumulation of myeloid derived immune cells in oral carcinogen induced Mif−/− mice.
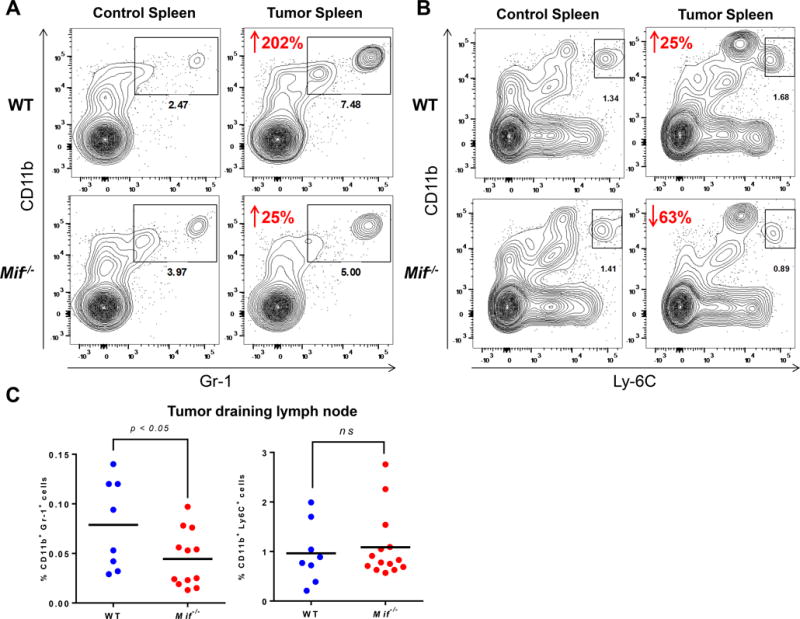
(A and B) Representative flow cytometric contour plots, showing spleens of control and carcinogen treated C57BL/6 WT and Mif−/− mice. Splenocytes were stained with (A) CD11b and Gr-1 antibodies, and (B) CD11b, Gr-1 and Ly-6c. Numbers in red represent the percentage increase/decrease in cell populations of carcinogen induced mice compared to their respective controls. (C) Percentage of Gr-1+ CD11b+ and CD11b+ Ly-6c+ cells in the draining lymph nodes of WT and Mif−/− mice treated with oral carcinogen, as determined by flow cytometry.
Frequency of T cells in Mif−/− mice experimentally induced with oral cancer
T cell recruitment to oral tumors is vital to the generation of both tumor-promoting and antitumor adaptive immune responses6. We therefore examined whether the absence of MIF will affect CD4+ and CD8+ T cell recruitment during oral carcinogenesis. Flow cytometric analysis of draining lymph nodes of carcinogen-treated WT and Mif−/− mice revealed that CD4+ and CD8+ T cell accumulation were not impaired in the absence of MIF (Figure 5A and B). Surprisingly, the proportion of CD4+ CD25+ Foxp3+ regulatory T cells (Tregs) was higher in carcinogen treated Mif−/− mice compared to WT mice (Figure 5C). Tregs are generally associated with suppression of antitumor cytotoxic T cells responses34. However it appears that the moderately increased levels of Tregs in tumor bearing Mif−/− mice does not negatively impact oral carcinogenesis in these mice.
Figure 5. Frequency of T cells in mice experimentally induced with oral cancer.
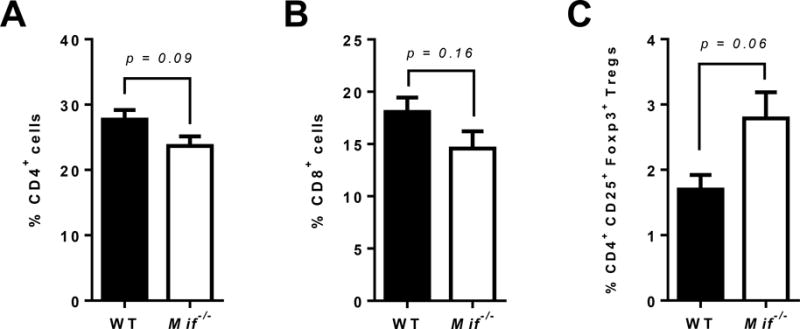
(A–D) Percentage of (A) CD4+ T cells, (B) CD8+ T cells and (C) regulatory T cells (CD4+ CD25+ Foxp3+) in the draining lymph nodes of carcinogen induced WT and Mif−/− mice, as determined by flow cytometry.
Effect of MIF on angiogenesis, invasion and p53 mediated apoptosis
Chronic inflammation can contribute to various cancer hallmark capabilities, such as angiogenesis, tumor invasion and metastasis, through the release of various bioactive molecules into the tumor microenvironment. Since the absence of MIF attenuates chronic inflammation in oral cancer, we determined whether genetic Mif deletion affected the expression of angiogenic markers as well as matrix degrading enzymes which facilitate tumor invasiveness. We observed that in oral carcinogen treated Mif−/− mice, transcriptional expression of VegFC was slightly lower than in WT mice, although the difference was not significant (Figure 6A). We observed no differences in VegFA expression between the two groups of mice (Figure 6B). Analysis of matrix metallo-proteinases (MMPs) revealed a significant downregulation of MMP1 in Mif−/− mice (Figure 6C), while MMP3 was slightly, but not significantly, downregulated (Figure 6D). Interestingly, as revealed by our pathway analysis of microarray data, MMP1 is associated with carcinogenesis in oral cancer patients (Figure 1).
Figure 6. Effect of MIF on angiogenesis, invasion and p53 mediated apoptosis.
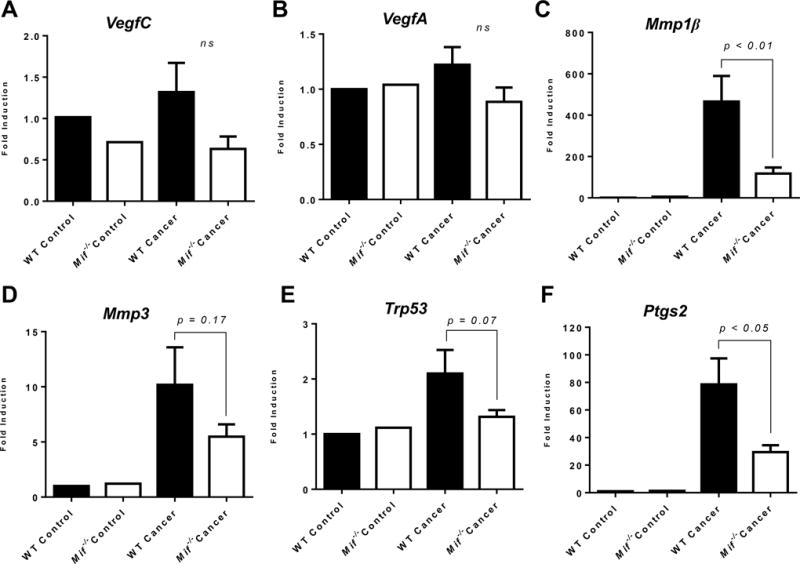
(A–D) Real time PCR analysis of (A) VegFC, (B) VegFA, (C) Mmp1β, (D) Mmp3, (E) Trp53 and (F) Ptgs2 in C57BL/6 WT and Mif−/− mice treated with the oral carcinogen 4NQO. Data are expressed as fold induction over untreated control mice using β-actin as housekeeping gene.
In a number of cancer models, MIF has been shown to inhibit the expression and function of the tumor suppressor protein p53, which induces apoptosis of damaged cells16. We therefore determined levels of p53 expression as a measure of apoptosis in oral carcinogen treated WT and Mif−/− mice. While levels of p53 were elevated in tumor bearing WT mice compared to controls, expression in Mif−/− mice remained largely unchanged. However, the reduced gene expression levels of p53 in tumor bearing Mif−/− compared to WT mice were not significant (Figure 6E).
Inflammatory cells are also known to release reactive oxygen species (ROS), which contribute to malignancy35. Since MIF contributes to enhanced chronic inflammation in oral carcinogenesis, we determined the effect of MIF on Ptgs2, an enzyme involved in the generation of ROS. Levels of Ptgs2 were significantly lower in Mif−/− mice treated with 4NQO, compared to WT counterparts (Figure 6F).
Taken together, our results demonstrate that genetic deletion of Mif−/− downregulates the expression of Mmp1 and Ptgs2, which potentially contribute to the reduction in invasiveness and malignancy during oral carcinogenesis.
DISCUSSION
Results from this study clearly implicate a role for MIF in the pathogenesis of oral cancer. This is the first report which demonstrates that genetic deletion of Mif reduces the incidence and severity of oral carcinogenesis. Our data have significant implications in oral cancer prevention and treatment, for they suggest that targeting the pro-inflammatory cytokine MIF is a promising and viable strategy in the management of oral cancer.
Chronic inflammation has become recognized as an enabling characteristic which promotes tumorigenesis due to its ability to modulate key hallmarks of cancer including cellular proliferation, angiogenesis, anti-apoptotic mechanisms and, probably most importantly, suppression of anti-tumor immune responses35, 36. Results from our study strongly suggest that during oral carcinogenesis, MIF plays a major role in initiation and amplification of this chronic inflammatory tumor microenvironment which favors tumor progression. This is especially relevant to the human disease, given that some major causes of oral carcinogenesis – tobacco smoking, alcohol consumption and HPV – are linked to chronic inflammation.
As shown by our clinical trial data, it is evident that in addition to MIF protein expression, there is an active up-regulation of MIF mRNA transcripts during oral carcinogenesis. This suggests that de novo synthesis of MIF which amplifies tumor-promoting inflammation drives oral malignant progression. It should be noted however, that levels of MIF mRNA expression may fluctuate during the multi-step process of oral carcinogenesis and may vary between patients. Indeed, not all patients displayed significant MIF mRNA upregulation in their tumors compared to adjacent non-cancerous tissue. More studies will be required to determine at what points during multistep oral carcinogenesis MIF actively plays a role. Our studies using the mouse model seems to suggest that MIF is critical during the initiation stages of oral carcinogenesis. Supportive of this hypothesis, is the observation that tumor sizes were comparable between WT and Mif−/− groups, whereas the number of tumors which progressed to squamous cell carcinomas were greater in WT mice. Further, the fact that Mif deletion significantly reduced the levels of other pro-inflammatory cytokines (Tnfα, Il-1β, Cxcl1, Ccl3) suggests that MIF acts upstream during carcinogen-induced chronic inflammation and is an early target for chemoprevention of oral carcinogenesis. We are currently addressing the signaling mechanisms behind MIF-mediated downregulation of these pro-inflammatory immune mediators in carcinogen induced Mif−/− mice. Our data suggests that the MIF-CD74 axis which leads to the activation of ERK1/2 MAP kinase signaling pathway is a major factor that contributes to the expression of these mediators. Oral epithelial cells express CD74 and MIF induces ERK1/2 phosphorylation in oral cancer cells in vitro. MIF also mediates NFκB activation and we see evidence for this in oral cancer cells. Other reports also show that MIF activates the PI3K/Akt signaling pathway, leading to survival of tumor cells. Moreover, as shown by Zhu et al.37, we cannot rule out the involvement of CXCR2 and CXCR4 in MIF-mediated recruitment of tumor promoting myeloid immune cell populations into the oral tumor microenvironment, which could further amplify the expression of these mediators. These multiple mechanisms further emphasize the multi-functional role of MIF in promoting oral carcinogenesis.
Numerous studies demonstrate that MIF contributes to tumorigenesis in colon, breast and melanoma and bladder cancer14, 38. Although the mechanisms behind the observed MIF-mediated contribution to tumorigenesis in the above mentioned cancers are multiple, there exists some similarities and differences between these mechanisms and those of MIF mediated oral carcinogenesis. For example, in bladder cancer, MIF promotes cellular proliferation and angiogenesis39, mechanisms which are also important in oral carcinogenesis. In contrast, although MIF is known to inhibit p53 expression in a number of cancer models14, 40, we did not observe a reduction in p53 expression in oral carcinogen induced WT mice as compared to Mif−/− mice. Interestingly, the promotion of inflammation characterized by the recruitment of tumor promoting innate immune cells appears to be a common mechanism by which MIF contributes to tumor progression in most cancers14, 41, 42. Summarily, the multiple mechanisms observed in MIF-mediated contribution to oral carcinogenesis represent multiple intervention points, which make the targeting of MIF a viable strategy in oral cancer prevention and treatment.
A significant finding in this study was the impact of MIF on splenic accumulation of myeloid cell populations, as well as its contribution to a chronic pro-inflammatory oral microenvironment. The absence of MIF caused a significant reduction in the presence of CD11b+ Gr-1high cells and CD11b+ Gr-1low Ly6Chi cells in the spleens of oral tumor bearing mice. Although the proportions of these myeloid cell populations in oral tumor bearing WT mice are not as high as in other solid tumors, these levels are comparable to those seen in other studies of 4NQO induced mouse oral cancer, where these cells have been shown to play a role in oral tumor development33. It is therefore not surprising that inhibiting the accumulation of these cells by targeting MIF suppresses oral carcinogenesis.
Myeloid derived suppressor cells (MDSCs) are immature myeloid cells defined by their ability to suppress adaptive immune responses. In cancer, they have been shown to suppress anti-tumor CTL and NK cell activity43, thereby presenting a major obstacle to anti-tumor immunity and immunotherapy44. These cells are of particular interest in oral carcinogenesis because they contribute to oral cancer progression and represent a potential target for therapy against oral carcinogenesis33. Targeting MDSCs has been a major focus of anti-cancer research in recent years. Our data suggests that genetic deletion of MIF attenuates MDSC recruitment. It should be noted that MDSCs are defined by their functional capacity to suppress innate and adaptive immune responses so further studies will be required to characterize the CD11b+ Gr-1+ cells we observed in our study. Nevertheless, this myeloid cell population promotes oral carcinogenesis, and based on our data, it is evident that a major cellular mechanism by which Mif deletion inhibits oral carcinogenesis is the reduction of tumor-promoting myeloid cell recruitment to the oral cancer microenvironment.
Regulatory T cells (Tregs) are a subset of T cells that contribute to tumor progression by inhibiting anti-tumor effector T cell immune responses6, 36. The involvement of Tregs in promoting tumor progression has been established for lung, prostrate, breast and other cancers45, 46. In a recent study, the absence of MIF was shown to decrease the circulating Treg population in colorectal cancer47, which was associated with decreased tumor burden. It was therefore surprising to observe in our study, an increase in the Treg population in the draining lymph nodes of carcinogen induced Mif−/− mice compared to WT mice. Interestingly, in the colorectal cancer study, the researchers showed that splenic T cells from Mif−/− mice generated significantly greater amounts of inducible Tregs compared to WT splenic T cells when stimulated in vitro47. It appears that the cellular and molecular pathways by which MIF affects the Treg population during oral carcinogenesis are complex. Further, it should be noted that the role of Tregs during oral carcinogenesis still remain unclear and conflicting reports exist. For example, in recent studies, it was observed that increased Treg population was associated with better loco-regional control of oral tumors48 and improved survival in oral cancer patients49. It is therefore possible that different subsets of Tregs may play different roles during oral carcinogenesis49, and some Tregs could suppress the damaging inflammation that favors oral tumor progression48. It is noteworthy that the antitumor effector T cell responses observed in our study do not appear to be inhibited in oral carcinogen treated Mif−/− mice, as CD25+ CD4+ T cells were significantly greater in these mice compared to WT counterparts. This subset of effector T cells have been shown to initiate and maintain antitumor immune responses and have been associated with a favorable prognosis in head and neck squamous cell carcinoma (HNSCC) patients50.
In conclusion, we demonstrate for the first time, that deletion of MIF reduces the incidence and severity of oral carcinogenesis, by inhibiting the expression inflammatory cytokines and infiltration of tumor promoting immune cells. Therefore, targeting MIF is a promising strategy for prevention or therapy of oral cancers.
Supplementary Material
Novelty and Impact.
Macrophage migration inhibitory factor (MIF) is expressed in oral cancer patients, but its precise role in oral carcinogenesis is not completely understood. In this study, the authors examined the contribution of MIF to oral carcinogenesis using a well-established mouse model. Their findings show that MIF deletion reduces the incidence and severity of oral carcinogenesis, by inhibiting the expression of chronic pro-inflammatory immune mediators. Targeting MIF is therefore a potentially viable strategy for oral cancer management.
Acknowledgments
Funding for this project was obtained from NIH grants 3U01CA18825002S1 awarded to SO and CW.
Abbreviations
- 4NQO
4-nitroquinoline-1-oxide
- MIF
Macrophage migration inhibitory factor
- WT
Wild type
- MDSC
myeloid derived suppressor cells
Footnotes
DISCLOSURE/CONFLICTS OF INTEREST:
No conflicts of interest are declared.
References
- 1.Bagan J, Sarrion G, Jimenez Y. Oral cancer: clinical features. Oral Oncol. 2010;46:414–7. doi: 10.1016/j.oraloncology.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 2.Choi S, Myers JN. Molecular pathogenesis of oral squamous cell carcinoma: implications for therapy. J Dent Res. 2008;87:14–32. doi: 10.1177/154405910808700104. [DOI] [PubMed] [Google Scholar]
- 3.Johnson NW, Jayasekara P, Amarasinghe AA. Squamous cell carcinoma and precursor lesions of the oral cavity: epidemiology and aetiology. Periodontol 2000. 2011;57:19–37. doi: 10.1111/j.1600-0757.2011.00401.x. [DOI] [PubMed] [Google Scholar]
- 4.Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6:963–8. doi: 10.1002/1097-0142(195309)6:5<963::aid-cncr2820060515>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 5.Braakhuis BJ, Bloemena E, Leemans CR, Brakenhoff RH. Molecular analysis of surgical margins in head and neck cancer: more than a marginal issue. Oral Oncol. 2010;46:485–91. doi: 10.1016/j.oraloncology.2010.01.019. [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Conroy H, Mawhinney L, Donnelly SC. Inflammation and cancer: macrophage migration inhibitory factor (MIF)–the potential missing link. QJM. 2010;103:831–6. doi: 10.1093/qjmed/hcq148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bacher M, Metz CN, Calandra T, Mayer K, Chesney J, Lohoff M, Gemsa D, Donnelly T, Bucala R. An essential regulatory role for macrophage migration inhibitory factor in T-cell activation. Proc Natl Acad Sci U S A. 1996;93:7849–54. doi: 10.1073/pnas.93.15.7849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu X, Wang B, Ye C, Yao C, Lin Y, Huang X, Zhang Y, Wang S. Overexpression of macrophage migration inhibitory factor induces angiogenesis in human breast cancer. Cancer Lett. 2008;261:147–57. doi: 10.1016/j.canlet.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 10.He XX, Chen K, Yang J, Li XY, Gan HY, Liu CY, Coleman TR, Al-Abed Y. Macrophage migration inhibitory factor promotes colorectal cancer. Mol Med. 2009;15:1–10. doi: 10.2119/molmed.2008.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meyer-Siegler KL, Iczkowski KA, Leng L, Bucala R, Vera PL. Inhibition of macrophage migration inhibitory factor or its receptor (CD74) attenuates growth and invasion of DU-145 prostate cancer cells. J Immunol. 2006;177:8730–9. doi: 10.4049/jimmunol.177.12.8730. [DOI] [PubMed] [Google Scholar]
- 12.Ren Y, Tsui HT, Poon RT, Ng IO, Li Z, Chen Y, Jiang G, Lau C, Yu WC, Bacher M, Fan ST. Macrophage migration inhibitory factor: roles in regulating tumor cell migration and expression of angiogenic factors in hepatocellular carcinoma. Int J Cancer. 2003;107:22–9. doi: 10.1002/ijc.11287. [DOI] [PubMed] [Google Scholar]
- 13.Kamimura A, Kamachi M, Nishihira J, Ogura S, Isobe H, Dosaka-Akita H, Ogata A, Shindoh M, Ohbuchi T, Kawakami Y. Intracellular distribution of macrophage migration inhibitory factor predicts the prognosis of patients with adenocarcinoma of the lung. Cancer. 2000;89:334–41. [PubMed] [Google Scholar]
- 14.Martin J, Duncan FJ, Keiser T, Shin S, Kusewitt DF, Oberyszyn T, Satoskar AR, VanBuskirk AM. Macrophage migration inhibitory factor (MIF) plays a critical role in pathogenesis of ultraviolet-B (UVB) -induced nonmelanoma skin cancer (NMSC) FASEB J. 2009;23:720–30. doi: 10.1096/fj.08-119628. [DOI] [PubMed] [Google Scholar]
- 15.Mitchell RA, Liao H, Chesney J, Fingerle-Rowson G, Baugh J, David J, Bucala R. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc Natl Acad Sci U S A. 2002;99:345–50. doi: 10.1073/pnas.012511599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jung H, Seong HA, Ha H. Critical role of cysteine residue 81 of macrophage migration inhibitory factor (MIF) in MIF-induced inhibition of p53 activity. J Biol Chem. 2008;283:20383–96. doi: 10.1074/jbc.M800050200. [DOI] [PubMed] [Google Scholar]
- 17.Santarpia L, Lippman SM, El-Naggar AK. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:103–19. doi: 10.1517/14728222.2011.645805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baugh JA, Gantier M, Li L, Byrne A, Buckley A, Donnelly SC. Dual regulation of macrophage migration inhibitory factor (MIF) expression in hypoxia by CREB and HIF-1. Biochemical and Biophysical Research Communications. 2006;347:895–903. doi: 10.1016/j.bbrc.2006.06.148. [DOI] [PubMed] [Google Scholar]
- 19.Winner M, Koong AC, Rendon BE, Zundel W, Mitchell RA. Amplification of Tumor Hypoxic Responses by Macrophage Migration Inhibitory Factor–Dependent Hypoxia-Inducible Factor Stabilization. Cancer Research. 2007;67:186–93. doi: 10.1158/0008-5472.CAN-06-3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.França CM, Batista AC, Borra RC, Ventiades-Flores JA, Mendonça EF, Deana AM, Mesquita-Ferrari RA, de Natali Caly D, de Mello Rode S, Faria MR. Macrophage migration inhibitory factor and oral cancer. Journal of Oral Pathology & Medicine. 2013;42:368–73. doi: 10.1111/jop.12011. [DOI] [PubMed] [Google Scholar]
- 21.Kindt N, Lechien J, Decaestecker C, Rodriguez A, Chantrain G, Remmelink M, Laurent G, Gabius HJ, Saussez S. Expression of macrophage migration-inhibitory factor is correlated with progression in oral cavity carcinomas. Anticancer Res. 2012;32:4499–505. [PubMed] [Google Scholar]
- 22.MB DES, Curioni OA, Kanda JL, MB DEC. Serum and salivary macrophage migration inhibitory factor in patients with oral squamous cell carcinoma. Oncol Lett. 2014;8:2267–75. doi: 10.3892/ol.2014.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeng J, Quan J, Xia X. Transient transfection of macrophage migration inhibitory factor small interfering RNA disrupts the biological behavior of oral squamous carcinoma cells. Mol Med Rep. 2016;13:174–80. doi: 10.3892/mmr.2015.4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanojia D, Vaidya MM. 4-nitroquinoline-1-oxide induced experimental oral carcinogenesis. Oral Oncol. 2006;42:655–67. doi: 10.1016/j.oraloncology.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 25.Ohkoshi A, Suzuki T, Ono M, Kobayashi T, Yamamoto M. Roles of Keap1-Nrf2 system in upper aerodigestive tract carcinogenesis. Cancer Prev Res (Phila) 2013;6:149–59. doi: 10.1158/1940-6207.CAPR-12-0401-T. [DOI] [PubMed] [Google Scholar]
- 26.Warner BM, Casto BC, Knobloch TJ, Accurso BT, Weghorst CM. Chemoprevention of oral cancer by topical application of black raspberries on high at-risk mucosa. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;118:674–83. doi: 10.1016/j.oooo.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang XH, Knudsen B, Bemis D, Tickoo S, Gudas LJ. Oral cavity and esophageal carcinogenesis modeled in carcinogen-treated mice. Clin Cancer Res. 2004;10:301–13. doi: 10.1158/1078-0432.ccr-0999-3. [DOI] [PubMed] [Google Scholar]
- 28.Fujino H, Chino T, Imai T. Experimental production of labial and lingual carcinoma by local application of 4-nitroquinoline N-oxide. J Natl Cancer Inst. 1965;35:907–18. [PubMed] [Google Scholar]
- 29.Trellakis S, Bruderek K, Dumitru CA, Gholaman H, Gu X, Bankfalvi A, Scherag A, Hutte J, Dominas N, Lehnerdt GF, Hoffmann TK, Lang S, et al. Polymorphonuclear granulocytes in human head and neck cancer: enhanced inflammatory activity, modulation by cancer cells and expansion in advanced disease. Int J Cancer. 2011;129:2183–93. doi: 10.1002/ijc.25892. [DOI] [PubMed] [Google Scholar]
- 30.Wang N, Feng Y, Wang Q, Liu S, Xiang L, Sun M, Zhang X, Liu G, Qu X, Wei F. Neutrophils infiltration in the tongue squamous cell carcinoma and its correlation with CEACAM1 expression on tumor cells. PLoS One. 2014;9:e89991. doi: 10.1371/journal.pone.0089991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Glogauer J, Sun CX, Bradley G, Magalhaes MA. Neutrophils Increase Oral Squamous Cell Carcinoma Invasion Through An Invadopodia-Dependent Pathway. Cancer Immunol Res. 2015 doi: 10.1158/2326-6066.CIR-15-0017. [DOI] [PubMed] [Google Scholar]
- 32.Magalhaes MA, Glogauer JE, Glogauer M. Neutrophils and oral squamous cell carcinoma: lessons learned and future directions. J Leukoc Biol. 2014;96:695–702. doi: 10.1189/jlb.4RU0614-294R. [DOI] [PubMed] [Google Scholar]
- 33.Chu M, Su YX, Wang L, Zhang TH, Liang YJ, Liang LZ, Liao GQ. Myeloid-derived suppressor cells contribute to oral cancer progression in 4NQO-treated mice. Oral Dis. 2012;18:67–73. doi: 10.1111/j.1601-0825.2011.01846.x. [DOI] [PubMed] [Google Scholar]
- 34.Mougiakakos D, Choudhury A, Lladser A, Kiessling R, Johansson CC. Regulatory T cells in cancer. Adv Cancer Res. 2010;107:57–117. doi: 10.1016/S0065-230X(10)07003-X. [DOI] [PubMed] [Google Scholar]
- 35.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30:1073–81. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- 37.Zhu G, Tang Y, Geng N, Zheng M, Jiang J, Li L, Li K, Lei Z, Chen W, Fan Y, Ma X, Li L, et al. HIF-alpha/MIF and NF-kappaB/IL-6 axes contribute to the recruitment of CD11b+Gr-1+ myeloid cells in hypoxic microenvironment of HNSCC. Neoplasia. 2014;16:168–79. doi: 10.1593/neo.132034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagarajan P, Tober KL, Riggenbach JA, Kusewitt DF, Lehman AM, Sielecki T, Pruitt J, Satoskar AR, Oberyszyn TM. MIF antagonist (CPSI-1306) protects against UVB-induced squamous cell carcinoma. Mol Cancer Res. 2014;12:1292–302. doi: 10.1158/1541-7786.MCR-14-0255-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choudhary S, Hegde P, Pruitt JR, Sielecki TM, Choudhary D, Scarpato K, DeGraff DJ, Pilbeam CC, Taylor JA. Macrophage migratory inhibitory factor promotes bladder cancer progression via increasing proliferation and angiogenesis. Carcinogenesis. 2013;34:2891–99. doi: 10.1093/carcin/bgt239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brock SE, Rendon BE, Xin D, Yaddanapudi K, Mitchell RA. MIF family members cooperatively inhibit p53 expression and activity. PLoS One. 2014;9:e99795. doi: 10.1371/journal.pone.0099795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gordon-Weeks AN, Lim SY, Yuzhalin AE, Jones K, Muschel R. Macrophage migration inhibitory factor: A key cytokine and therapeutic target in colon cancer. Cytokine Growth Factor Rev. 2015 doi: 10.1016/j.cytogfr.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 42.Simpson KD, Templeton DJ, Cross JV. Macrophage migration inhibitory factor promotes tumor growth and metastasis by inducing myeloid-derived suppressor cells in the tumor microenvironment. J Immunol. 2012;189:5533–40. doi: 10.4049/jimmunol.1201161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parker KH, Beury DW, Ostrand-Rosenberg S. Chapter Three - Myeloid-Derived Suppressor Cells: Critical Cells Driving Immune Suppression in the Tumor Microenvironment. In: Xiang-Yang W, Paul BF, editors. Advances in Cancer Researched. Vol. 128. Academic Press; 2015. pp. 95–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fox SB, Launchbury R, Bates GJ, Han C, Shaida N, Malone PR, Harris AL, Banham AH. The number of regulatory T cells in prostate cancer is associated with the androgen receptor and hypoxia-inducible factor (HIF)-2α but not HIF-1α. The Prostate. 2007;67:623–29. doi: 10.1002/pros.20538. [DOI] [PubMed] [Google Scholar]
- 46.Okita R, Saeki T, Takashima S, Yamaguchi Y, Toge T. CD4+CD25+ regulatory T cells in the peripheral blood of patients with breast cancer and non-small cell lung cancer. Oncol Rep. 2005;14:1269–73. [PubMed] [Google Scholar]
- 47.Choi S, Kim HR, Leng L, Kang I, Jorgensen WL, Cho CS, Bucala R, Kim WU. Role of macrophage migration inhibitory factor in the regulatory T cell response of tumor-bearing mice. J Immunol. 2012;189:3905–13. doi: 10.4049/jimmunol.1102152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Badoual C, Hans S, Rodriguez J, Peyrard S, Klein C, Agueznay NEH, Mosseri V, Laccourreye O, Bruneval P, Fridman WH, Brasnu DF, Tartour E. Prognostic Value of Tumor-Infiltrating CD4+ T-Cell Subpopulations in Head and Neck Cancers. Clinical Cancer Research. 2006;12:465–72. doi: 10.1158/1078-0432.CCR-05-1886. [DOI] [PubMed] [Google Scholar]
- 49.Lim KP, Chun NA, Ismail SM, Abraham MT, Yusoff MN, Zain RB, Ngeow WC, Ponniah S, Cheong SC. CD4+CD25hiCD127low regulatory T cells are increased in oral squamous cell carcinoma patients. PLoS One. 2014;9:e103975. doi: 10.1371/journal.pone.0103975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loose D, Signore A, Bonanno E, Vermeersch H, Dierckx R, Deron P, Van de Wiele C. Prognostic value of CD25 expression on lymphocytes and tumor cells in squamous-cell carcinoma of the head and neck. Cancer Biother Radiopharm. 2008;23:25–33. doi: 10.1089/cbr.2007.0373. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.