

Abstract
Bioethanol is becoming increasingly important in energy supply and economic development. However, the low yield of bioethanol and the insufficiency of high-efficient genetic manipulation approaches limit its application. In this study, a novel transcription activator-like effector nuclease (TALEN) vector containing the left and right arms of TALEN was electroporated into Saccharomyces cerevisiae strain As2.4 to sequence the alcohol dehydrogenase gene ADH2 and the hygromycin-resistant gene hyg. Western blot analysis using anti-FLAG monoclonal antibody proved the successful expression of TALE proteins in As2.4 strains. qPCR and sequencing demonstrated the accurate knockout of the 17 bp target gene with 80% efficiency. The TALEN vector and ADH2 PCR product were electroporated into ΔADH2 to complement the ADH2 gene (ADH2+ As2.4). LC–MS and GC were employed to detect ethanol yields in the native As2.4, ΔADH2 As2.4, and ADH2+ As2.4 strains. Results showed that ethanol production was improved by 52.4 ± 5.3% through the disruption of ADH2 in As2.4. The bioethanol yield of ADH2+ As2.4 was nearly the same as that of native As2.4. This study is the first to report on the disruption of a target gene in S. cerevisiae by employing Fast TALEN technology to improve bioethanol yield. This work provides a novel approach for the disruption of a target gene in S. cerevisiae with high efficiency and specificity, thereby promoting the improvement of bioethanol production in S. cerevisiae by metabolic engineering.
Keywords: Saccharomyces cerevisiae, Fast TALEN technology, disruption, complement, bioethanol production
Introduction
With the development of economics and the constant increase in energy consumption, fossil fuels could not meet the requirements of humankind. Bioethanol is becoming increasingly important in energy supply and economic development. However, the traditional bioethanol production has low conversion rate, high cost, and low yield. Saccharomyces cerevisiae is a key microorganism that could produce bioethanol; however, the insufficiency of high-efficient genetic manipulation methods for S. cerevisiae limits the wide application of bioethanol. Therefore, genetic engineering approaches with high efficiency for S. cerevisiae must be developed to improve bioethanol yield.
Native S. cerevisiae can only produce ethanol by utilizing glucose; the disadvantages of low ethanol yield and unavailability of other carbon sources, such as cellulose and starch, prevent the wide use of native S. cerevisiae in producing ethanol. Therefore, considerable efforts have been exerted to obtain a genetically engineered S. cerevisiae strain that would improve bioethanol yield. Introduction of the exogenous gene encoding xylose isomerase endows S. cerevisiae with the ability to utilize xylose for ethanol production (Kuyper et al., 2005; Matsushika et al., 2009). The GDP1 gene encoding glyceraldehyde-phosphate dehydrogenase was integrated with the genome of S. cerevisiae to facilitate NAPDH regeneration, thereby promoting bioethanol production from xylose through the pentose pathway (Verho et al., 2003).
Aside from utilizing different carbon sources, genes involved in the ethanol production pathway were also regulated or disrupted to improve bioethanol yield. Alcohol dehydrogenase Adh2p encoded by the ADH2 gene could catalyze ethanol into aldehyde, and the affinity of Adh2p to ethanol is approximately 10 times higher than that of other isozymes (Wiebe et al., 2007). Thus, many scholars have attempted to regulate or disrupt the expression of the ADH2 gene. The yeast regulatory protein ADR1 could activate the expression of the ADH2 gene; thus, the expression of ADH2 is repressed by controlling the synthesis of the ADR1 protein (Vallari et al., 1992). Many genetic engineering approaches have been employed to delete ADH2 in S. cerevisiae. However, PCR-mediated homologous recombination is only suitable for the genetic manipulation of S. cerevisiae with a clear genetic background (Beier et al., 1985; Gray and Honigberg, 2001). The Cre/loxP recombination system possesses a short homologous arm but is accompanied by a relatively low efficiency and residual loxP site, which may result in the knockout of non-target genes (Gueldener et al., 2002; Fang et al., 2011). Therefore, novel genetic engineering approaches with high efficiency and specificity must be developed to regulate the expression of ADH2 in S. cerevisiae.
Transcription activator-like effector nuclease (TALEN) is a hybrid protein containing DNA-binding and FokI cleavage domain. The DNA-binding domain consists of a variable number of tandem repeats of a 34-amino-acid monomer, which specifies the DNA-binding sequence by its 12 and 13th repeat-variable di-residues (RVDs, which specifically recognize a single nucleotide; Bogdanove and Voytas, 2011; Grau et al., 2013; Nakajima and Yaoita, 2013). TALEN technology has been widely used in the genome editing of various species, including human, plant, and zebrafish, because of its high specificity and efficiency (Joung and Sander, 2013; Sakuma et al., 2013; Zhang et al., 2013; Zu et al., 2013; Uhde-stone et al., 2014). However, only few studies have applied TALEN technology in microorganisms because of its relatively complicated reconstruction procedure for TALEN vectors. The establishment of Fast TALEN technology widens the application of TALEN in different fields (Bogdanove and Voytas, 2011; Grau et al., 2013).
In this study, a novel recombinant TALEN vector targeting the 17 bp ADH2 gene was electroporated into S. cerevisiae As2.4. SDS-PAGE and Western blot were performed to confirm the successful introduction of the TALEN vector. The results of sequencing, qRT-PCR, and enzymatic activity assay demonstrated the accurate knockout of the target gene. The disruption of ADH2 significantly improved the bioethanol yield of S. cerevisiae As2.4; the complement of ADH2 was also used to confirm the function of this gene in S. cerevisiae As2.4. This study is the first to report on the disruption of ADH2 in S. cerevisiae using Fast TALEN technology to improve bioethanol yield. The results of this study could widen the application of bioethanol and promote the development of genetic engineering in yeast.
Materials and Methods
Strain, Plasmid, and Growth Medium
Saccharomyces cerevisiae As2.4 (GIM 2.167) was provided by Microbial Culture Collection Center of Guangdong Institute of Microbiology. The Fast TALEN Assembly kit and TALEN backbone vector p1301M1 were purchased from SIDANSAI (Shanghai, China). Yeast cells were grown at 30°C in YPD medium (1% yeast extract, 2% peptone, and 2% glucose). The vector maps of Ptalen L48 and Ptalen R36 are shown in Figure 1A. Anti-FLAG monoclonal antibody was purchased from CST (Danvers, MA, USA). All primers used in this study are listed in Table 1.
FIGURE 1.
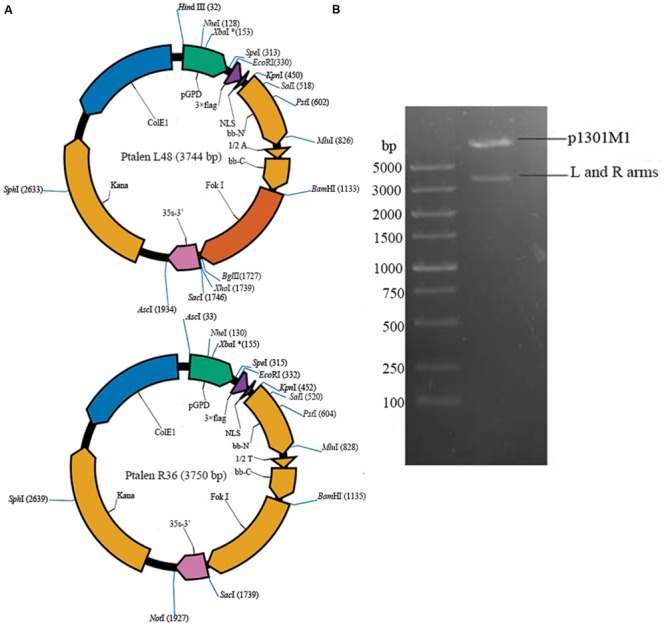
Construction of recombinant TALEN vector. (A) Vector map of Ptalen L48 and Ptalen R36; (B) Digestion of recombinant TALEN vector p1301M1-TALENs with restriction enzymes HindIII, SacI, and AscI.
Table 1.
Primers for the construction of recombinant TALEN vectors.
| Primers | Sequence |
|---|---|
| F1 | 5′-ggaattccatatgtctattccagaaactcaaaaag-3′ |
| R1 | 5′-ggaattccatatgtctattccagaaactcaaaaag-3′ |
| F2 | 5′-ctattcctttgccctcggacgag-3′ |
| R2 | 5′-atgaaaaagcctgaactcac-3′ |
| F3 | 5′-ccattatcttctacgaatccaacggc-3′ |
| R3 | 5′-tgggctttggctttggaactgggat-3′ |
Construction of Recombinant TALEN Vector
Primers F1 and R1 were used to obtain the full sequence of ADH2. The 17 bp target sequence of AGTTGGAGCATAAGGAT in ADH2 was selected using E-TALEN1. The left and right arms of sequence-specific TALENs (Ptalen L48 and Ptalen R36) targeting 17 bp sequences adjacent to AGTTGGAGCATAAGGAT were constructed through one-step ligation using the FastTALETM TALEN Assembly Kit (SIDANSAI) in accordance with the manufacturer’s instructions. PCR and sequencing confirmed the successful construction of recombinant Ptalen L48 and Ptalen R36. Recombinant Ptalen L48 was digested with restriction enzymes HindIII and AscI, and Ptalen R36 was digested with restriction enzymes AscI and SacI. Subcloning vector p1301 M1 containing the hygromycin phosphotransferase (hyg) gene was digested with HindIII and SacI. The resultant larger fragments were recovered, ligated by T4 DNA ligase (Fermentas, USA) at 22°C for 16 h, and then transformed into Escherichia coli Top10 competent cells. Positive clones were screened via 100 μg/mL hygromycin and further confirmed through restriction enzyme digestion (HindIII, AscI, and SacI) and sequencing.
Introduction of Recombinant TALEN Vector
Saccharomyces cerevisiae As2.4 was cultivated in YPD medium and collected at the early stage of the logarithmic phase to prepare competent cells. As2.4 cells were collected at an OD600 of approximately 1.0 via centrifugation at 6000 rpm at 4°C. After incubation on ice for 10 min, the cells were washed twice with ice-cold sterilized water and then resuspended in sterilized water. The recombinant plasmid p1301 M1-TALENs was electroporated into As2.4-competent cells under 2.0 kV voltage and 5.0 ms pulse duration across a 2 mm cuvette. Positive clones were selected through PCR using primers F2 and R2 to amplify the hyg gene and further confirmed by sequencing.
The PCR products of the ADH2 gene and recombinant plasmid p1301M1-TALENs were electroprated into ΔADH2 As2.4. PCR and sequencing were performed to confirm the successful complementation of ADH2 in ΔADH2 As2.4 (ADH2+ As2.4).
qPCR and Western Blot of Recombinant As2.4 Strain
After 24 h of incubation, the total RNAs of native As2.4, ΔADH2 As2.4, and ADH2+ As2.4 were extracted, and cDNAs were obtained using 5× All-In-One MasterMix (Abm, Canada). Primers F3 and R3 combined with cDNAs and 2× SYBR green qPCR Mix (Fermentas, USA) were used to amplify a 70 bp fragment containing the target sequence. GAPDH was employed as a reference gene. qPCR was performed in accordance with the manufacturer’s instructions. Each reaction was conducted in biological triplicate. The CT values obtained were used as the original data to calculate the relative expression level of the target gene to the GAPDH gene by the 2-ΔΔCT method. The qRT-PCR product was also identified by 2% agarose gel electrophoresis.
The total proteins of As2.4 strains were extracted by sonication (50 W at 3 s on, and 3 s off), and the supernatants were obtained by centrifugation at 6000 rpm at 4°C. The supernatants were loaded to 12% SDS-PAGE and then transferred to an NC membrane at a voltage of 100 V for 70 min. The membrane was blocked by 5% non-fat milk and then incubated with anti-FLAG mouse monoclonal antibody with a dilution factor of 1:2000 at 4°C overnight. The membrane was washed with TBST buffer and then incubated with rabbit anti-mouse IgG with a dilution factor of 1:2000. The target bands were visualized with an ECL kit (Thermo Fisher Scientific, USA) in accordance with the manufacturer’s instructions.
Alcohol Dehydrogenase Activity Assay
The cells of native As2.4, ΔADH2 As2.4, and ADH2+ As2.4 strains were collected via centrifugation, and the proteins were released by a high-pressure cell disruption system (Constant Systems, Warwick, UK) under 1100 bar for three cycles. Supernatants were obtained after centrifugation. NAD I (5.5 mM) was dissolved in 7.5 g/L glycine buffer (pH 9.0) with a volume ratio of 1:5 as a substrate. Supernatants (5 μL) from different As2.4 strains were added to 100 μL of the substrate, and 5 μL of purified water was added as blank. After incubation at 37°C for 2 min, 20 μL of ethanol was added to the mixture and then incubated at 37°C for 5 min. The absorbance of the reaction mix at 340 nm was detected on a microplate reader (Thermo scientific, USA). Enzymatic activity was defined as 1 μg of NADH released by the catalysis of alcohol dehydrogenase in one minute (Beier et al., 1985).
Analysis of Ethanol Yield in As2.4 Strains
The native As2.4, ΔADH2 As2.4, and ADH2+ As2.4 strains were inoculated into 150 mL of YPD medium in a 250 mL flask and then cultivated at 30°C at 180 rpm. After 72 h of cultivation, the fermentation liquid was loaded onto LC–MS (Agilent 6430, American) at a flow rate of 0.2 mL/min using C18 column (2 mm × 150 mm).
Fermentation liquids from the different As2.4 strains in head-space bottles were incubated at 80°C for 30 min. After reaching the equilibrium, a predetermined amount of the head-space of the vial was flushed into the gas chromatograph; isopropanol mixed with ethanol in different ratios was employed as the internal standard. Ethanol yield was identified according to the peak areas of test samples and standard substances. Each analysis was carried out in triplicates.
Results
Construction of p1301 M1-TALENs
The nucleotide sequences encoding repeats of 34 amino acids were assembled using Fast TALEN Assembly kit, inserted into Ptalen L48 and Ptalen R36 (Figure 1A), and then further confirmed by sequencing and restriction enzyme digestion. Digested TALEN arms were ligated with the backbone vector p1301M1. Two bands of approximately 8600 and 4000 bp were obtained after the recombinant plasmid was digested with HindIII, SacI, and AscI, which demonstrated the successful construction of p1301M1-TALENs (Figure 1B).
Introduction of p1301M1-TALENs
Hygromycin was used to select positive clones, and the successful amplification of approximately 1000 bp hyg gene indicated the successful introduction of p1301 M1-TALENs into the native S. cerevisiae strain As2.4 (Figure 2A). The acquirement of hyg and ADH2 demonstrated the successful introduction of p1301M1-TALENS combined with ADH2 into ΔADH2 As2.4.
FIGURE 2.
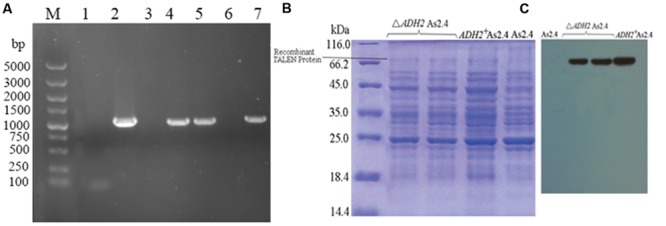
Introduction of recombinant TALEN vector. (A) Amplification of the hyg gene in ΔADH2 As2.4 and ADH2+ As2.4: Lane 1, PCR product without a template; Lane 2, PCR product using hyg gene fragment as a template; Lanes 3–7, PCR products using genomes of recombinant As2.4 colonies picked from YPD medium as templates. (B) SDS-PAGE analysis of total proteins from different As2.4 strains. (C) Western blot analysis of total proteins from different As2.4 strains using anti-FLAG antibody.
The proteins of native As2.4, ΔADH2 As2.4, and ADH2+ As2.4 were extracted, and the supernatants were loaded onto 12% SDS-PAGE. As shown in Figure 2B, bands of approximately 68 kD corresponding to repeats consisting of 34 amino acids were detected in the ΔADH2 As2.4 strain and complement strain, whereas no corresponding band was detected in native As2.4.
As shown in Figure 2C, when anti-FLAG monoclonal antibody was used to perform Western blot analysis, target bands of 68 kD were also visualized in ΔADH2 As2.4 and ADH2+ As2.4, whereas no corresponding band was detected in native As2.4. The result further confirmed the successful introduction of p1301M1-TALENs into the As2.4 strains.
Disruption of ADH2 in As2.4
The total RNAs of native As2.4, ΔADH2 As2.4, and ADH2+ As2.4 were extracted to amplify cDNAs. Primers F3 and R3 were used to amplify a 70 bp fragment containing the target sequence. As shown in Figure 3A, results of qRT-PCR analysis indicated that the relative expression levels of the target sequences were much higher in native As2.4 and ADH2+ As2.4 than in ΔADH2 As2.4. The 70 bp bands were detected in the qRT-PCR products of native As2.4 and ADH2+ As2.4, whereas, no corresponding band was detected in ΔADH2 As2.4 (Figure 3B). When F1 and R1 were used to perform PCR, a slightly smaller fragment was detected in ΔADH2 As2.4 compared with those in native As2.4 and ADH2+ As2.4 (Figure 3C). The relative expression levels in native As2.4 and ADH2+ As2.4 were also much higher than that in ΔADH2 As2.4. The PCR products using primers F1, R1, and genomes of native As2.4, ΔADH2 As2.4, and ADH2+ As2.4 were sequenced, and alignment results indicated that the 17 bp target sequence AGTTGGAGCATAAGGAT was knocked out accurately (Figure 4A). Twenty colonies on YPD medium were selected, and the ADH2 gene fragment was amplified and sequenced; the target sequence in 16 colonies was knocked out successfully, indicating that ADH2 in As2.4 was knocked out using TALEN technology with 80% efficiency.
FIGURE 3.
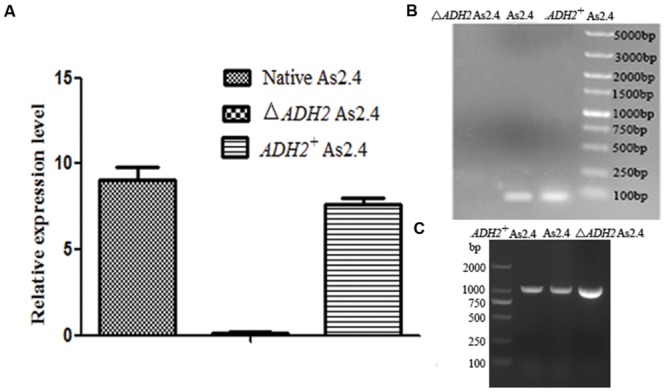
Disruption and complementation of the ADH2 gene in As2.4 strains. (A) Relative expression level of 70 bp target product in different As2.4 strains. (B) Detection of qRT-PCR products by agarose gel electrophoresis. (C) Amplification of the ADH2 gene using genomes of native As2.4, ΔADH2 As2.4, and ADH2+ As2.4 strains as templates.
FIGURE 4.
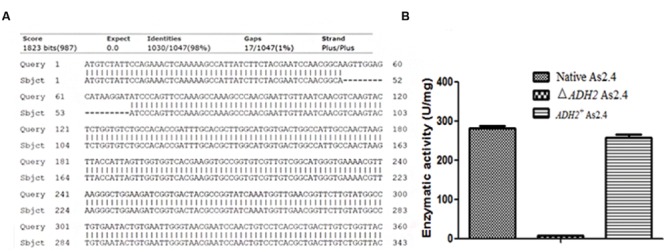
Knockout of 17 bp target sequence. (A) Sequence alignments of ADH2 gene and PCR products of ΔADH2 As2.4. (B) Alcohol dehydrogenase activities of supernatants from different As2.4 strains after cell disruption.
The alcohol dehydrogenase activities of supernatants from the different As2.4 strains were assayed, as shown in Figure 4B; supernatants of native As2.4, ΔADH2 As2.4, and ADH2+ As2.4 showed enzymatic activities of 282 ± 10.3, 5.6 ± 0.5, and 258 ± 12.5 U/mg, respectively. The results indicated that ΔADH2 As2.4 rarely showed any alcohol dehydrogenase activity after ADH2 in As2.4 was disrupted by the recombinant TALEN vector.
Ethanol Yields in Different As2.4 Strains
The ethanol yields in the different As2.4 strains were detected by LC–MS and GC. As shown in Supplementary Figure S1, the anion peak of 45.0 corresponding to ethanol was detected in fermentation liquids in the different As2.4 strains and ethanol standards. This result indicates that ethanol was successfully produced by the different As2.4 strains. The peak area of ethanol in ΔADH2 As2.4 was significantly larger than those in native As2.4 and ADH2+ As2.4 (Figure 5A). The ethanol yields in native As2.4, ΔADH2 As2.4, and ADH2+ As2.4 were 9.6 ± 1.51, 14.6 ± 1.35, and 10.1 ± 1.22 g/L, respectively, according to the peak areas of different internal standard substances. Ethanol production was improved by 52.4 ± 5.3% through the disruption of ADH2 in As2.4 and decreased to the same level as that in native As2.4 after ADH2 was complemented into ΔADH2 As2.4 (Figure 5B).
FIGURE 5.
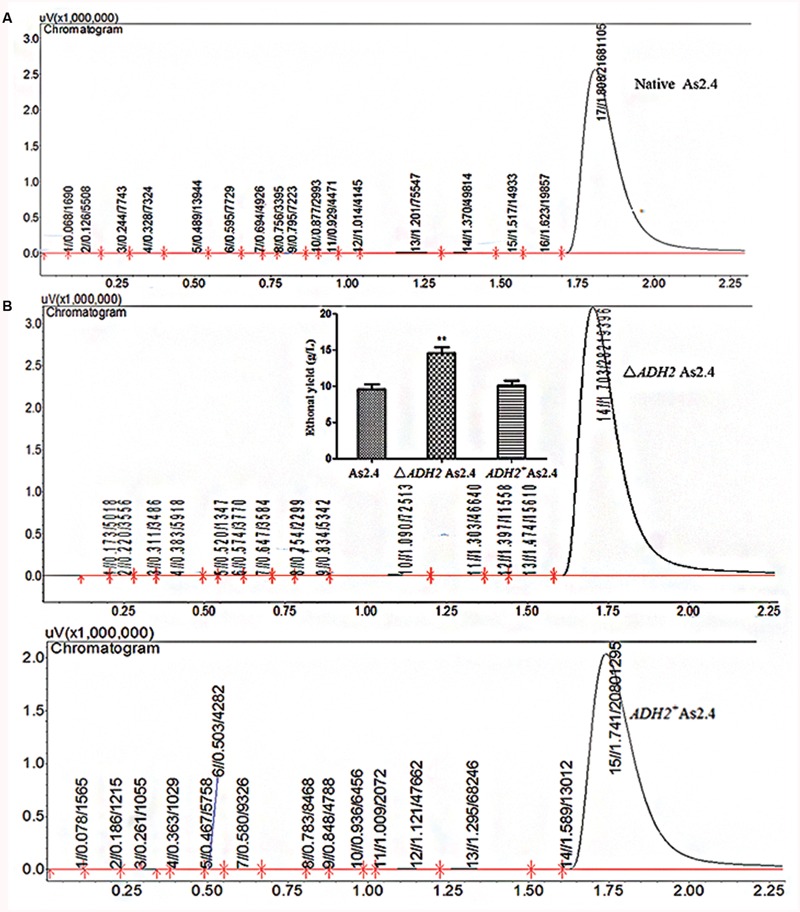
Detection of ethanol yields in different As2.4 strains. (A) Detection of ethanol in As2.4 strains by gas chromatography. (B) Ethanol yields in native As2.4, ΔADH2 As2.4, and ADH2+ As2.4.
Discussion
In this study, the ADH2 gene in As2.4 was first disrupted using Fast TALEN technology. The ethanol yield in As2.4 was significantly improved by the disruption of ADH2. The illustration of improvement of ethanol yield in S. cerevisiae strain using Fast TALEN technology was shown in Supplementary Figure S2. The results of this study would lay a foundation for the metabolic engineering of S. cerevisiae, thereby promoting the application of bioethanol in different industries.
Alcohol dehydrogenase encoded by ADH2 could catalyze ethanol to aldehyde. In consequence, different approaches, including PCR-mediated homologous recombination and Cre-loxP system, were employed to knock out ADH2 and improve ethanol yield (Ding et al., 2014; Zheng et al., 2015). However, these approaches showed low efficiency, low specificity, and detectable alcohol dehydrogenase II activity in ADH2 gene depletion strain (Wang et al., 2009). Moreover, ethanol yield was only improved by 5–20% through the disruption of ADH2. The relatively lower improvement probably resulted from the residual alcohol dehydrogenase II activity after ADH2 was knocked out (Wang et al., 2009; Krivoruchko et al., 2011). In the present study, ADH2 in As2.4 was disrupted by a novel recombinant TALEN vector with 80% efficiency, and the enzymatic activity assay results revealed that the total proteins from ΔADH2 As2.4 rarely showed any alcohol dehydrogenase activity toward alcohol. Moreover, the sequencing result revealed no off-target effect during the knockout of ADH2 in native S. cerevisiae strain As2.4. The ethanol yield in As2.4 was improved by 52.4 ± 5.3% through the disruption of ADH2; the increasing ratio of ethanol yield was much higher than those previously reported (Wang et al., 2009; Krivoruchko et al., 2011). ADH2 was also knocked into ΔADH2 As2.4 using Fast TALEN technology. The bioethanol level produced in ADH2+ As2.4 was almost the same as that in native As2.4, which further demonstrated the function of ADH2 during bioethanol production in S. cerevisiae strain. This Fast TALEN technology for the disruption of ADH2 in native As2.4 showed high efficiency and specificity compared with previously reported methods.
Conclusion
In this study, ADH2 in S. cerevisiae As2.4 strain was disrupted and complemented. Ethanol yield was improved by 52.4 ± 5.3% through the disruption of ADH2 in As2.4 using Fast TALEN technology. The complement of ADH2 could decrease the ethanol yield in ADH2+ As2.4 to the same level as that in native As2.4. To the best of our knowledge, this study is the first to report on the disruption of a target gene in S. cerevisiae using Fast TALEN technology. This study would provide a novel approach for the disruption of a target gene in S. cerevisiae with high efficiency and specificity, thereby promoting the improvement of ethanol production in S. cerevisiae by metabolic engineering.
Author Contributions
WY: design the experiment, WZ: direct the experiment, WY, TL, GT, HL, ZH: did the experiment.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge financial support from National Natural Science Foundation of China (31500037), Natural Science Foundation of Guangdong Province (2015A030310103), Science and Technology Planning Project of Guangdong Province (2016A020222022, 2015A030302061).
Footnotes
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01067
Detection of ethanol yields in native As2.4, ΔADH2 As2.4, and ADH2+ As2.4 strains by LC–MS.
The illustration of improvement of ethanol yield in S. cerevisiae strain using Fast TALEN technology.
References
- Beier D. R., Sledziewski A., Young E. T. (1985). Deletion analysis identifies a region, upstream of the ADH2 gene of Saccharomyces cerevisiae, which is required for ADR1-mediated derepression. Mol. Cell. Biol. 5 1743–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanove A. J., Voytas D. F. (2011). TAL effectors: customizable proteins for DNA targeting. Science 333 1843–1846. 10.1126/science.1204094 [DOI] [PubMed] [Google Scholar]
- Ding Y., Yu A. Q., Li C. L., Fang J., Zeng Y., Li D. S. (2014). TALEN-mediated Nanog disruption results in less invasiveness, more chemosensitivity and reversal of EMT in Hela cells. Oncotarget 5 8393–8401. 10.18632/oncotarget.2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang F., Salmon K., Shen M. W., Aeling K. A., Ito E., Irwin B., et al. (2011). A vector set for systematic metabolic engineering in Saccharomyces cerevisiae. Yeast 28 123–136. 10.1002/yea.1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau J., Boch J., Posch S. (2013). TALEN offer: genome-wide TALEN off-target prediction. Bioinformatics 29 2931–2932. 10.1093/bioinformatics/btt501 [DOI] [PubMed] [Google Scholar]
- Gray M., Honigberg S. M. (2001). Effect of chromosomal locus, GC content and length of homology on PCR-mediated targeted gene replacement in Saccharomyces. Nucleic Acids Res. 29 5156–5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueldener U., Heinisch J., Koehler G. J., Voss D., Hegemann J. H. (2002). A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 30:e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung J. K., Sander J. D. (2013). TALENs: a widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 14 49–55. 10.1038/nrm3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivoruchko A., Siewers V., Nielsen J. (2011). Opportunities for yeast metabolic engineering: lessons from synthetic biology. Biotechnol. J. 6 262–276. 10.1002/biot.201000308 [DOI] [PubMed] [Google Scholar]
- Kuyper M., Hartog M. M., Toirkens M. J., Almering M. J., Winkler A. A., Dijken J. P., et al. (2005). Metabolic engineering of a xylose-isomerase-expressing Saccharomyces cerevisiae strain for rapid anaerobic xylose fermentation. FEMS Yeast Res. 5 399–409. 10.1016/j.femsyr.2004.09.010 [DOI] [PubMed] [Google Scholar]
- Matsushika A., Inoue H., Watanabe S., Kodaki T., Makino K., Wawayama S. (2009). Ethanol production from xylose in engineered Saccharomyces cerevisiae strains: current state and perspectives. Appl. Microbiol. Biotechnol. 84 37–53. 10.1007/s00253-009-2101-x [DOI] [PubMed] [Google Scholar]
- Nakajima K., Yaoita Y. (2013). Comparison of TALEN scaffolds in Xenopus tropicalis. Biology Open 2 1364–1370. 10.1242/bio.20136676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma T., Hosoi S., Woltjen K., Suzuki K., Kashiwagi K., Wada H., et al. (2013). Efficient TALEN construction and evaluation methods for human cell and animal applications. Genes Cells 18 315–326. 10.1111/gtc.12037 [DOI] [PubMed] [Google Scholar]
- Uhde-stone C., Cheung E., Lu B. (2014). TALE activators regulate gene expression in a position- and strand-dependent manner in mammalian cells. Biochem. Biophys. Res. Commun. 443 1189–1194. 10.1016/j.bbrc.2013.12.111 [DOI] [PubMed] [Google Scholar]
- Vallari R. C., Cook W. J., Audino D. C., Morgan M. J., Jensen D. E., Laudano A. P., et al. (1992). Glucose repression of the yeast ADH2 gene occurs through multiple mechanisms, including control of the protein synthesis of its transcriptional activator, ADR1. Mol. Cell. Biol. 12 1663–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verho R., Londesborough J., Penttilä M., Richard P. (2003). Engineering redox cofactor regeneration for improved pentose fermentation in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 69 5892–5897. 10.1128/AEM.69.10.5892-5897.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. Y., Wang J. J., Liu X. F., He X. P., Zhang B. R. (2009). Recombinant industrial brewing yeast strains with ADHII interruption using self-cloning GSH1+CUP1 cassette. FEMS Yeast Res. 9 574–581. 10.1111/j.1567-1364.2009.00502.x [DOI] [PubMed] [Google Scholar]
- Wiebe M. G., Rintala E., Tamminen A., Simolin H., Salusjärvi L., Toivari M., et al. (2007). Central carbon metabolism of Saccharomyces cerevisiae in anaerobic, oxygen-limited and fully aerobic steady-state conditions and following a shift to anaerobic conditions. FEMS Yeast Res. 8 140–154. 10.1111/j.1567-1364.2007.00234.x [DOI] [PubMed] [Google Scholar]
- Zhang Y., Zhang F., Li X., Baller J. A., Qi Y., Starker C. G., et al. (2013). Transcription activator-like effector nucleases enable efficient plant genome engineering. Plant Physiol. 161 20–27. 10.1104/pp.112.205179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L., Gu T. P., Weber A. R., Shen J. Z., Li B. Z., Xie Z. G., et al. (2015). Gadd45a promotes DNA demethylation through TDG. Nucleic Acids Res. 43 3986–3997. 10.1093/nar/gkv283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu Y., Tong X. J., Wang Z. X., Liu D., Pan R. C., Li Z., et al. (2013). TALEN-mediated precise genome modification by homologous recombination in zebrafish. Nat. Methods 10 329–331. 10.1038/nmeth.2374 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detection of ethanol yields in native As2.4, ΔADH2 As2.4, and ADH2+ As2.4 strains by LC–MS.
The illustration of improvement of ethanol yield in S. cerevisiae strain using Fast TALEN technology.