

Abstract
Many disease-related single nucleotide polymorphisms (SNPs) have been inferred from genome-wide association studies (GWAS) in recent years. Numerous studies have shown that some SNPs located in protein-coding regions are associated with numerous diseases by affecting gene expression. However, in noncoding regions, the mechanism of how SNPs contribute to disease susceptibility remains unclear. Enhancer elements are functional segments of DNA located in noncoding regions that play an important role in regulating gene expression. The SNPs located in enhancer elements may affect gene expression and lead to disease. We presented a method for identifying liver cancer-related enhancer SNPs through integrating GWAS and histone modification ChIP-seq data. We identified 22 liver cancer-related enhancer SNPs, 9 of which were regulatory SNPs involved in distal transcriptional regulation. The results highlight that these enhancer SNPs may play important roles in liver cancer.
1. Introduction
Single nucleotide polymorphism (SNP) is a variation at a single nucleotide in a DNA sequence [1]. In the last decade, a large number of genome-wide association studies (GWAS) have been published, indicating that thousands of SNPs are associated with diseases. Linkage disequilibrium is the nonrandom association of alleles at different genome locations [2]. There are many SNPs in LD with the causal SNP at specific GWAS locus [3, 4]. Over 90% of these GWAS variants are located in noncoding regions, and approximately 10% are in LD with a protein-coding variant [5, 6]. In protein-coding regions, many studies have shown that some SNPs are associated with numerous diseases by affecting gene expression [7, 8]. However, in noncoding regions, the mechanism of how SNPs contribute to disease susceptibility remains unclear.
Enhancers are the core regulatory components of the genome that act over a distance to positively regulate gene expression [9]. It is estimated that 400,000 to 1 million putative enhancers exist in the human genome [10, 11]. Recently, some studies have shown that disease-related GWAS SNPs are correlated with enhancers marked with special histone modifications [12–15]. Therefore, through integrating GWAS and histone modification ChIP-seq data in a given disorder, we can identify disease-related enhancer SNPs.
We provided a method for identifying liver cancer-related enhancer SNPs through integrating liver cancer GWAS and histone modification ChIP-seq data. We identified 22 enhancer SNPs associated with liver cancer, 9 of which were regulatory SNPs involved in distal transcriptional regulation. The results highlight that these enhancer SNPs may play important roles in liver cancer.
2. Results
2.1. Pipeline of Identifying Liver Cancer-Related Enhancer SNPs
As shown in Figure 1, the pipeline consists of four steps. Firstly, we downloaded liver cancer-related GWAS SNPs from the GRASP [16] database and used LD data from HapMap [17] to infer liver cancer-related LD SNPs. Secondly, we identified enhancer regions in liver cancer through integrating histone modification ChIP-seq data in the HepG2 cell line. Thirdly, we mapped the liver cancer-related LD SNPs to the identified enhancers in liver cancer and obtained liver cancer-related enhancer SNPs. Finally, we used a curated regulatory SNP database named rVarBase [18] to validate our results.
Figure 1.
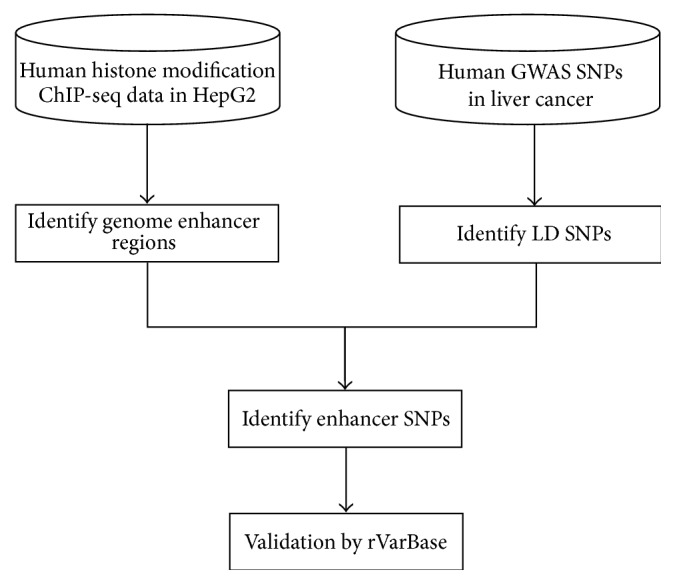
Flowchart identifying liver cancer-related enhancer SNPs. LD SNPs: SNPs located in linkage disequilibrium blocks of GWAS SNPs. Enhancer SNPs: SNPs identified in enhancer regions. rVarBase: a database for variant's regulatory features.
2.2. Linkage Disequilibrium Analysis with Liver Cancer-Associated SNPs
We obtained 45 liver cancer-associated SNPs from GRASP (Table 1). These SNPs are the raw potential liver cancer-related SNPs. Then, we used LD data from HapMap to achieve liver cancer-associated LD SNPs. The total number of potential liver cancer-related SNPs is 340.
Table 1.
Summary of liver cancer-associated SNPs from GRASP database.
| SNP ID | p value | Chromosome | Populations | PMID |
|---|---|---|---|---|
| rs17401966 | 1.20E − 19 | 1 | CHB | 20676096 |
| rs1249458 | 8.30E − 06 | 2 | CHB | 22807686 |
| rs1714259 | 1.10E − 06 | 2 | CHB | 22807686 |
| rs2396470 | 5.10E − 07 | 2 | CHB | 20676096 |
| rs7424161 | 8.80E − 06 | 2 | CHB | 22807686 |
| rs7574865 | 1.70E − 11 | 2 | CHB | 23242368 |
| rs3905886 | 3.70E − 06 | 3 | CHB | 22807686 |
| rs1073547 | 6.80E − 06 | 4 | CHB | 22807686 |
| rs17081345 | 3.70E − 07 | 6 | CHB | 20676096 |
| rs9272105 | 3.30E − 23 | 6 | CHB | 22807686 |
| rs9275319 | 8.70E − 19 | 6 | CHB | 23242368 |
| rs9494257 | 1.10E − 14 | 6 | CHB | 20676096 |
| rs12682266 | 6.70E − 06 | 8 | CHB | 22174901 |
| rs1573266 | 7.40E − 06 | 8 | CHB | 22174901 |
| rs2275959 | 6.40E − 06 | 8 | CHB | 22174901 |
| rs7821974 | 7.00E − 06 | 8 | CHB | 22174901 |
| rs7898005 | 7.00E − 08 | 10 | CHB | 20676096 |
| rs10160758 | 6.00E − 06 | 11 | CHB | 22807686 |
| rs10896464 | 6.50E − 06 | 11 | CHB | 22807686 |
| rs2611145 | 9.30E − 06 | 11 | CHB | 22174901 |
| rs3825023 | 3.10E − 06 | 11 | CHB | 22807686 |
| rs7119426 | 3.50E − 06 | 11 | CHB | 22807686 |
| rs402071 | 8.60E − 06 | 19 | CHB | 22807686 |
| rs3092194 | 4.40E − 06 | 20 | CHB | 22807686 |
| rs368007 | 9.90E − 06 | 20 | CHB | 22807686 |
| rs455804 | 4.40E − 10 | 21 | CHB | 22807686 |
| rs1980215 | 2.30E − 06 | 3 | JPT | 21499248 |
| rs2596542 | 4.20E − 13 | 6 | JPT | 21499248 |
| rs9275572 | 1.40E − 09 | 6 | JPT | 21499248 |
| rs1568658 | 6.90E − 06 | 7 | JPT | 21499248 |
| rs952656 | 2.80E − 06 | 8 | JPT | 21499248 |
| rs4363614 | 4.20E − 07 | 11 | JPT | 21499248 |
| rs1957496 | 4.60E − 06 | 14 | JPT | 21499248 |
| rs8019534 | 3.90E − 06 | 14 | JPT | 21499248 |
| rs1794304 | 3.60E − 06 | 16 | JPT | 21725309 |
| rs2208456 | 3.80E − 06 | 20 | JPT | 21499248 |
| rs1012068 | 1.30E − 14 | 22 | JPT | 21725309 |
| rs11703779 | 4.20E − 06 | 22 | JPT | 21725309 |
| rs4820994 | 3.00E − 06 | 22 | JPT | 21725309 |
| rs4820996 | 4.20E − 06 | 22 | JPT | 21725309 |
| rs5753816 | 4.90E − 06 | 22 | JPT | 21725309 |
| rs5753818 | 9.40E − 06 | 22 | JPT | 21725309 |
| rs5998152 | 1.20E − 07 | 22 | JPT | 21725309 |
| rs7287054 | 3.80E − 06 | 22 | JPT | 21725309 |
| rs737084 | 5.90E − 06 | 22 | JPT | 21725309 |
2.3. Identification of Liver Cancer-Related Enhancer SNPs
Previous studies indicated that the enhancer regions are marked by a strong H3K4me1 signal and a relatively weak H3K4me3 signal [19, 20]. Thus, we used histone modification ChIP-seq data to recognize the enhancer regions in liver cancer. Then, we mapped the liver cancer-related GWAS SNPs to the enhancer regions and obtained 22 enhancer SNPs in liver cancer (Table 2).
Table 2.
Summary of predicted enhancer SNPs in liver cancer.
| SNP ID | Chromosome | Start | End | Chain | Populations |
|---|---|---|---|---|---|
| rs12751375 | chr1 | 10291873 | 10291874 | + | CHB |
| rs6700866 | chr1 | 10306037 | 10306038 | + | CHB |
| rs9494257 | chr6 | 135827471 | 135827472 | + | CHB |
| rs17064474 | chr6 | 135680137 | 135680138 | + | CHB |
| rs17721919 | chr6 | 135748923 | 135748924 | + | CHB |
| rs17721931 | chr6 | 135749377 | 135749378 | + | CHB |
| rs6903949 | chr6 | 135821065 | 135821066 | + | CHB |
| rs6996881 | chr8 | 37407919 | 37407920 | + | CHB |
| rs4739519 | chr8 | 37412858 | 37412859 | + | CHB |
| rs6988263 | chr8 | 37414659 | 37414660 | + | CHB |
| rs12156293 | chr8 | 37419921 | 37419922 | + | CHB |
| rs6928810 | chr6 | 31410523 | 31410524 | + | JPT |
| rs3869132 | chr6 | 31410947 | 31410948 | − | JPT |
| rs2596562 | chr6 | 31354594 | 31354595 | − | JPT |
| rs2523475 | chr6 | 31361709 | 31361710 | − | JPT |
| rs2523467 | chr6 | 31362929 | 31362930 | − | JPT |
| rs9501387 | chr6 | 31364458 | 31364459 | + | JPT |
| rs1568658 | chr7 | 29141557 | 29141558 | − | JPT |
| rs1794304 | chr16 | 12625394 | 12625395 | + | JPT |
| rs5994449 | chr22 | 32304178 | 32304179 | + | JPT |
| rs5753816 | chr22 | 32312841 | 32312842 | + | JPT |
| rs5749339 | chr22 | 32315734 | 32315735 | + | JPT |
2.4. Validation as Regulatory SNPs
rVarBase is a database that provides reliable, comprehensive, and user-friendly annotations on variant's regulatory features [18]. It includes regulatory SNPs (rSNPs), LD-proxies of rSNPs, and genes that are potentially regulated by rSNPs. We used rVarBase to analyze these 22 enhancer SNPs in liver cancer and found that 14 SNPs have evidence of regulatory SNPs and 9 SNPs (rs9494257, rs6903949, rs6996881, rs4739519, rs6988263, rs12156293, rs1568658, rs5994449, and rs5753816) are involved in distal transcriptional regulation (Table 3). Table 4 shows the potential target genes of these 9 SNPs.
Table 3.
Summary of liver cancer-related regulatory SNPs validated by rVarBase.
| SNP ID | Regulatory SNP | Distal regulation | Chromatin state | Related regulatory elements |
|---|---|---|---|---|
| rs12751375 | Yes | No | Inactive region | n/a |
| rs6700866 | Yes | No | Weak transcription; ZNF genes and repeats; strong transcription; enhancers | n/a |
| rs9494257 | Yes | Yes | Enhancers; flanking active TSS; weak transcription | Chromatin interactive region |
| rs17064474 | Yes | No | Weak transcription; active TSS; flanking active TSS; enhancers | n/a |
| rs17721919 | Yes | No | Weak transcription | n/a |
| rs17721931 | Yes | No | Weak transcription | n/a |
| rs6903949 | Yes | Yes | Weak transcription; enhancers | TF binding region; chromatin interactive region |
| rs6996881 | Yes | Yes | Weak transcription; enhancers | Chromatin interactive region |
| rs4739519 | Yes | Yes | Enhancers; weak transcription | Chromatin interactive region |
| rs6988263 | Yes | Yes | Enhancers; weak transcription; genic enhancers; bivalent enhancer; flanking active TSS | Chromatin interactive region |
| rs12156293 | Yes | Yes | Enhancers; weak transcription; bivalent enhancer; genic enhancers | Chromatin interactive region |
| rs1568658 | Yes | Yes | Weak transcription; enhancers; strong transcription | Chromatin interactive region |
| rs5994449 | Yes | Yes | Weak transcription; strong transcription; ZNF genes and repeats | Chromatin interactive region |
| rs5753816 | Yes | Yes | Weak transcription; enhancers; flanking active TSS | Chromatin interactive region |
Table 4.
Summary of liver cancer-related regulatory SNPs and potential target genes validated by rVarBase.
| SNP ID | Gene symbol | Ensemble ID | Regulation type |
|---|---|---|---|
| rs9494257 | BCLAF1 | ENSG00000029363 | Distal transcriptional regulation |
| rs9494257 | AHI1 | ENSG00000135541 | Distal transcriptional regulation |
| rs9494257 | LINC00271 | ENSG00000231028 | Distal transcriptional regulation |
| rs6903949 | MYB | ENSG00000118513 | Distal transcriptional regulation |
| rs6903949 | BCLAF1 | ENSG00000029363 | Distal transcriptional regulation |
| rs6903949 | AHI1 | ENSG00000135541 | Distal transcriptional regulation |
| rs6903949 | LINC00271 | ENSG00000231028 | Distal transcriptional regulation |
| rs6996881 | ZNF703 | ENSG00000183779 | Distal transcriptional regulation |
| rs6996881 | ERLIN2 | ENSG00000147475 | Distal transcriptional regulation |
| rs6996881 | Null | ENSG00000183154 | Distal transcriptional regulation |
| rs6996881 | Null | ENSG00000253161 | Distal transcriptional regulation |
| rs4739519 | ZNF703 | ENSG00000183779 | Distal transcriptional regulation |
| rs4739519 | Null | ENSG00000254290 | Distal transcriptional regulation |
| rs6988263 | ZNF703 | ENSG00000183779 | Distal transcriptional regulation |
| rs6988263 | Null | ENSG00000254290 | Distal transcriptional regulation |
| rs12156293 | ZNF703 | ENSG00000183779 | Distal transcriptional regulation |
| rs12156293 | Null | ENSG00000254290 | Distal transcriptional regulation |
| rs12156293 | ERLIN2 | ENSG00000147475 | Distal transcriptional regulation |
| rs12156293 | Null | ENSG00000183154 | Distal transcriptional regulation |
| rs1568658 | Null | ENSG00000228421 | Distal transcriptional regulation |
| rs1568658 | TRIL | ENSG00000176734 | Distal transcriptional regulation |
| rs1568658 | Null | ENSG00000255690 | Distal transcriptional regulation |
| rs5994449 | DEPDC5 | ENSG00000100150 | Distal transcriptional regulation |
| rs5994449 | FBXO7 | ENSG00000100225 | Distal transcriptional regulation |
| rs5994449 | SYN3 | ENSG00000185666 | Distal transcriptional regulation |
| rs5994449 | PRR14L | ENSG00000183530 | Distal transcriptional regulation |
| rs5994449 | PISD | ENSG00000241878 | Distal transcriptional regulation |
| rs5994449 | EIF4ENIF1 | ENSG00000184708 | Distal transcriptional regulation |
| rs5994449 | RNU6-28 | ENSG00000199248 | Distal transcriptional regulation |
| rs5994449 | SFI1 | ENSG00000198089 | Distal transcriptional regulation |
| rs5753816 | YWHAH | ENSG00000128245 | Distal transcriptional regulation |
| rs5753816 | C22orf24 | ENSG00000128254 | Distal transcriptional regulation |
| rs5753816 | PISD | ENSG00000241878 | Distal transcriptional regulation |
| rs5753816 | DEPDC5 | ENSG00000100150 | Distal transcriptional regulation |
| rs5753816 | RNU6-28 | ENSG00000199248 | Distal transcriptional regulation |
| rs5753816 | SFI1 | ENSG00000198089 | Distal transcriptional regulation |
| rs5753816 | EIF4ENIF1 | ENSG00000184708 | Distal transcriptional regulation |
| rs5753816 | RFPL3S | ENSG00000205853 | Distal transcriptional regulation |
| rs5753816 | Null | ENSG00000230736 | Distal transcriptional regulation |
| rs5753816 | Null | ENSG00000243519 | Distal transcriptional regulation |
| rs5753816 | Null | ENSG00000241954 | Distal transcriptional regulation |
| rs5753816 | Null | ENSG00000232218 | Distal transcriptional regulation |
| rs5753816 | SYN3 | ENSG00000185666 | Distal transcriptional regulation |
3. Materials and Methods
3.1. GWAS and LD Datasets
We downloaded the human liver cancer-related GWAS SNPs from GRASP. The database includes 26 and 19 liver cancer-associated SNPs (p < 10−5) from Han Chinese in Beijing, China (CHB), and Japanese in Tokyo, Japan (JPT), respectively. The URL is https://grasp.nhlbi.nih.gov/Overview.aspx. We obtained all SNPs in LD with GWAS-lead SNPs using LD blocks identified with publicly available HapMap data on the CHB and JPT populations. The LD data can be downloaded from http://hapmap.ncbi.nlm.nih.gov/index.html/.
3.2. Histone Modification Datasets
We downloaded the human histone modification ChIP-seq datasets in the HepG2 cell line from the ENCODE Production Data/Broad Institute. The URL is http://genome.ucsc.edu/ENCODE/downloads.html.
3.3. Linkage Disequilibrium Analysis
In the genome, SNPs located in close proximity tend to be in linkage disequilibrium with each other. The International HapMap Project has established linkage disequilibrium of human genome SNPs. We used LD data from HapMap to achieve liver cancer-associated LD SNPs (R 2 > 0.8).
3.4. Identify Enhancer Regions and Enhancer SNPs
Firstly, we downloaded the human histone modification BAM files (H3K4me1 and H3K4me3) in the HepG2 cell line from the ENCODE project. Then, we used BEDtools [21] to count read coverage for every position of the genome. Through calculating the ratio H3K4me1/H3K4me3 and picking up the regions with log2(H3K4me1/H3K4me3) > 1.2, we identified the potential enhancer regions. Finally, we mapped the potential LD SNPs to these enhancer regions and achieved liver cancer-related enhancer SNPs.
4. Discussion
Through integrating liver cancer GWAS SNPs from GRASP, LD data from HapMap, and histone modification ChIP-seq data from ENCODE, we explored liver cancer-related enhancer SNPs. We compared our results with rVarBase and found that 9 SNPs (rs9494257, rs6903949, rs6996881, rs4739519, rs6988263, rs12156293, rs1568658, rs5994449, and rs5753816) were regulatory SNPs involved in distal transcriptional regulation. The results highlight that these enhancer SNPs may play important roles in liver cancer.
Compared with protein-coding regions in the human genome, noncoding regions contain much more genetic variations. Some important regulation regions, such as enhancers, have great influence on target gene expression. SNPs located in these regions may disturb gene expression and even cause diseases. Thus, the identification of SNPs in enhancer regions is helpful to understand the mechanism of association between SNPs and diseases.
We presented a method to identify disease-related SNPs located in enhancer regions that gives a new solution to investigate the relationship between SNPs and diseases. The presented method can also be applied to other diseases and will enable biologists to investigate the mechanism of disease risk associated with SNPs.
Acknowledgments
This work was partially supported by the National High-Tech Research and Development Program (863) of China (2012AA02A601, 2012AA02A602, 2012AA020404, 2012AA020409, 2012AA02A604, 2014AA021505, 2015AA020101, and 2015AA020108), National Science and Technology Major Project [no. 2013ZX03005012], and the National Natural Science Foundation of China (61571152, 31301089).
Competing Interests
The authors declare that there are no competing interests regarding the publication of this paper.
Authors' Contributions
Tianjiao Zhang collected the data, designed the computational experiments, carried out the statistical analysis, and wrote the paper. Qinghua Jiang participated in the design of the study. Yang Hu, Xiaoliang Wu, and Rui Ma participated in the revision of this paper. Yadong Wang gave comments and revisions to the final version of this paper. All authors read and approved the final paper. Tianjiao Zhang and Yang Hu equally contributed to this paper.
References
- 1.Sachidanandam R., Weissman D., Schmidt S. C., et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409(6822):928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- 2.Slatkin M. Linkage disequilibrium—understanding the evolutionary past and mapping the medical future. Nature Reviews Genetics. 2008;9(6):477–485. doi: 10.1038/nrg2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peng J., Uygun S., Kim T., Wang Y., Rhee S. Y., Chen J. Measuring semantic similarities by combining gene ontology annotations and gene co-function networks. BMC Bioinformatics. 2015;16(1, article 44) doi: 10.1186/s12859-015-0474-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peng J., Wang T., Wang J., Wang Y., Chen J. Extending gene ontology with gene association networks. Bioinformatics. 2016;32(8):1185–1194. doi: 10.1093/bioinformatics/btv712. [DOI] [PubMed] [Google Scholar]
- 5.Maurano M. T., Humbert R., Rynes E., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337(6099):1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schaub M. A., Boyle A. P., Kundaje A., Batzoglou S., Snyder M. Linking disease associations with regulatory information in the human genome. Genome Research. 2012;22(9):1748–1759. doi: 10.1101/gr.136127.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hindorff L. A., Sethupathy P., Junkins H. A., et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(23):9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zou Q., Li J., Hong Q., et al. Prediction of microRNA-disease associations based on social network analysis methods. BioMed Research International. 2015;2015:9. doi: 10.1155/2015/810514.810514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banerji J., Rusconi S., Schaffner W. Expression of a β-globin gene is enhanced by remote SV40 DNA sequences. Cell. 1981;27(2):299–308. doi: 10.1016/0092-8674(81)90413-x. [DOI] [PubMed] [Google Scholar]
- 10.Buecker C., Wysocka J. Enhancers as information integration hubs in development: lessons from genomics. Trends in Genetics. 2012;28(6):276–284. doi: 10.1016/j.tig.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie W., Ren B. Developmental biology. Enhancing pluripotency and lineage specification. Science. 2013;341(6143):245–247. doi: 10.1126/science.1236254. [DOI] [PubMed] [Google Scholar]
- 12.Heinz S., Benner C., Spann N., et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular Cell. 2010;38(4):576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ernst J., Kheradpour P., Mikkelsen T. S., et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473(7345):43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akhtar-Zaidi B., Cowper-Sallari R., Corradin O., et al. Epigenomic enhancer profiling defines a signature of colon cancer. Science. 2012;336(6082):736–739. doi: 10.1126/science.1217277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trynka G., Sandor C., Han B., et al. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nature Genetics. 2013;45(2):124–130. doi: 10.1038/ng.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leslie R., O'Donnell C. J., Johnson A. D. GRASP: analysis of genotype-phenotype results from 1390 genome-wide association studies and corresponding open access database. Bioinformatics. 2014;30(12):i185–i194. doi: 10.1093/bioinformatics/btu273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.The International HapMap Consortium. The international HapMap project. Nature. 2003;426(6968):789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 18.Guo L., Du Y., Qu S., Wang J. rVarBase: an updated database for regulatory features of human variants. Nucleic Acids Research. 2016;44(1):D888–D893. doi: 10.1093/nar/gkv1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Santa F., Barozzi I., Mietton F., et al. A large fraction of extragenic RNA Pol II transcription sites overlap enhancers. PLoS Biology. 2010;8(5, article e1000384) doi: 10.1371/journal.pbio.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marques A. C., Hughes J., Graham B., Kowalczyk M. S., Higgs D. R., Ponting C. P. Chromatin signatures at transcriptional start sites separate two equally populated yet distinct classes of intergenic long noncoding RNAs. Genome Biology. 2013;14(11, article R131) doi: 10.1186/gb-2013-14-11-r131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quinlan A. R. UNIT 11.12 BEDTools: the swiss-army tool for genome feature analysis. Current Protocols in Bioinformatics. 2014;47:11.12.1–11.12.34. doi: 10.1002/0471250953.bi1112s47. [DOI] [PMC free article] [PubMed] [Google Scholar]