

Abstract
CX3C chemokine ligand 1 (CX3CL1) is an intriguing chemokine belonging to the CX3C family. CX3CL1 is secreted by neurons and plays an important role in modulating glial activation in the central nervous system after binding to its sole receptor CX3CR1 which mainly is expressed on microglia. Emerging data highlights the beneficial potential of CX3CL1-CX3CR1 in the pathogenesis of Alzheimer's disease (AD), a common progressive neurodegenerative disease, and in the progression of which neuroinflammation plays a vital role. Even so, the importance of CX3CL1/CX3CR1 in AD is still controversial and needs further clarification. In this review, we make an attempt to present a concise map of CX3CL1-CX3CR1 associated with AD to find biomarkers for early diagnosis or therapeutic interventions.
1. Introduction
Alzheimer's disease (AD), a common progressive neurodegenerative disease, is the most frequent cause of cognitive decline and dementia, which affects more than 46 million people worldwide. The etiology of AD is still unclear now. One of the main pathological characteristics is extracellular deposits of β-amyloid (Aβ) peptides in senile plaques. Aβ cascade-inflammatory hypothesis has been elucidated to look forward to seeking treatment for AD [1]. Some scholars believe that Aβ-burdened neurons may play a crucial role in initiating microglial activation and eliciting chronic inflammation which lead to synaptic dysfunction, neurotoxicity, and behavioral deficits in the progression of AD [2–6]. Reactive microglia is also related to driving tau pathology and correlating with the spread of tau pathology [7], which induces neurofibrillary tangles (NFT), another major pathological characteristic of AD. Consistently, depleting microglia dramatically suppressed the propagation of tau in the brain [8].
CX3C chemokine ligand 1 (CX3CL1, also named fractalkine) plays an important role in reducing neuroinflammation and is highly expressed in the main area of pathological changes in AD, such as the hippocampus and cerebral cortex, and the expression level of CX3CL1 reflects the progression of the disease [9]. CX3CL1 has been demonstrated to play a neuroprotective role in CNS by reducing neurotoxicity and microglial activation [10–12]. Consistent with this is the fact that treatment of aged rats with CX3CL1 attenuates the age-related increase in microglial activation [13]. Moreover, CX3CL1 also has an effect on Aβ clearance and p-tau accumulation in AD [14]. All the above show that CX3CL1 has a major role in the progression of AD. In this review, we summarize the multiple roles of CX3CL1 in neuroinflammation, neurotoxicity, and synaptic plasticity in AD pathogenesis.
2. CX3CL1/CX3CR1 and Microglia
CX3CL1 is a large cytokine protein of 373 amino acids with an extended mucin-like stalk and a chemokine domain on top. It is the only member of CX3C family which belongs to the large family of small secreted chemotactic cytokines. CX3CL1 is expressed with particularly high levels in hippocampal and cortical neurons constitutively but none on microglia [15]. It exists in both secreted and membrane-bound form and its membrane-tethered mucin stalk acts as a cell adhesion molecule adhering to microglia during an inflammatory reaction [16]. The membrane-bound form can be cleaved in the condition of cathepsin S, ADAM-10, and ADAM-17; then the soluble one can serve as a signaling molecule mediating neural/microglial interactions via its sole receptor CX3CR1 that is mainly expressed on microglia and partly on astrocyte as well as on neurons in the CNS [17–19]. These suggest that CX3CL1/CX3CR1 is an important bridge to connect neuron and microglia.
Microglia, resident mononuclear phagocytes in the CNS, intimately involved in the development of the nervous system, are highly active in their presumed resting state, continually surveying their microenvironment with extremely motile processes and protrusions [20, 21]. It has been demonstrated that Aβ burdened neurons inducing microglial activation may be an early phenomenon in the procession of AD [22]. However, microglia activation in AD is suggested to be heterogeneous: beneficial or harmful [23]. This may be associated with microglia activation phenotype which includes M1 (iNOS+ microglia) and M2 (Arg+ microglia); iNOS+ microglia induce production of neuroinflammation factors while Arg+ microglia have enhanced phagocytic activity. In accordance with this, greater numbers of Arg+ microglia containing Aβ were found when compared to iNOS+ microglia in the inflamed hemisphere [24]. Moreover, amounts of evidence indicate that microglia phenotype changes from M2 to M1 in the progression of AD [25].
Neuronal soluble CX3CL1 is likely to alter the microglial state to a more neuroprotective one by acting on CX3CR1 in microglia [26]. This also has been confirmed that disruption of CX3CL1-CX3CR1 leads to dysregulate microglial responses and neuronal damage [12, 18]. Besides, hAPP-CX3CR1−/− mice as well as hTau-CX3CR1−/− mice showed increased expression of inflammatory factors, enhanced tau phosphorylation, and exacerbated plaque-independent neuronal dysfunction and cognitive deficits [27, 28], while researches also demonstrated that both APP-PS1/CX3CR1−/− and CRND8/CX3CR1−/− mice showed reduction in Aβ deposition with increased number of microglia [29, 30]. Moreover, the suppression of CX3CL1-CX3CR1 alleviated Aβ-induced neurotoxicity and memory deficiency [31, 32]. Well, CX3CL1/CX3CR1 may play a beneficial role in controlling the progression of AD by inhibiting the inflammation and tau phosphorylation but at a cost of the increased Aβ deposition. Overexpression of soluble CX3CL1 by adeno-associated viral (AAV) vectors plays an active role in reducing tau pathology and neuron loss, while it has no effect on Aβ deposition indicating that additional CX3CL1 signaling has no additive effect on Aβ deposition [26, 33]. Surprisingly, neither enhanced tau phosphorylation nor reduced Aβ deposition in CX3CL1-deficient APP-PS1 animals was altered by soluble CX3CL1 isoform, which was introduced by bacterial artificial chromosome (BAC) transgene encoding truncated CX3CL1 [34]. Thus making the function of soluble CX3CL1 is full of doubt. A possible explanation is that AAV vectors might make soluble CX3CL1 build the required local gradient and it should suffice, while the only soluble CX3CL1 can be diluted rapidly [35]. This needs to be further clarified.
The expression of CX3CL1 is decreased in cerebral cortex and hippocampus of APP transgenic mice while it is increased in tau-injured neurons [36, 37]. Moreover, the level of plasma soluble CX3CL1 is significantly greater in the patients with mild to moderate AD than in the patients with severe AD, and the level of CX3CL1 is inversely correlated to AD severity [38]. Together, these studies suggest that CX3CL1/CX3CR1 associated with neuroinflammation, neurotoxicity, and synaptic plasticity plays variable roles in different stages of AD pathogenesis. Considering this, we conjecture that mild decreased CX3CL1-CX3CR1 due to intraneuronal Aβ accumulation in the early stage of AD increases clearance of Aβ deposition by enhancing the phagocytosis of microglia while resulting in tau hyperphosphorylation and severe downgraded CX3CL1-CX3CR1 signal gives rise to deregulated microglia and abnormally excited neuron which lead to neuron damage and loss in the progression of AD.
3. CX3CL1/CX3CR1 and Neuroinflammation
Neuroinflammation is classically attributed to Aβ deposition and plays a vital role in the pathological progress of AD [5, 39]. It is always correlated with increased levels of proinflammatory cytokines including tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), IL-1β, interferon gamma (IFN-γ), and chemokine (C–C motif) ligand 2 (CCL2) and C–X–C motif chemokine 10 (CXCL10/IP-10) [40]. CX3CL1, which is identified inhibiting the production of TNF-α, nitric oxide (NO), and superoxide in neuron-glial cell cultures [41], has been implicated as an endogenous neuronal modulator and may limit microgliosis in AD by reducing the inflammatory reaction [37, 42, 43].
TNF-α, a prototypic proinflammatory cytokine, is mainly released by activated microglia, colocalized with Aβ deposition, and is elevated in the cortex of animal models and human with AD [44–46]. It has been shown that glial TNF-α enhances Aβ deposition through inhibiting BACE1 expression and Aβ clearance and promotes neuronal cell cycle events which are toxic for terminally differentiated neurons in the pathogenesis of AD [47, 48]. Besides, Lourenco et al. have proved that Aβ oligomers lead to synapse loss and memory impairment in a TNFR1 dependent manner [49]. TNF-α actives TNFR1 leading to neuron death while TNFR2 which is expressed primarily by microglia [50] is beneficial to control microglia activity in the progression of AD [51].
Fewer Aβ plaques and Aβ-related lesions developed in APP23/TNFR1−/− mice when compared with APP23/TNFR1+/+ littermates [52]. However, Barger et al. suggested that TNF-α protects hippocampal neurons against Aβ toxicity [53]. Both 3xTg-AD lacking TNF-R1+R2 and 3xTg-ADxTNF-R1/R2 knock-out exhibit enhanced Aβ and tau-related pathological features by the age of 15 months, in stark contrast to age-matched 3xTg-AD counterparts [54]. Loss of opposing TNFR2 leads to a stage-independent increase in Iba-1 positive microglia, and TNFR1 mediated exacerbation of Aβ and tau pathology in aged 3xTg-AD mice [55]. Thus suggesting the role of CX3CL1/CX3CR1 which inhibits TNF-α secretion [56] may be divaricated dependent on TNFR. But in view of the fact that TNFR1 is increased by 17–28% and TNFR2 is significantly decreased by 35–43% in AD brains [57], CX3CL1/CX3CR1 inclines to play a beneficial role in the pathogenesis of AD.
The expression of another inflammatory cytokine IL-1β is also increased in the CX3CR1-deficient APP/PS1 animals [29]. The major role of increased IL-1β in neuroinflammation and subsequent induction of the microglial autophagy potentially are contributed to AD [58, 59]. CX3CR1 deficiency promotes impairment of cognitive function, synaptic plasticity, and tau hyperphosphorylation via increasing action of IL-1β and the impairment could be reversed by infusion with IL-1β receptor antagonist significantly [28, 42]. On the other hand, the upregulated expression of chronic IL-1β increases plaque-associated microglia and ameliorates amyloid pathology in the APP/PS1 mouse model of AD [60, 61]. The generation of this contradiction is likely to depend on the stage of AD, which may be coordinated with CX3CL1 functions in different period.
In addition, CX3CL1 dose-dependently suppressed the production of nitric oxide (NO) [10]. NO, related to the increased levels of IFN-γ and TNF-α [62], has been involved in neuroinflammation with increased expression of inducible NO synthase (iNOS) at mild and severe stages of AD [63]. Inhibition of iNOS which mediates CNS inflammatory processes reduces the risk of AD [64]. In all, CX3CL1-CX3CR1 inhibits microglia activity via controlling the overproduction of inflammatory mediators. The distinctly decreased expression of CX3CL1 gives rise to dysregulated microglia, leading to neuroinflammation. Drugs that attenuate neuronal degeneration and improve learning and memory ability are accompanied by reduced TNF-α, IL-1β, TGF-β, and NO levels induced by Aβ in CSF in mouse models and patients with AD [65–70]. Apart from AD, CX3CL1/CX3CR1 is also involved in other neuroinflammation disorders, including Parkinson's Disease (PD) [71, 72], multiple sclerosis (MS) [73], tauopathies [33], and age-related macular degeneration (ARMD) [74]. These neurodegenerative disorders are all associated with chronic neuroinflammation caused by activated microglia [75], indicating that CX3CL1/CX3CR1 may have the similar mechanisms between AD and other neurodegenerative disorders in regulating neuroinflammation. The complex roles of CX3CL1/CX3CR1 are still being studied.
4. CX3CL1/CX3CR1 Regulates Synaptic Plasticity
Synaptic plasticity plays an important role in learning and memory, and Aβ-induced synaptic dysfunction is strongly associated with AD [76]. CX3CL1 is upregulated in the rat hippocampus during memory-associated synaptic plasticity [77]. It is considered as a potent neuromodulator of the evoked excitatory synaptic transmission and plays a major role in synaptic plasticity and neuroprotection [78]. Furthermore, the functions of CX3CL1 rely on CX3CR1, as long-lasting-enriched environment failed to affect hippocampal-dependent plasticity in the absence of CX3CR1 [79]. Although the underlying mechanisms have been underexplored, CX3CL1/CX3CR1 may mediate synaptic plasticity and cognitive function mainly by regulating long-term potentiation (LTP) [80], NO signaling, and production of brain-derived neurotrophic factor (BDNF) [81].
LTP is thought to be related to the storage of declarative memory in the mammalian brain [82]. CX3CL1 clearly interferes with LTP mechanisms and its modulation of neuronal plasticity appears to be mediated through activation of adenosine [80]. Adenosine acts as a neuromodulator with four types of G protein-coupled receptors, termed A1, A2A, A2B, and A3, and exerts important functions in the synaptic plasticity [83]. The downstream pathways branch because of the different types of adenosine receptor. Intracerebroventricular injection of Aβ 1–42 inhibited not only NMDA receptor-dependent LTP but also voltage-activated Ca2+-dependent LTP induced by strong conditioning stimulation during NMDAR blockade [84], indicating that there is a non-NMDAR-dependent but Ca2+-dependent pathway involved in synaptic dysfunction in AD. CX3CL1 increases NMDA-fast excitatory postsynaptic potentials by a mechanism involving the activity of the adenosine receptor type A2 (A2AR) and the release of the NMDAR coagonist D-serine [85]. NMDAR activation affects the threshold for LTP induction which is strongly influenced by the recent history of synaptic activity [86]. An increased density of A2AR on microglia has been detected in human cortex from AD patients [87]. Thus indicating CX3CL1/CX3CR1 may activate A2AR by increasing adenosine and promote the release of D-serine; then D-serine enhances the function of NMDAR and facilitates LTP. Moreover, CX3CL1 causes a reversible depression of excitatory postsynaptic current (EPSC), which is abolished by the A3R antagonist [88], and the inhibition failed to occur in CX3CR1 null mice [80]. Stimulation of A3R induces an intracellular signaling that increases calcium concentrations [89]. The phosphorylation of CAMKII and cyclic adenosine monophosphate response element-binding protein (CREB) is important to hippocampal long-term synaptic plasticity [90]. αCaMKII autophosphorylation is also required for synaptic plasticity induced by a short and precise stimulus, but maybe not for a longer and stronger stimulation [91]. Besides, the reduction of CREB activation also leads to memory impairment [92]. Based on the information given above, we can hypothesize the way CX3CL1 affects LTP; that is, CX3CL1 acts with CX3CR1 on the surface of the microglia and stimulates the release of adenosine; adenosine then activates A2AR and promotes synaptic facilitation by NMDAR-dependent pathway, activates A3R simultaneity, and induces synaptic inhibition by a Ca2+-dependent pathway.
Brain-derived neurotrophic factor (BDNF), an important growth factor in the CNS, is of great significance for neurons to maintain the survival, growth, differentiation, repair, and regeneration after nerve injury as well as increasing synaptic plasticity. A clinical study involving 535 old participants who underwent annual cognitive assessments and brain autopsy at death showed that higher brain BDNF expression is associated with slower cognitive decline and BDNF may also reduce the deleterious effects of AD pathology on cognitive decline [93]. Studies have shown that Aβ induces decreased anterograde as well as retrograde transport of BDNF vesicles in hippocampal neurons of various AD models [94]. Upregulation of BDNF by activating of ERK/CREB pathway can ameliorate Aβ-induced neurons loss and dendritic atrophy [95]. Restoration of normal neuronal BDNF expression levels in the cerebral hippocampi and cortices ameliorates the impairment in recognition memory and associative learning in mice of AD [96]. Importantly, BDNF concentrations are associated with CX3CL1 [97]. Chronic injection of CX3CL1 rescues the hippocampal-dependent memory deficits and reverses the decreased hippocampal neurogenesis in genetic BDNF variation mice [98].
In addition, NO is also consistently involved in recognition memory [99]. As mentioned before, CX3CL1/CX3CR1 inhibits the expression of NO in activated microglia cells [10, 100] and may induce synaptic inhibition. NO signaling through neuronal NO synthase (nNOS) prior to the appearance of cognitive symptoms focuses on early developments of AD [63]. NO/soluble GC (sGC)/cGMP-PKG and ERK signaling is important for modulating synaptic transmission and plasticity in the hippocampus and cerebral cortex, which are critical for learning and memory [101]. Recruitment of NO is serving a compensatory role to boost synaptic transmission and plasticity during early AD stage [102]. NO inhibitors ameliorate overexpressed NMDA receptor subunit NR2B which plays a role in memory formation in an inflammatory model of AD [103]. Besides, endothelial NO deficiency also promotes AD pathology [104].
5. CX3CL1/CX3CR1 Reduces Excitotoxicity
It has been verified that CX3CL1 released from hippocampal cells after excitotoxic insult has an essential role in brain protection by reducing against glutamate mediated excitotoxicity [105]. As mentioned before, microglia shape their neuronal environment actively thanks to their ability to trigger neuronal death [106–108]. Apart from regulating neuroinflammation, CX3CL1/CX3CR1 negatively modulates the function of AMPA receptor at active glutamatergic synapses [109]. CX3CL1 reduces the glutamate mediated excitotoxicity by reducing the influx of Ca2+ [105]. Calcium channel blockers also exhibit cognitive enhancing abilities and reduce the risk of dementia genuinely [110]. Moreover, the application of ion channel blockers with specific antagonists of the NR2B subunit could reduce neurotoxicity significantly [111]. Besides, CX3CL1 mediated neuroprotection by increasing glutamate transporter-1 (GLT-1) activity on astrocytes is dependent on the presence and the activity of A1 adenosine receptor (A1R), which can be blocked by the specific antagonist DPCPX and absent in A1R−/− astrocytes [112, 113]. Consistently, hippocampal neurons obtained from A1R−/− mice are not protected by CX3CL1 against Glu excitotoxicity [114]. Collectively, these data indicate that CX3CL1/CX3CR1 reduces excitotoxicity by modulating glutamatergic transmission and may play an important role in cognitive functions in AD.
6. Conclusions and Perspectives
There is persistent neuroinflammation throughout the progression of AD associated with neurotoxicity and synaptic dysfunction [115, 116]. The expression of CX3CL1 is significantly decreased in AD and inversely correlated to AD severity. As shown in Figure 1, CX3CL1/CX3CR1 may regulate the activation of microglia by controlling the release of inflammatory cytokines and synaptic plasticity and cognitive functions by modulating receptors in neurons directly or indirectly. The involvement of CX3CL1/CX3CR1 in AD suggests that CX3CL1/CX3CR1 contributes positively to neuron protective as well as detrimental role in the course of the disease. Therefore, targeting CX3CL1 and/or CX3CR1 may provide novel opportunities for treatment of AD. In particular, the development stage of the disease should be considered to better analyze the functions of CX3CL1/CX3CR1 in the progression of AD.
Figure 1.
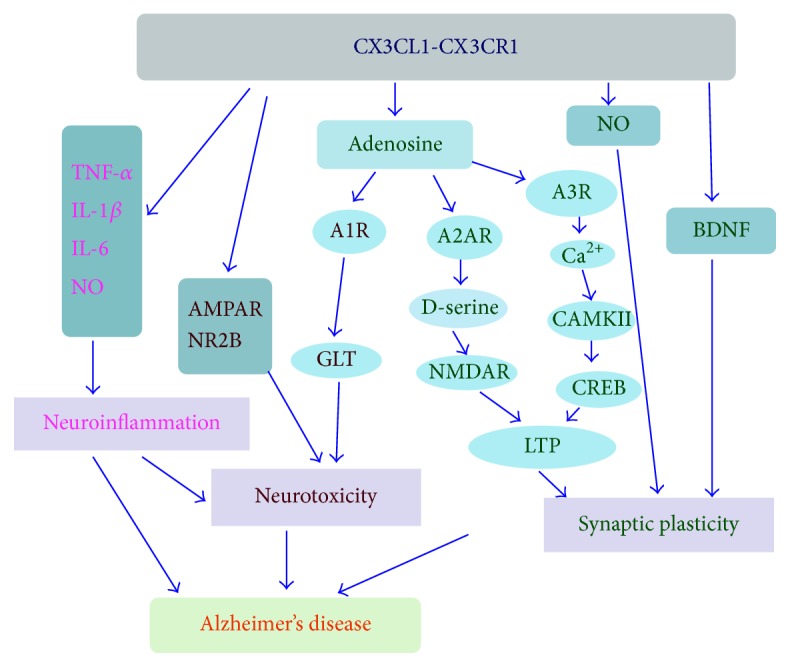
The effect of CX3CL1/CX3CR1 in Alzheimer's disease (AD). CX3CL1 binds to CX3CR1 which is its sole receptor and mainly expressed by microglia: (1) regulating introduction of inflammation cytokine (TNF-α, IL-1β, IL-6, NO, etc.) and reducing neuroinflammation in AD; (2) negatively modulating the function of AMPAR and NR2B, increasing GLT activity through the mechanism dependent on A1R, and then decreasing the neurotoxicity induced by Glu; (3) stimulating the release of adenosine; adenosine then activates A2AR and promotes synaptic facilitation by NMDAR-dependent pathway and simultaneity activates A3R and induces synaptic inhibition by Ca2+-dependent pathway. TNF-α: tumor necrosis factor-alpha; IL-1β: interleukin-1β; IL-6: interleukin-6; NO: nitric oxide; A1R: adenosine 1 receptor; A2AR: adenosine A2a receptor; A3R: adenosine 3 receptor; GLT: glutamate transporter; LTP: long-term potentiation; CREB: cyclic adenosine monophosphate response element-binding protein; BNDF: brain-derived neurotrophic factor.
In addition, experiment evidences have described the active involvement of CX3CL1/CX3CR1 in many other diseases, such as atherosclerotic, allergic asthma and rhinitis, renal diseases, rheumatoid arthritis (RA), Sjögren's syndrome (SS), systemic lupus erythematosus (SLE), scleroderma, colorectal cancer, and breast cancer [117–123]. For example, a genetically defined less active CX3CL1/CX3CR1 pathway is associated with a reduced risk of atherosclerotic disease in humans and the blockade of the CX3CL1/CX3CR1 pathway ameliorates the severity of atherosclerosis [124, 125]. Moreover, insulin resistance (IR) increases atherosclerotic lesion vulnerability, and this is related to the augment of CX3CL1/CX3CR1 axis [126]. All these indicate that any pharmacological agent that alters CX3CL1 signaling in AD should take into account any other potential effects.
Acknowledgments
This study was supported by the grants from the Natural Science Foundation of China (NSFC) (Grant no. 81471232), the Natural Science Foundation of Shanghai Science and Technology Commission, China (Grant no. 14431901400), and the scientific research fund of Shanghai Jiaotong University (Grant no. 14X130040002).
Competing Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.McGeer P. L., McGeer E. G. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathologica. 2013;126(4):479–497. doi: 10.1007/s00401-013-1177-7. [DOI] [PubMed] [Google Scholar]
- 2.Miyata S., Nishimura Y., Nakashima T. Perineuronal nets protect against amyloid β-protein neurotoxicity in cultured cortical neurons. Brain Research. 2007;1150(1):200–206. doi: 10.1016/j.brainres.2007.02.066. [DOI] [PubMed] [Google Scholar]
- 3.Perry V. H., Nicoll J. A. R., Holmes C. Microglia in neurodegenerative disease. Nature Reviews Neurology. 2010;6(4):193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- 4.Hyman B. T. Amyloid-dependent and amyloid-independent stages of alzheimer disease. Archives of Neurology. 2011;68(8):1062–1064. doi: 10.1001/archneurol.2011.70. [DOI] [PubMed] [Google Scholar]
- 5.Hanzel C. E., Pichet-Binette A., Pimentel L. S. B., et al. Neuronal driven pre-plaque inflammation in a transgenic rat model of Alzheimer's disease. Neurobiology of Aging. 2014;35(10):2249–2262. doi: 10.1016/j.neurobiolaging.2014.03.026. [DOI] [PubMed] [Google Scholar]
- 6.Noda M., Doi Y., Liang J., et al. Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression. Journal of Biological Chemistry. 2011;286(3):2308–2319. doi: 10.1074/jbc.M110.169839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maphis N., Xu G., Kokiko-Cochran O. N., et al. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain: A Journal of Neurology. 2015;138, part 6:1738–1755. doi: 10.1093/brain/awv081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asai H., Ikezu S., Tsunoda S., et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nature Neuroscience. 2015;18(11):1584–1593. doi: 10.1038/nn.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strobel S., Grünblatt E., Riederer P., et al. Changes in the expression of genes related to neuroinflammation over the course of sporadic Alzheimer’s disease progression: CX3CL1, TREM2, and PPARγ . Journal of Neural Transmission. 2015;122(7):1069–1076. doi: 10.1007/s00702-015-1369-5. [DOI] [PubMed] [Google Scholar]
- 10.Mizuno T., Kawanokuchi J., Numata K., Suzumura A. Production and neuroprotective functions of fractalkine in the central nervous system. Brain Research. 2003;979(1-2):65–70. doi: 10.1016/S0006-8993(03)02867-1. [DOI] [PubMed] [Google Scholar]
- 11.Pabon M. M., Bachstetter A. D., Hudson C. E., Gemma C., Bickford P. C. CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson's disease. Journal of Neuroinflammation. 2011;8, article 9 doi: 10.1186/1742-2094-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Febinger H. Y., Thomasy H. E., Pavlova M. N., et al. Time-dependent effects of CX3CR1 in a mouse model of mild traumatic brain injury. Journal of Neuroinflammation. 2015;12(1, article 154) doi: 10.1186/s12974-015-0386-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lyons A., Lynch A. M., Downer E. J., et al. Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attentuates microglial activation in vivo and in vitro. Journal of Neurochemistry. 2009;110(5):1547–1556. doi: 10.1111/j.1471-4159.2009.06253.x. [DOI] [PubMed] [Google Scholar]
- 14.Merino J., Muñetón-Gómez V., Alvárez M., Toledano-Díaz A. Effects of CX3CR1 and fractalkine chemokines in amyloid beta clearance and p-Tau accumulation in Alzheimer's Disease (AD) rodent models: is fractalkine a systemic biomarker for AD? Current Alzheimer Research. 2016;13(4):403–412. doi: 10.2174/1567205013666151116125714. [DOI] [PubMed] [Google Scholar]
- 15.Harrison J. K., Jiang Y., Chen S., et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(18):10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hermand P., Pincet F., Carvalho S., et al. Functional adhesiveness of the CX3CL1 chemokine requires its aggregation: role of the transmembrane domain. Journal of Biological Chemistry. 2008;283(44):30225–30234. doi: 10.1074/jbc.m802638200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chapman G. A., Moores K., Harrison D., Campbell C. A., Stewart B. R., Strijbos P. J. Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. Journal of Neuroscience. 2000;20(15, article RC87):5. doi: 10.1523/JNEUROSCI.20-15-j0004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cardona A. E., Pioro E. P., Sasse M. E., et al. Control of microglial neurotoxicity by the fractalkine receptor. Nature Neuroscience. 2006;9(7):917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 19.Meucci O., Fatatis A., Simen A. A., Miller R. J. Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(14):8075–8080. doi: 10.1073/pnas.090017497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nimmerjahn A., Kirchhoff F., Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 21.Helmut K., Hanisch U.-K., Noda M., Verkhratsky A. Physiology of microglia. Physiological Reviews. 2011;91(2):461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 22.Zimmer E. R., Leuzy A., Benedet A. L., Breitner J., Gauthier S., Rosa-Neto P. Tracking neuroinflammation in Alzheimer's disease: the role of positron emission tomography imaging. Journal of Neuroinflammation. 2014;11, article 120 doi: 10.1186/1742-2094-11-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tejera D., Heneka M. Microglia in Alzheimer's disease: the good, the bad and the ugly. Current Alzheimer Research. 2016;13(4):370–380. doi: 10.2174/1567205013666151116125012. [DOI] [PubMed] [Google Scholar]
- 24.Cherry J. D., Olschowka J. A., O'Banion M. K. Arginase 1+ microglia reduce Aβ plaque deposition during IL-1β-dependent neuroinflammation. Journal of Neuroinflammation. 2015;12(1, article 203) doi: 10.1186/s12974-015-0411-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varnum M. M., Ikezu T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in alzheimer's disease brain. Archivum Immunologiae et Therapiae Experimentalis. 2012;60(4):251–266. doi: 10.1007/s00005-012-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nash K. R., Moran P., Finneran D. J., et al. Fractalkine over expression suppresses α-synuclein-mediated neurodegeneration. Molecular Therapy. 2015;23(1):17–23. doi: 10.1038/mt.2014.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cho S.-H., Sun B., Zhou Y., et al. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. The Journal of Biological Chemistry. 2011;286(37):32713–32722. doi: 10.1074/jbc.m111.254268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhaskar K., Konerth M., Kokiko-Cochran O. N., Cardona A., Ransohoff R. M., Lamb B. T. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68(1):19–31. doi: 10.1016/j.neuron.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee S., Varvel N. H., Konerth M. E., et al. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. The American Journal of Pathology. 2010;177(5):2549–2562. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Z., Condello C., Schain A., Harb R., Grutzendler J. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-β phagocytosis. Journal of Neuroscience. 2010;30(50):17091–17101. doi: 10.1523/jneurosci.4403-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dworzak J., Renvoisé B., Habchi J., et al. Neuronal Cx3cr1 deficiency protects against amyloid β-induced neurotoxicity. PLoS ONE. 2015;10(6) doi: 10.1371/journal.pone.0127730.e0127730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu J., Bie B., Yang H., Xu J. J., Brown D. L., Naguib M. Suppression of central chemokine fractalkine receptor signaling alleviates amyloid-induced memory deficiency. Neurobiology of Aging. 2013;34(12):2843–2852. doi: 10.1016/j.neurobiolaging.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 33.Nash K. R., Lee D. C., Hunt J. B., et al. Fractalkine overexpression suppresses tau pathology in a mouse model of tauopathy. Neurobiology of Aging. 2013;34(6):1540–1548. doi: 10.1016/j.neurobiolaging.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee S., Xu G., Jay T. R., et al. Opposing effects of membrane-anchored CX3CL1 on amyloid and tau pathologies via the p38 MAPK pathway. The Journal of Neuroscience. 2014;34(37):12538–12546. doi: 10.1523/jneurosci.0853-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim K.-W., Vallon-Eberhard A., Zigmond E., et al. In vivo structure/function and expression analysis of the CX3C chemokine fractalkine. Blood. 2011;118(22):e156–e167. doi: 10.1182/blood-2011-04-348946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duan R.-S., Yang X., Chen Z.-G., et al. Decreased fractalkine and increased IP-10 expression in aged brain of APPswe transgenic mice. Neurochemical Research. 2008;33(6):1085–1089. doi: 10.1007/s11064-007-9554-z. [DOI] [PubMed] [Google Scholar]
- 37.Lastres-Becker I., Innamorato N. G., Jaworski T., et al. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain. 2014;137(1):78–91. doi: 10.1093/brain/awt323. [DOI] [PubMed] [Google Scholar]
- 38.Kim T.-S., Lim H.-K., Lee J. Y., et al. Changes in the levels of plasma soluble fractalkine in patients with mild cognitive impairment and Alzheimer's disease. Neuroscience Letters. 2008;436(2):196–200. doi: 10.1016/j.neulet.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 39.Halliday G., Robinson S. R., Shepherd C., Kril J. Alzheimer's disease and inflammation: a review of cellular and therapeutic mechanisms. Clinical and Experimental Pharmacology and Physiology. 2000;27(1-2):1–8. doi: 10.1046/j.1440-1681.2000.03200.x. [DOI] [PubMed] [Google Scholar]
- 40.Zaheer S., Thangavel R., Wu Y., Khan M. M., Kempuraj D., Zaheer A. Enhanced expression of glia maturation factor correlates with glial activation in the brain of triple transgenic Alzheimer's disease mice. Neurochemical Research. 2013;38(1):218–225. doi: 10.1007/s11064-012-0913-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mattison H. A., Nie H., Gao H., Zhou H., Hong J.-S., Zhang J. Suppressed pro-inflammatory response of microglia in CX3CR1 knockout mice. Journal of Neuroimmunology. 2013;257(1-2):110–115. doi: 10.1016/j.jneuroim.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rogers J. T., Morganti J. M., Bachstetter A. D., et al. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. The Journal of Neuroscience. 2011;31(45):16241–16250. doi: 10.1523/jneurosci.3667-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Biber K., Neumann H., Inoue K., Boddeke H. W. G. M. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends in Neurosciences. 2007;30(11):596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 44.Fillit H., Ding W., Buee L., et al. Elevated circulating tumor necrosis factor levels in Alzheimer's disease. Neuroscience Letters. 1991;129(2):318–320. doi: 10.1016/0304-3940(91)90490-K. [DOI] [PubMed] [Google Scholar]
- 45.Tarkowski E., Blennow K., Wallin A., Tarkowski A. Intracerebral production of tumor necrosis factor-α, a local neuroprotective agent, in Alzheimer disease and vascular dementia. Journal of Clinical Immunology. 1999;19(4):223–230. doi: 10.1023/A:1020568013953. [DOI] [PubMed] [Google Scholar]
- 46.Kang H.-J., Kim J.-M., Kim S.-W., et al. Associations of cytokine genes with Alzheimer's disease and depression in an elderly Korean population. Journal of Neurology, Neurosurgery & Psychiatry. 2015;86(9):1002–1007. doi: 10.1136/jnnp-2014-308469. [DOI] [PubMed] [Google Scholar]
- 47.Yamamoto M., Kiyota T., Horiba M., et al. Interferon-γ and tumor necrosis factor-α regulate amyloid-β plaque deposition and β-secretase expression in Swedish mutant APP transgenic mice. American Journal of Pathology. 2007;170(2):680–692. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhaskar K., Maphis N., Xu G., et al. Microglial derived tumor necrosis factor-α drives Alzheimer's disease-related neuronal cell cycle events. Neurobiology of Disease. 2014;62:273–285. doi: 10.1016/j.nbd.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lourenco M. V., Clarke J. R., Frozza R. L., et al. TNF-α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer's β-amyloid oligomers in mice and monkeys. Cell Metabolism. 2013;18(6):831–843. doi: 10.1016/j.cmet.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 50.McCoy M. K., Tansey M. G. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. Journal of Neuroinflammation. 2008;5, article 45 doi: 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheng X., Shen Y., Li R. Targeting TNF: a therapeutic strategy for Alzheimer's disease. Drug Discovery Today. 2014;19(11):1822–1827. doi: 10.1016/j.drudis.2014.06.029. [DOI] [PubMed] [Google Scholar]
- 52.He P., Zhong Z., Lindholm K., et al. Deletion of tumor necrosis factor death receptor inhibits amyloid β generation and prevents learning and memory deficits in Alzheimer's mice. The Journal of Cell Biology. 2007;178(5):829–841. doi: 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barger S. W., Hörster D., Furukawa K., Goodman Y., Krieglstein J., Mattson M. P. Tumor necrosis factors alpha and beta protect neurons against amyloid beta-peptide toxicity: evidence for involvement of a kappa B-binding factor and attenuation of peroxide and Ca2+ accumulation. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(20):9328–9332. doi: 10.1073/pnas.92.20.9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Montgomery S. L., Mastrangelo M. A., Habib D., et al. Ablation of TNF-RI/RII expression in Alzheimer's disease mice leads to an unexpected enhancement of pathology: Implications for chronic pan-TNF-α suppressive therapeutic strategies in the brain. American Journal of Pathology. 2011;179(4):2053–2070. doi: 10.1016/j.ajpath.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Montgomery S. L., Narrow W. C., Mastrangelo M. A., Olschowka J. A., O'Banion M. K., Bowers W. J. Chronic neuron- and age-selective down-regulation of TNF receptor expression in triple-transgenic alzheimer disease mice leads to significant modulation of amyloid- and Tau-related pathologies. American Journal of Pathology. 2013;182(6):2285–2297. doi: 10.1016/j.ajpath.2013.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zujovic V., Benavides J., Vigé X., Carter C., Taupin V. Fractalkine modulates TNF-α secretion and neurotoxicity induced by microglial activation. Glia. 2000;29(4):305–315. doi: 10.1002/(sici)1098-1136(20000215)29:4<305::aid-glia2>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 57.Cheng X., Yang L., He P., Li R., Shen Y. Differential activation of tumor necrosis factor receptors distinguishes between brains from Alzheimer's disease and non-demented patients. Journal of Alzheimer's Disease. 2010;19(2):621–630. doi: 10.3233/JAD-2010-1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang P., Yu X., Guan P. P., et al. Magnesium ion influx reduces neuroinflammation in Aβ precursor protein/Presenilin 1 transgenic mice by suppressing the expression of interleukin-1β . Cellular and Molecular Immunology. 2015 doi: 10.1038/cmi.2015.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.François A., Terro F., Janet T., Bilan A. R., Paccalin M., Page G. Involvement of interleukin-1β in the autophagic process of microglia: relevance to Alzheimer's disease. Journal of Neuroinflammation. 2013;10, article 151 doi: 10.1186/1742-2094-10-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rivera-Escalera F., Matousek S. B., Ghosh S., Olschowka J. A., O'banion M. K. Interleukin-1β mediated amyloid plaque clearance is independent of CCR2 signaling in the APP/PS1 mouse model of Alzheimer's disease. Neurobiology of Disease. 2014;69:124–133. doi: 10.1016/j.nbd.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matousek S. B., Ghosh S., Shaftel S. S., Kyrkanides S., Olschowka J. A., O'Banion M. K. Chronic IL-1β-mediated neuroinflammation mitigates amyloid pathology in a mouse model of alzheimer's disease without inducing overt neurodegeneration. Journal of Neuroimmune Pharmacology. 2012;7(1):156–164. doi: 10.1007/s11481-011-9331-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Belkhelfa M., Rafa H., Medjeber O., et al. IFN-γ and TNF-α are involved during Alzheimer disease progression and correlate with nitric oxide production: a study in Algerian patients. Journal of Interferon and Cytokine Research. 2014;34(11):839–847. doi: 10.1089/jir.2013.0085. [DOI] [PubMed] [Google Scholar]
- 63.Balez R., Ooi L. Getting to NO Alzheimer's disease: neuroprotection versus neurotoxicity mediated by nitric oxide. Oxidative Medicine and Cellular Longevity. 2016;2016:8. doi: 10.1155/2016/3806157.3806157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jiang P., Li C., Xiang Z., Jiao B. Tanshinone IIA reduces the risk of Alzheimer's disease by inhibiting iNOS, MMP-2 and NF-κBp65 transcription and translation in the temporal lobes of rat models of Alzheimer's disease. Molecular Medicine Reports. 2014;10(2):689–694. doi: 10.3892/mmr.2014.2254. [DOI] [PubMed] [Google Scholar]
- 65.Shi S., Liang D., Chen Y., et al. Gx-50 reduces β-amyloid-induced TNF-α, IL-1β, NO, and PGE2 expression and inhibits NF-kappaB signaling in a mouse model of Alzheimer's disease. European Journal of Immunology. 2016;46(3):665–676. doi: 10.1002/eji.201545855. [DOI] [PubMed] [Google Scholar]
- 66.Li W., Zhang J.-W., Lu F., et al. Effects of telmisartan on the level of Aβ1–42, interleukin-1β, tumor necrosis factor α and cognition in hypertensive patients with Alzheimer's disease. Zhonghua Yi Xue Za Zhi. 2012;92(39):2743–2746. doi: 10.3760/cma.j.issn.0376-2491.2012.39.003. [DOI] [PubMed] [Google Scholar]
- 67.Zhang Y. Y., Fan Y. C., Wang M., et al. Atorvastatin attenuates the production of IL-1β, IL-6, and TNF-α in the hippocampus of an amyloid β1–42-induced rat model of Alzheimer's disease. Journal of Clinical Interventions in Aging. 2013;8:103–110. doi: 10.2147/CIA.S40405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cheng X., Wang Q., Li N., Zhao H. Effects of resveratrol on hippocampal astrocytes and expression of TNF-α in Alzheimer's disease model rate. Wei Sheng Yan Jiu. 2015;44(4):610–614. [PubMed] [Google Scholar]
- 69.Sachdeva A. K., Chopra K. Lycopene abrogates Aβ(1-42)-mediated neuroinflammatory cascade in an experimental model of Alzheimer's disease. Journal of Nutritional Biochemistry. 2015;26(7):736–744. doi: 10.1016/j.jnutbio.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 70.Xuan A.-G., Pan X.-B., Wei P., et al. Valproic acid alleviates memory deficits and attenuates amyloid-β deposition in transgenic mouse model of Alzheimer's disease. Molecular Neurobiology. 2014;51(1):300–312. doi: 10.1007/s12035-014-8751-4. [DOI] [PubMed] [Google Scholar]
- 71.Thome A. D., Standaert D. G., Harms A. S. Fractalkine signaling regulates the inflammatory response in an α-synuclein model of Parkinson disease. PLoS ONE. 2015;10(10) doi: 10.1371/journal.pone.0140566.e0140566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morganti J. M., Nash K. R., Grimmig B. A., et al. The soluble isoform of CX3CL1 is necessary for neuroprotection in a mouse model of Parkinson's disease. The Journal of Neuroscience. 2012;32(42):14592–14601. doi: 10.1523/jneurosci.0539-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stojković L., Djurić T., Stanković A., et al. The association of V249I and T280M fractalkine receptor haplotypes with disease course of multiple sclerosis. Journal of Neuroimmunology. 2012;245(1-2):87–92. doi: 10.1016/j.jneuroim.2011.12.028. [DOI] [PubMed] [Google Scholar]
- 74.Falk M. K., Singh A., Faber C., Nissen M. H., Hviid T., Sørensen T. L. CX3CL1/CX3CR1 and CCL2/CCR2 chemokine/chemokine receptor complex in patients with AMD. PLoS ONE. 2014;9(12) doi: 10.1371/journal.pone.0112473.e112473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frank-Cannon T. C., Alto L. T., McAlpine F. E., Tansey M. G. Does neuroinflammation fan the flame in neurodegenerative diseases? Molecular Neurodegeneration. 2009;4, article 47 doi: 10.1186/1750-1326-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang Z.-C., Zhao J., Li S. Dysregulation of synaptic and extrasynaptic N-methyl-D-aspartate receptors induced by amyloid-β . Neuroscience Bulletin. 2013;29(6):752–760. doi: 10.1007/s12264-013-1383-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sheridan G. K., Wdowicz A., Pickering M., et al. CX3CL1 is up-regulated in the rat hippocampus during memory-associated synaptic plasticity. Frontiers in Cellular Neuroscience. 2014;8, article 233 doi: 10.3389/fncel.2014.00233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bertollini C., Ragozzino D., Gross C., Limatola C., Eusebi F. Fractalkine/CX3CL1 depresses central synaptic transmission in mouse hippocampal slices. Neuropharmacology. 2006;51(4):816–821. doi: 10.1016/j.neuropharm.2006.05.027. [DOI] [PubMed] [Google Scholar]
- 79.Maggi L., Scianni M., Branchi I., D'Andrea I., Lauro C., Limatola C. CX3CR1 deficiency alters hippocampal-dependent plasticity phenomena blunting the effects of enriched environment. Frontiers in Cellular Neuroscience. 2011;5(22) doi: 10.3389/fncel.2011.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Maggi L., Trettel F., Scianni M., et al. LTP impairment by fractalkine/CX3CL1 in mouse hippocampus is mediated through the activity of adenosine receptor type 3 (A3R) Journal of Neuroimmunology. 2009;215(1-2):36–42. doi: 10.1016/j.jneuroim.2009.07.016. [DOI] [PubMed] [Google Scholar]
- 81.Parkhurst C. N., Yang G., Ninan I., et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155(7):1596–1609. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Squire L. R., Zola-Morgan S. The medial temporal lobe memory system. Science. 1991;253(5026):1380–1386. doi: 10.1126/science.1896849. [DOI] [PubMed] [Google Scholar]
- 83.Fredholm B. B., Abbracchio M. P., Burnstock G., et al. Nomenclature and classification of purinoceptors. Pharmacological Reviews. 1994;46(2):143–156. [PMC free article] [PubMed] [Google Scholar]
- 84.Klyubin I., Ondrejcak T., Hayes J., et al. Neurotransmitter receptor and time dependence of the synaptic plasticity disrupting actions of Alzheimer's disease Aβ in vivo. Philosophical Transactions of the Royal Society B: Biological Sciences. 2014;369(1633) doi: 10.1098/rstb.2013.0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Scianni M., Antonilli L., Chece G., et al. Fractalkine (CX3CL1) enhances hippocampal N-methyl-d-aspartate receptor (NMDAR) function via d-serine and adenosine receptor type A2 (A2AR) activity. Journal of Neuroinflammation. 2013;10, article 108 doi: 10.1186/1742-2094-10-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huang Y.-Y., Colino A., Selig D. K., Malenka R. C. The influence of prior synaptic activity on the induction of long-term potentiation. Science. 1992;255(5045):730–733. doi: 10.1126/science.1346729. [DOI] [PubMed] [Google Scholar]
- 87.Albasanz J. L., Perez S., Barrachina M., Ferrer I., Martín M. Up-regulation of adenosine receptors in the frontal cortex in Alzheimer's disease. Brain Pathology. 2008;18(2):211–219. doi: 10.1111/j.1750-3639.2007.00112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Piccinin S., Di Angelantonio S., Piccioni A., et al. CX3CL1-induced modulation at CA1 synapses reveals multiple mechanisms of EPSC modulation involving adenosine receptor subtypes. Journal of Neuroimmunology. 2010;224(1-2):85–92. doi: 10.1016/j.jneuroim.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 89.Olah M. E., Stiles G. L. Adenosine receptor subtypes: characterization and therapeutic regulation. Annual Review of Pharmacology and Toxicology. 1995;35:581–606. doi: 10.1146/annurev.pa.35.040195.003053. [DOI] [PubMed] [Google Scholar]
- 90.Qiao F., Gao X.-P., Yuan L., Cai H.-Y., Qi J.-S. Apolipoprotein E4 impairs in vivo hippocampal long-term synaptic plasticity by reducing the phosphorylation of CaMKIIα and CREB. Journal of Alzheimer's Disease. 2014;41(4):1165–1176. doi: 10.3233/jad-140375. [DOI] [PubMed] [Google Scholar]
- 91.Villers A., Giese K. P., Ris L. Long-term potentiation can be induced in the CA1 region of hippocampus in the absence of αCaMKII T286-autophosphorylation. Learning and Memory. 2014;21(11):616–626. doi: 10.1101/lm.035972.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Teich A. F., Nicholls R. E., Puzzo D., et al. Synaptic therapy in Alzheimer’s disease: a CREB-centric approach. Neurotherapeutics. 2015;12(1):29–41. doi: 10.1007/s13311-014-0327-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Buchman A. S., Yu L., Boyle P. A., Schneider J. A., De Jager P. L., Bennett D. A. Higher brain BDNF gene expression is associated with slower cognitive decline in older adults. Neurology. 2016;86(8):735–741. doi: 10.1212/wnl.0000000000002387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Seifert B., Eckenstaler R., Rönicke R., et al. Amyloid-beta induced changes in vesicular transport of BDNF in hippocampal neurons. Neural Plasticity. 2016;2016:15. doi: 10.1155/2016/4145708.4145708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Meng C., He Z., Xing D. Low-level laser therapy rescues dendrite atrophy via upregulating BDNF expression: implications for Alzheimer's disease. The Journal of Neuroscience. 2013;33(33):13505–13517. doi: 10.1523/jneurosci.0918-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fukumoto K., Mizoguchi H., Takeuchi H., et al. Fingolimod increases brain-derived neurotrophic factor levels and ameliorates amyloid β-induced memory impairment. Behavioural Brain Research. 2014;268:88–93. doi: 10.1016/j.bbr.2014.03.046. [DOI] [PubMed] [Google Scholar]
- 97.Pedraz M., Martín-Velasco A. I., García-Marchena N., et al. Plasma concentrations of BDNF and IGF-1 in abstinent cocaine users with high prevalence of substance use disorders: relationship to psychiatric comorbidity. PLoS ONE. 2015;10(3) doi: 10.1371/journal.pone.0118610.e0118610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang D.-D., Tian T., Dong Q., et al. Transcriptome profiling analysis of the mechanisms underlying the BDNF Val66Met polymorphism induced dysfunctions of the central nervous system. Hippocampus. 2014;24(1):65–78. doi: 10.1002/hipo.22204. [DOI] [PubMed] [Google Scholar]
- 99.Pitsikas N. The role of nitric oxide in the object recognition memory. Behavioural Brain Research. 2015;285:200–207. doi: 10.1016/j.bbr.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 100.Zhu X.-J., Song Y.-F., Zhang Q.-Y., Cao Y.-P., Xu W.-Q., Su D.-H. Effects of fractalkine on the expression of inflammatory substances in LPS-activated microglia cells. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2011;27(12):1298–1300. [PubMed] [Google Scholar]
- 101.Zhihui Q. Modulating nitric oxide signaling in the CNS for Alzheimer's disease therapy. Future Medicinal Chemistry. 2013;5(12):1451–1468. doi: 10.4155/fmc.13.111. [DOI] [PubMed] [Google Scholar]
- 102.Chakroborty S., Kim J., Schneider C., West A. R., Stutzmann G. E. Nitric oxide signaling is recruited as a compensatory mechanism for sustaining synaptic plasticity in Alzheimer’s disease mice. Journal of Neuroscience. 2015;35(17):6893–6902. doi: 10.1523/JNEUROSCI.4002-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Maher A., El-Sayed N. S.-E., Breitinger H.-G., Gad M. Z. Overexpression of NMDAR2B in an inflammatory model of Alzheimer's disease: modulation by NOS inhibitors. Brain Research Bulletin. 2014;109:109–116. doi: 10.1016/j.brainresbull.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 104.Austin S. A., Santhanam A. V., Hinton D. J., Choi D.-S., Katusic Z. S. Endothelial nitric oxide deficiency promotes Alzheimer's disease pathology. Journal of Neurochemistry. 2013;127(5):691–700. doi: 10.1111/jnc.12334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Limatola C., Lauro C., Catalano M., et al. Chemokine CX3CL1 protects rat hippocampal neurons against glutamate-mediated excitotoxicity. Journal of Neuroimmunology. 2005;166(1-2):19–28. doi: 10.1016/j.jneuroim.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 106.Marín-Teva J. L., Dusart I., Colin C., Gervais A., Van Rooijen N., Mallat M. Microglia promote the death of developing Purkinje cells. Neuron. 2004;41(4):535–547. doi: 10.1016/s0896-6273(04)00069-8. [DOI] [PubMed] [Google Scholar]
- 107.Tyler C. M., Boulanger L. M. Complement-mediated microglial clearance of developing retinal ganglion cell axons. Neuron. 2012;74(4):597–599. doi: 10.1016/j.neuron.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wakselman S., Béchade C., Roumier A., Bernard D., Triller A., Bessis A. Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. The Journal of Neuroscience. 2008;28(32):8138–8143. doi: 10.1523/jneurosci.1006-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ragozzino D., Di Angelantonio S., Trettel F., et al. Chemokine fractalkine/CX3CL1 negatively modulates active glutamatergic synapses in rat hippocampal neurons. Journal of Neuroscience. 2006;26(41):10488–10498. doi: 10.1523/jneurosci.3192-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Saravanaraman P., Chinnadurai R. K., Boopathy R. Why calcium channel blockers could be an elite choice in the treatment of Alzheimer's disease: A comprehensive review of evidences. Reviews in the Neurosciences. 2014;25(2):231–246. doi: 10.1515/revneuro-2013-0056. [DOI] [PubMed] [Google Scholar]
- 111.Zádori D., Veres G., Szalárdy L., Klivényi P., Toldi J., Vécsei L. Glutamatergic dysfunctioning in Alzheimer's disease and related therapeutic targets. Journal of Alzheimer's Disease. 2014;42:S177–S187. doi: 10.3233/JAD-132621. [DOI] [PubMed] [Google Scholar]
- 112.Catalano M., Lauro C., Cipriani R., et al. CX3CL1 protects neurons against excitotoxicity enhancing GLT-1 activity on astrocytes. Journal of Neuroimmunology. 2013;263(1-2):75–82. doi: 10.1016/j.jneuroim.2013.07.020. [DOI] [PubMed] [Google Scholar]
- 113.Giunta S., Andriolo V., Castorina A. Dual blockade of the A1 and A2A adenosine receptor prevents amyloid beta toxicity in neuroblastoma cells exposed to aluminum chloride. International Journal of Biochemistry and Cell Biology. 2014;54:122–136. doi: 10.1016/j.biocel.2014.07.009. [DOI] [PubMed] [Google Scholar]
- 114.Lauro C., Cipriani R., Catalano M., et al. Adenosine A1 receptors and microglial cells mediate CX3CL1-induced protection of hippocampal neurons against glu-induced death. Neuropsychopharmacology. 2010;35(7):1550–1559. doi: 10.1038/npp.2010.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fan Z., Okello A. A., Brooks D. J., Edison P. Longitudinal influence of microglial activation and amyloid on neuronal function in Alzheimer's disease. Brain. 2015;138(12):3685–3698. doi: 10.1093/brain/awv288. [DOI] [PubMed] [Google Scholar]
- 116.Rao J. S., Kellom M., Kim H.-W., Rapoport S. I., Reese E. A. Neuroinflammation and synaptic loss. Neurochemical Research. 2012;37(5):903–910. doi: 10.1007/s11064-012-0708-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shah R., Matthews G. J., Shah R. Y., et al. Serum fractalkine (CX3CL1) and cardiovascular outcomes and diabetes: findings from the chronic renal insufficiency cohort (CRIC) study. American Journal of Kidney Diseases. 2015;66(2):266–273. doi: 10.1053/j.ajkd.2015.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang X., Feng X., Cai W., et al. Chemokine CX3CL1 and its receptor CX3CR1 are associated with human atherosclerotic lesion volnerability. Thrombosis Research. 2015;135(6):1147–1153. doi: 10.1016/j.thromres.2015.03.020. [DOI] [PubMed] [Google Scholar]
- 119.Ferretti E., Pistoia V., Corcione A. Role of fractalkine/CX3CL1 and its receptor in the pathogenesis of inflammatory and malignant diseases with emphasis on B cell malignancies. Mediators of Inflammation. 2014;2014:10. doi: 10.1155/2014/480941.480941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jones B., Koch A. E., Ahmed S. Pathological role of fractalkine/CX3CL1 in rheumatic diseases: a unique chemokine with multiple functions. Frontiers in Immunology. 2012;2, article 82 doi: 10.3389/fimmu.2011.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Huo L. W., Ye Y. L., Wang G. W., Ye Y. G. Fractalkine (CX3CL1): a biomarker reflecting symptomatic severity in patients with knee osteoarthritis. Journal of Investigative Medicine. 2015;63(4):626–631. doi: 10.1097/jim.0000000000000158. [DOI] [PubMed] [Google Scholar]
- 122.Astorri E., Scrivo R., Bombardieri M., et al. CX3CL1 and CX3CR1 expression in tertiary lymphoid structures in salivary gland infiltrates: fractalkine contribution to lymphoid neogenesis in Sjögren's syndrome. Rheumatology. 2014;53(4):611–620. doi: 10.1093/rheumatology/ket401. [DOI] [PubMed] [Google Scholar]
- 123.Flierl U., Bauersachs J., Schafer A. Modulation of platelet and monocyte function by the chemokine fractalkine (CX3 CL1) in cardiovascular disease. European Journal of Clinical Investigation. 2015;45(6):624–633. doi: 10.1111/eci.12443. [DOI] [PubMed] [Google Scholar]
- 124.Apostolakis S., Spandidos D. Chemokines and atherosclerosis: focus on the CX3CL1/CX3CR1 pathway. Acta Pharmacologica Sinica. 2013;34(10):1251–1256. doi: 10.1038/aps.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang H., Guo C., Wu D., et al. Hydrogen sulfide inhibits the development of atherosclerosis with suppressing CX3CR1 and CX3CL1 expression. PLoS ONE. 2012;7(7) doi: 10.1371/journal.pone.0041147.e41147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Martinez-Hervas S., Vinue A., Nunez L., et al. Insulin resistance aggravates atherosclerosis by reducing vascular smooth muscle cell survival and increasing CX3CL1/CX3CR1 axis. Cardiovascular Research. 2014;103(2):324–336. doi: 10.1093/cvr/cvu115. [DOI] [PubMed] [Google Scholar]