

“Crucial challenges remain in adapting peptide triazoles for therapeutic use.”
Effective HIV-1 cell entry blockers remain a critical unmet need in the effort to prevent virus infection and to suppress virus production in individuals who are already infected. The Env gp120 protein is an appealing and yet elusive target for suppressing infection due to its exposure on the virus surface and obligatory requirement for binding to host cell receptors. In the face of the continuing need for gp120-targeting therapies, we have discovered a class of peptide triazoles (PTs) that bind HIV-1 Env gp120 from a broad range of virus subtypes with submicromolar to nanomolar affinities, suppress gp120 interactions to host cells at both its CD4 and coreceptor binding sites, cause gp120 shedding from the virion particle and, as a consequence, inactivate the virus and prevent cell infection. Furthermore, thiol-containing peptide triazoles and their multivalent conjugates on gold nanoparticles enhance HIV-1 breakdown and inactivation by disrupting the virus membrane and releasing luminal p24. The additional function of the lytic derivatives to suppress active virus formation from infected cells with surface-expressed Env is being evaluated. Recent investigations combining protein mutagenesis, PT synthetic variation and computational modeling have revealed that PTs bind to a conserved two-cavity region overlapping the CD4 binding site found in both monomeric and trimeric Env. In turn, this binding model has led to emerging designs of macrocyclic peptide triazoles with enhanced potency and, very importantly, resistance to proteases. Overall, studies with PTs have shown how gp120 targeting can hijack intrinsic metastability properties of the Env protein spike and of the virus particle as a whole to achieve Env antagonism and irreversible virus inactivation. And, the advent of stable and low molecular weight macrocyclic peptidomimetics derived from PTs offers a precedent to develop clinically useful drug-like HIV-1 inactivators that can intervene with the virus before host cell encounter to suppress infection and destroy viruses already present in infected individuals.
The fundamental challenge of blocking the HIV-1 cell entry machine
There is currently no cure for HIV-1 infection once acquired, and methods to prevent infection by either a vaccine or microbicide have not yet been developed. The global prevalence of HIV-1 infection continues to challenge pursuit of new approaches to combat the spread of AIDS disease [1,2]. The Env protein on the surface of the virus is an appealing target, since inhibiting this protein offers the opportunity to suppress the initial cell infection process. The Env protein spike itself is the virus homing component that recognizes and binds to host cells through a virological synapse with two cell receptors, CD4 and a 7-transmembrane coreceptor, most commonly CCR5 or CXCD4. Env is a trimer of heterodimers, containing three outer gp120 subunits and three inner gp41 subunits [3]. Initial CD4 binding to gp120 triggers structural rearrangements in the Env spike. CD4 binding leads to enhanced gp120 binding to coreceptor and then to exposure and rearrangement of the three gp41 subunits, which bind to cell membrane through the fusion peptide domains and form a 6-helix bundle that brings the virus and cell membranes together for fusion and entry of virus RNA into the host cell [4]. The conformational rearrangements of Env and ultimately virus–cell fusion fundamentally are driven by an energy gradient from the high energy and metastable native unliganded Env spike to low energy 6-helix bundle, and the energy derived from this gradient is used to transform the membrane for the otherwise-unfavorable fusion step [5].
In spite of the promise of Env gp120 for virus-specific targeting, antagonizing virus entry by gp120 binding agents has proven elusive due to such factors as Env sequence variability, flexibility, conformational masking and an extensive glycan shield, all of which limit the occurrence and access of conserved sites of inhibition [6-8]. These structural challenges limit the ability to formulate inhibitors that will function effectively against a wide range of virus subtypes that exist globally. Hence, there is a continuing need to identify molecular agents that can target conserved vulnerabilities to block virus cell entry [1].
We have explored strategies for irreversible HIV-1 antagonism by targeting the virus Env before host cell encounter. These strategies go beyond receptor binding blockade to additionally cause irreversible Env spike protein inactivation by hijacking the conformational change program that is intrinsic to the virus and normally used to promote both virus-to-cell and cell-to-cell fusion. We have discovered several molecular constructs [9-12] that can bind to gp120 and inactivate the virus, including by lytic transformation of the virus membrane. These agents function in the absence of host cells and hence can result in irreversible virus inactivation before the virus encounters cells to either initiate infection or propagate an existing infection. The ability of multiple gp120-targeting strategies to inactivate the HIV-1 virion argues for the general usefulness of the overall metastability-hijacking strategy for virus killing before cell encounter. Among the agents we have investigated, a class that offers an appealing medicinal chemistry approach to small molecule virus inactivators of therapeutic and preventive interest is PTs.
Early investigations leading to PTs
PTs were derived from an initially-reported peptide-denoted 12p1, which was identified in a phage display screen and found to inhibit gp120 binding to both CD4 and the coreceptor surrogate mAb 17b [13,14]. The dual receptor site inhibition function was appealing, since it offered the ability to suppress CD4 binding without activating the coreceptor site, an undesired consequence of Env gp120 binding by CD4 and many CD4-mimicking compounds. However, 12p1 itself possessed only low (>micromolar) Envbinding potency. Investigation of 12p1 derivatization led to high-potency PTs, by conversion of the central Pro to azido-Pro, that was then used for click reaction [15] using copper-catalyzed 3+2 cycloaddition conditions [16] (Figure 1).
Figure 1.
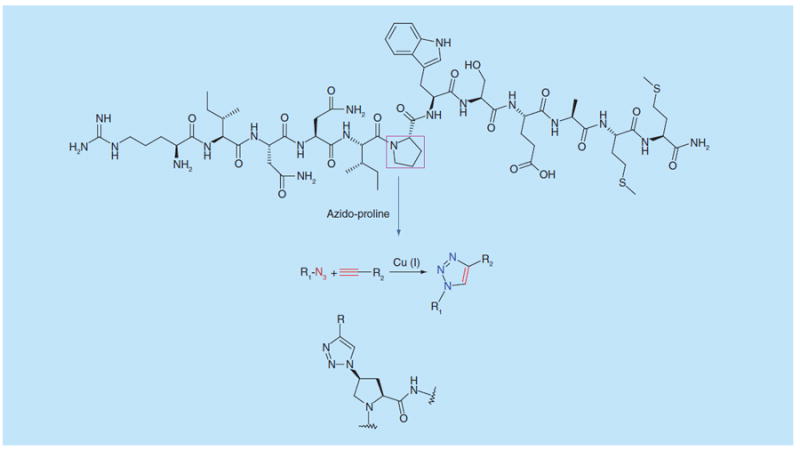
Origin of peptide triazoles, from the 12p1 identified by phage display screening, by 3+2 copper-catalyzed cycloaddition on central azidoproline incorporated in starting synthetic peptide. See [13-15].
A series of PT conjugates with improved binding affinity for gp120 identified the importance of large hydrophobic triazole substitutions for enhanced potency [17,18]. A key high affinity candidate identified was the ferrocenyl triazole conjugate HNG-156, which was found to bind to monomeric gp120 with of 7 nM, in an equilibrium dissociation constant KD value of the parent pepcontrast to the 2600 nM KD tide 12p1. Furthermore, like 12p1, HNG-156 inhibited binding of gp120 to both CD4 and the coreceptor surrogate mAb 17b, but the PT did so with sub-μM potency. And, potency enhancement applied also to antiviral potency, as HNG-156 neutralized viral infection by subtype A, B, C and D HIV-1 isolates (IC50 range = 0.08–62.5 μM), but not infection by negative control viruses pseudotyped with VSV-G (vesicular stomatitis virus G). The peptide also exhibited no detectable toxicity in a tissue explant model at concentrations up to 100 μM. Importantly, antiviral activity was observed with viruses of both R5 and X4 tropisms [17,19,20].
The encouraging retention of function of HNG-156 led to efforts to minimize the structure of the inhibitor. Indeed, active core fragments were identified, including six residue variants [21]. These cores and structure-activity investigations demonstrated the presence of a tripartite Ile-TriazolePro-Trp pharmacophore as shown in Figure 2.
Figure 2.
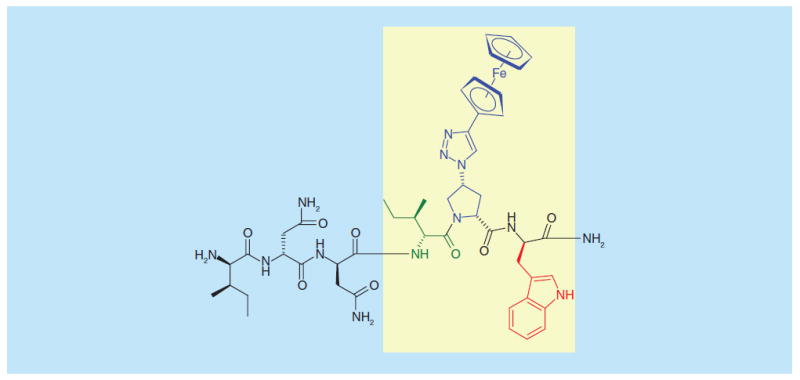
Structure of an active core of peptide triazoles, denoting the central Ile-ferrocenyltriazole Pro-Trp pharmacophore. See [21].
Direct virus inactivation by PTs discovered
While the initial peptide triazole functions of dual receptor site inhibition and infection inhibition were certainly encouraging, further investigation showed that PTs also have unique virus-killing functions (Figure 3). This class of compounds was found to induce shedding of gp120, thus inactivating the virus before cell encounter [9,10]. In addition, peptide triazole thiols (PTTs), PTs containing a free sulf-hydryl group (SH) on Cys at the C-terminus, were found to cause envelope disruption and p24 capsid protein release from the virus lumen [9,10]. The PTTs were initially made to conjugate to gold nanoparticles to form multivalent and consequently more potent inhibitors by providing the ability to interact with multiple Env proteins on single virions. While AuNP-PTs were indeed more potent [12], the PTTs themselves were found to have improved activity and, as noted above, virolytic activity [9,12]. Both gp120 shedding and lytic inactivation by PTs appear to occur by taking advantage of the intrinsic metastability of the Env protein that the virus normally uses to promote virus–cell fusion. The additional role of the PTT SH group in causing virus membrane disruption is currently being investigated [22]. One hypothesis is a covalent mode of inhibition through the reactivity of the SH group. Likely, gp120 shedding and lytic deformation of the virus induced by PT and PTT inhibitors are a consequence of gp120 conformational perturbation [21,23].
Figure 3.
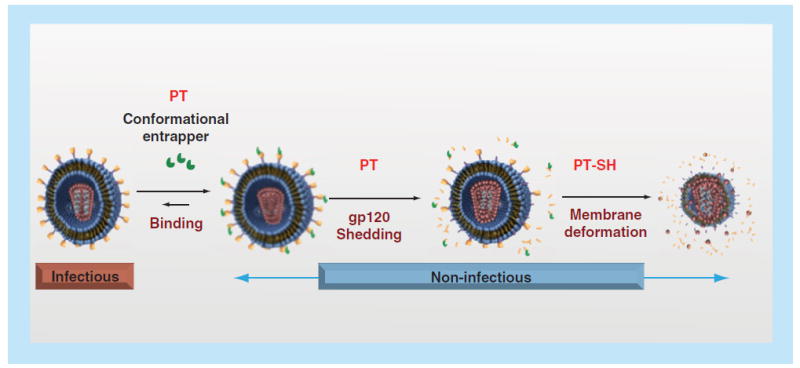
Functional phenotypes of peptide triazoles, including suppression of interactions at both receptor binding sites and virus inactivation through gp120 shedding and, for peptide triazole thiols, membrane lysis.
The overall phenotype of irreversible HIV-1 inactivation makes PTs appealing as HIV-1 antagonist leads. Nonetheless, the peptidic nature of these compounds has been a barrier to antagonist development due to the potential for proteolytic degradation and consequent short half-life. An improved structural understanding of the PT-gp120 encounter complex was needed to help define approaches to design more protease-resistant variants. We have addressed this problem by investigating structural mechanism and consequent design of protease-resistant macrocyclic PTs (cPTs).
Env gp120 cavity occupancy model & the derivation of cyclic peptide triazoles
We used coordinated mutagenesis, synthetic design and flexible docking to investigate the structural mechanism of Env gp120 encounter by peptide triazole inactivators of HIV-1 [24]. We found that PT encounters gp120 by engaging two inhibitor ring moieties at the junction between the inner and outer domains of the Env protein. PT binding was found to be sensitive to mutagenic modification of two closely related gp120 hot spots, one bounded by residues Thr257 and Ser375 associated with the CD4 Phe43 binding site cavity and the second at Trp112 in a hydrophobic cavity in the gp120 inner domain. Comparison of PT synthetic variant potencies with gp120 mutational variants argued that the critical Trp residue in the PT pharmacophore interacts at the Thr257/Ser375 site. Flexible docking confirmed that the PT Trp favors the Thr257/Ser375 cavity, while the aryl-triazole of the Ile-Triazole Pro-Trp pharma-cophore favors interaction at the inner domain hydrophobic cavity gated by Trp112. The results demonstrated how combined occupancy of both subsites of gp120 can coordinately suppress both receptor and coreceptor binding and conformationally entrap gp120 in a destabilized state [24]. Generally speaking, the two-cavity model provides a template to guide the design of peptidomimetic HIV-1 inactivators with prevention and intervention potential based on the PT pharmacophore.
Inspired by the two-cavity model (Figure 4, left), we recently established a synthetic methodology to derive macrocyclic HIV-1 antagonists (Figure 4, right) [25]. The cyclic PTs (cPTs) retained the ability of the parent PTs to dually inhibit binding of gp120 at both its CD4 and coreceptor binding sites and induce gp120 shedding from the virions, leading to irreversible inactivation. Furthermore, in preliminary studies, the virolysis effects of PT thiols have been recapitulated with a cyclized peptide triazole thiol [26]. These phenotypes argue that cyclization can lock the peptide conformation into an active form. The cPTs were able to inhibit HIV-1 pseu-dovirus cell infection at sub-micromolar concentrations, without host cell toxicity. The 6-residue cPT denoted AAR029b, the most active obtained so far, inhibited gp120 binding to both sCD4 and the core-ceptor surrogate, 17b, with IC50 ~30 nM. AAR029b also inhibited cell infection by both BaL.01 and JR-FL pseudovirus strains with IC50 values approximately 200 nM [25]. Importantly, these macrocycles were found to be stable to trypsin and chymotrypsin digestions, compared to a substantial susceptibility of corresponding linear PTs to these proteases. Overall, the cPTs are the first macrocyclic dual receptor site antagonists that target HIV-1 gp120, and represent promising new peptidomimetic leads for developing HIV-1 inactivators, combining the safety and selectivity profiles of peptides with the ease of chemical accessibility and high potency.
Figure 4.
Two-cavity model of peptide triazole pharmacophore binding to gp120 and the strategy of formation of cyclic peptide triazoles. See [25,26].
Reproduced with permission from [25] © American Chemical Society (2015).
Promise & challenges
We now know that peptide triazoles, upon binding to HIV-1 Env gp120, are able to not only block receptor interactions but to hijack the metastability of Env to inactivate the virus before host cell encounter. In addition, since infected cells also display the Env spike on their surface, PTs may be predicted to target cellular Env and suppress cell-to-cell infection as well as new virus formation. Preliminary data suggesting this potential have been obtained [27]. The small size, enhanced antiviral activity, proteolytic stability and ease of chemical synthesis of cPTs make these potentially important therapeutic leads. The constrained cPT conformation will also aid crystallization efforts to resolve high-resolution structures of the highly flexible HIV-1 Env with this class of inhibitors. In turn, such structures as well as computational models can provide guidance for further chemical optimization, and in addition may provide tools for computational screening to derive new classes of virus inactivators that bind in the PT site of gp120.
Crucial challenges remain in adapting peptide triazoles for therapeutic use. Resistance to proteases needs to be validated with enzymes from the tissue environments of initial virus exposure and proliferation, including circulation and mucosal tissues. Breadth of antiviral action for globally diverse viruses, sufficient resistance to virus mutational escape and efficacy in animal models will have to be established. Delivery may be required, in particular to mucosal sites where virus proliferates and can hide in latent reservoirs until activation. In spite of these challenges, the potential of cPTs as leads to clinically useful compositions argues for continued medicinal chemistry efforts to advance smaller and conformationally constrained cPTs and cPT nonpeptide surrogates.
Acknowledgments
Peptide triazole studies have been supported by Institutes of General Medical Sciences and Allergy and infectious Diseases, NIH; National Science Foundation; United States Agency for international Development; WW smith Charitable Trust; Schlumberger Foundation; and Drexel University.
Biographies

Irwin Chaiken

Adel A Rashad
Footnotes
Financial & competing interests disclosure
The authors have no other relevant affiliation or financial involvement with any organization or entity with a financial interest in or financial conflict matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Contributor Information
Irwin Chaiken, Department of Biochemistry & Molecular Biology, Drexel College of Medicine, Philadelphia, PA 19102, USA.
Adel A Rashad, Department of Biochemistry & Molecular Biology, Drexel University College of Medicine, Philadelphia, PA 19102, USA.
References
- 1.Acharya P, Lusvarghi S, Bewley CA, Kwong PD. HIV-1 gp120 as a therapeutic target: navigating a moving labyrinth. Expert Opin Ther Targets. 2015;19(6):765–783. doi: 10.1517/14728222.2015.1010513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Swindells S, Flexner C, Fletcher CV, Jacobson JM. The critical need for alternative antiretroviral formulations, and obstacles to their development. J Infect Dis. 2011;204(5):669–674. doi: 10.1093/infdis/jir370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wyatt R, Kwong PD, Desjardins E, et al. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature. 1998;393(6686):705–711. doi: 10.1038/31514. [DOI] [PubMed] [Google Scholar]
- 4.Jiao J, Rebane AA, Ma L, Gao Y, Zhang Y. Kinetically coupled folding of a single HIV-1 glycoprotein 41 complex in viral membrane fusion and inhibition. Proc Natl Acad Sci USA. 2015;112(22):E2855–E2864. doi: 10.1073/pnas.1424995112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doms RW. Beyond receptor expression: the influence of receptor conformation, density, and affinity in HIV-1 infection. Virology. 2000;276(2):229–237. doi: 10.1006/viro.2000.0612. [DOI] [PubMed] [Google Scholar]
- 6.Do Kwon Y, Pancera M, Acharya P, et al. Crystal structure, conformational fixation and entry-related interactions of mature ligand-free HIV-1 Env. Nat Struct Mol Biol. 2015;22(7):522–531. doi: 10.1038/nsmb.3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Julien JP, Cupo A, Sok D, et al. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science. 2013;342(6165):1477–1483. doi: 10.1126/science.1245625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munro JB, Gorman J, Ma X, et al. Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science. 2014;346(6210):759–763. doi: 10.1126/science.1254426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bastian AR, Contarino M, Bailey LD, et al. Interactions of peptide triazole thiols with Env gp120 induce irreversible breakdown and inactivation of HIV-1 virions. Retrovirology. 2013;10:153. doi: 10.1186/1742-4690-10-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bastian AR, Kantharaju, McFadden K, et al. Cell-free HIV-1 virucidal action by modified peptide triazole inhibitors of env gp120. ChemMedChem. 2011;6(8):1335–1339. doi: 10.1002/cmdc.201100177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Contarino M, Bastian AR, Kalyana Sundaram RV, et al. Chimeric cyanovirin-MPER recombinantly engineered proteins cause cell-free virolysis of HIV-1. Antimicrob Agents Chemother. 2013;57(10):4743–4750. doi: 10.1128/AAC.00309-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bastian AR, Nangarlia A, Bailey LD, et al. Mechanism of multivalent nanoparticle encounter with HIV-1 for potency enhancement of peptide triazole virus inactivation. J Biol Chem. 2015;290(1):529–543. doi: 10.1074/jbc.M114.608315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biorn AC, Cocklin S, Madani N, et al. Mode of action for linear peptide inhibitors of HIV-1 gp120 interactions. Biochemistry. 2004;43(7):1928–1938. doi: 10.1021/bi035088i. [DOI] [PubMed] [Google Scholar]
- 14.Ferrer M, Harrison SC. Peptide ligands to human immunodeficiency virus type 1 gp120 identified from phage display libraries. J Virol. 1999;73(7):5795–5802. doi: 10.1128/jvi.73.7.5795-5802.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gopi HN, Tirupula KC, Baxter S, Ajith S, Chaiken IM. Click chemistry on azidoproline: high-affinity dual antagonist for HIV-1 envelope glycoprotein gp120. ChemMedChem. 2006;1(1):54–57. doi: 10.1002/cmdc.200500037. [DOI] [PubMed] [Google Scholar]
- 16.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int Ed Engl. 2002;41(14):2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 17.Gopi H, Cocklin S, Pirrone V, et al. Introducing metallocene into a triazole peptide conjugate reduces its off-rate and enhances its affinity and antiviral potency for HIV-1 gp120. J Mol Recognit. 2009;22(2):169–174. doi: 10.1002/jmr.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gopi H, Umashankara M, Pirrone V, et al. Structural determinants for affinity enhancement of a dual antagonist peptide entry inhibitor of human immunodeficiency virus type-1. J Med Chem. 2008;51(9):2638–2647. doi: 10.1021/jm070814r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cocklin S, Gopi H, Baxter S, Chaiken I. A dual action peptide GP120 inhibitor that is effective against multiple HIV-1 clades. FASEB J. 2006;20(4):A465–A465. [Google Scholar]
- 20.McFadden K, Fletcher P, Rossi F, et al. Antiviral breadth and combination potential of peptide triazole HIV-1 entry inhibitors. Antimicrob Agents Chemother. 2012;56(2):1073–1080. doi: 10.1128/AAC.05555-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Umashankara M, McFadden K, Zentner I, et al. The active core in a triazole peptide dual-site antagonist of HIV-1 gp120. ChemMedChem. 2010;5(11):1871–1879. doi: 10.1002/cmdc.201000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bailey LB, Kalyana Sundram RV, Li H, et al. Disulfide sensitivity in the Env protein underlies lytic inactivation of HIV-1 by peptide triazole thiols. ACS Chem Biol. 2015 doi: 10.1021/acschembio.5b00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emileh A, Tuzer F, Yeh H, et al. A model of peptide triazole entry inhibitor binding to HIV-1 gp120 and the mechanism of bridging sheet disruption. Biochemistry. 2013;52(13):2245–2261. doi: 10.1021/bi400166b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aneja R, Rashad AA, Li H, et al. Peptide triazole inactivators of HIV-1 utilize a conserved two-cavity binding site at the junction of the inner and outer domains of Env gp120. J Med Chem. 2015;58(9):3843–3858. doi: 10.1021/acs.jmedchem.5b00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rashad AA, Kalyana Sundaram RV, Aneja R, Duffy C, Chaiken I. Macrocyclic envelope glycoprotein antagonists that irreversibly inactivate HIV-1 before host cell encounter. J Med Chem. 2015;58(18):7603–7608. doi: 10.1021/acs.jmedchem.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rashad AA, Kalyana Sundaram RV, Nangarlia A, Aneja R, Duffy C, Chaiken I. Macrocyclic HIV-1 envelope glycoprotein antagonists. Presented at: Structural Biology Related to HIV/AIDS; Natcher Conference Center; Bethesda, Maryland, USA. 18-19 June 2015. [Google Scholar]
- 27.Bastian AR. Irreversible breakdown of HIV-1 by peptide triazole thiols and multivalent gold nanoparticle conjugates [PhD thesis] Ann Arbor, Drexel University; MI, USA: 2014. [Google Scholar]