

Abstract
Tyrosyl-DNA phosphodiesterase 2 (TDP2) processes protein/DNA adducts resulting from abortive DNA topoisomerase II (Top2) activity. TDP2 inhibition could provide synergism with the Top2 poison class of chemotherapeutics. By virtual screening of the NCI diversity small molecule database, we identified selective TDP2 inhibitors and experimentally verified their selective inhibitory activity. Three inhibitors exhibited low-micromolar IC50 values. Molecular dynamics simulations revealed a common binding mode for these inhibitors, involving association to the TDP2 DNA-binding cleft. MM-PBSA per-residue energy decomposition identified important interactions of the compounds with specific TDP2 residues. These interactions could provide new avenues for synthetic optimization of these scaffolds.
Keywords: Tyrosyl-DNA phosphodiesterase 2, TDP2, inhibitor, virtual screening, rational drug design
Graphical Abstract

Tyrosyl-DNA phosphodiesterase (TDP) activity is necessary to cleave the tyrosyl-DNA linkage between a trapped topoisomerase and its substrate DNA1. In humans, there are two known TDPs: TDP1 cleaves 3’ type-IB topoisomerase-DNA linkages2, while TDP2 cleaves 5’ type-II topoisomerase-DNA linkages3,4.
Topoisomerase II (Top2) poisons act by trapping Top2 on its DNA substrate, causing the normally transient Top2-mediated double strand breaks to become permanent, resulting in cell death5. For this reason, Top2 poisons are widely used cancer therapeutics. TDP2 activity reduces the efficacy of Top2 poisons and is therefore an attractive anticancer drug target, with TDP2 deficient cells exhibiting extreme sensitivity to Top2 poisons3,6. The viability of TDP2 knockout mice indicates that TDP2 inhibition is theoretically possible without unacceptable side effects6. A TDP2 inhibitor could have great potential for synergistic effects when used in combination with Top2 poisons and could greatly increase the efficacy of such treatments.
Small-angle X-ray scattering analysis shows that TDP2 consists of a ~110-residue, disordered N-terminal domain and a 255-residue, globular catalytic domain7, 8. Only the catalytic domain is necessary for phosphodiesterase activity, while the N-terminal tail is thought to interact with cellular signaling machinery. Although no structures exist for human TDP2 (hTDP2), multiple crystal structures of TDP2 have been solved including C. elegans (cTDP2)8 , D. rerio (zTDP2)8 and M. musculus (mTDP2)7. mTDP2 serves as an excellent structural homologue to hTDP2, with the variants’ catalytic domains sharing 78% sequence identity and nearly 100% sequence similarity (Supplementary Figure S1). A structure of mTDP2* bound to a substrate analog (PDB accession code 4GZ1) shows a short DNA-binding cleft, contacting 3 DNA phosphates, leading directly into the active site (Supplementary Figure S2). Kinetic studies indicate that mammalian TDP2 is highly specific for 5’-tyrosine overhangs, as opposed to 3’ overhangs, blunt ends or other adducts, with the exception of p-nitrophenol, a compound frequently used as a DNA adduct in screening assays7,9. Interestingly, TDP2 shares very similar active site geometry and catalytic mechanism with the base excision repair enzyme apurinic/apyrimidinic endonuclease 1 (APE1), along with 14% sequence identity and 30% sequence similarity8. Despite these similarities, TDP2 does not show endonuclease activity. From mutational and structural data, Schellenberg, et al. proposed that hydrophobic contacts made to W307 and F325 by the substrate DNA backbone serve as the basis for the specificity toward 5’-tyrosine overhangs by forming favorable Van der Waals interactions with the substrate deoxyribose ring7. Crystal structures of the D. rerio and C. elegans TDP2 homologues in complex with substrate DNA provide further evidence that hydrophobic contacts are primarily responsible for substrate binding8. Binding of a 3’-tyrosine substrate would juxtapose a phosphate group in place of a ribose ring, unfavorably forcing a negatively charged group to contact hydrophobic sidechains. Moreover, the mutation of certain residues lining the DNA-binding cleft greatly alters catalytic activity in hTDP2. Of note are R231, R266, W297 and F315, all of which are important to the activity of TDP2 on 4-nucleotide overhang 5’-tyrosine substrate DNA, with mutations being very deleterious to catalysis7.
In contrast to TDP2, TDP1 cleaves 3’ adducts and is capable of acting on a relatively broad range of substrates10. TDP1 also displays some activity against 5’-tyrosine overhangs, causing specificity overlap with TDP211, 12. Designing a TDP2 inhibitor is complicated by the similar catalytic activity and substrate characteristics of TDP1. A potential inhibitor must be strongly selective for TDP2 to limit binding competition by TDP1. Because the TDP2 binding cleft may hold the key for TDP2 substrate specificity, it presents a promising region for targeting inhibitors that do not have activity against TDP1.
The discovery of selective TDP2 inhibitors based on toxoflavin and deazaflavin scaffolds has recently been reported, in which the authors began the inhibitor search with a high-throughput in vitro screening of 100,000 compounds13. Small molecule docking of this scaffold shows that inhibitors of this type are likely to bind in the active site of TDP2, directly blocking catalysis. While these compounds show promise, they exhibit slight inhibition of TDP1 at 100 µM. In addition, toxoflavins and deazaflavins also have undesirable characteristics for drug scaffolds. Toxoflavins are susceptible to redox activity and deazaflavins have poor cell permeability.
The difficulty in predicting the binding selectivity of a compound necessitates a high-throughput lead screening and optimization protocol. To this end, we have carried out an inhibitor discovery protocol (Figure 1) to identify selective TDP2 inhibitors. Our protocol exploits the large scale docking of 11,000 compounds from the 250,000-compound Open National Cancer Institute (NCI) Database14 followed by virtual screening (VS) and in vitro assays using whole cell extract (WCE). With this protocol, we have discovered three potent and selective small molecule inhibitors of TDP2. Results from molecular docking, molecular dynamics (MD), molecular mechanics – Poisson-Boltzmann surface area (MM-PBSA) calculations, biochemical and kinetics experiments provide evidence that our inhibitors bind in the DNA-binding cleft of TDP2, effectively blocking substrate binding and catalysis. The scaffolds of these inhibitors have the potential to be further optimized by targeting specific contacts made to residues in the TDP2 binding cleft, increasing both potency and selectivity for TDP2.
Figure 1.

Flowchart overview of our TDP2 inhibitor discovery process.
Compound 21 (Supplementary Figure S3), an APE1 inhibitor provided by Dr. Neamati, was chosen to initiate our screening protocol to identify hTDP2-selective inhibitors because it inhibited hTDP2 with an IC50 of 12, 20 µM (n=2) without affecting hTDP1 up to 200 µM (data not shown). To predict how a ligand will bind to hTDP2, a homology model of the catalytic domain was constructed from the substrate analog-bound mTDP2 structure (PDB accession code 4GZ1) using the program Modeller15. Although the homologues have extraordinary sequence similarity (Supplementary Figure S1), and therefore nearly identical secondary structure, minor alterations to the surface of the enzyme can influence preferred ligand binding positions. Compound 21 was first docked into the hTDP2 homology model (Supplementary Figure S3). Docking was performed with the program AutoDock416, after preparing a ligand/protein atom-pair interaction grid with AutoGrid4 within the search area. The search area was comprised of both the DNA binding cleft and active site, the two regions of the enzyme known to directly impact catalytic activity. The three best-scoring docking poses that were distinct from the others with respect to ligand conformation and ligand-protein contacts were chosen for further analysis. Each pose was simulated for 30ns in MD, using the NAMD2.9 package17. The MD trajectory which resulted in the most stable binding pose was clustered with respect to the root mean square deviation (rmsd) of the atomic coordinates of the small molecule using the hierarchical clustering algorithm (ε=10.0), as implemented in the ptraj18 MD analysis program. The representative frame from the most populated cluster was then used for VS. VS against this dominant conformation was performed as follows: sets of fifty random conformers for each compound in the NCI database were first constructed using the Openeye Software program Omega19 and screened for structural and electrostatic similarity to the dominant MD conformer using Openeye’s ROCS20 and EON21 programs, respectively. The 11,000 top hits from the VS, as determined by averaging structural and electrostatic Tanimoto overlaps with the lead compound, were then docked using the AutoDock4 program into our hTDP2 homology model.
From the top 500 docking hits, a set of 178 was selected for optimal structural diversity. From these 178 compounds, 95 were available and were tested against recombinant human TDP2 (hTDP2) at a single concentration of 1 mM (Figure 1). 48 active compounds were further tested at a single concentration of 100 µM against hTDP2. 11 TDP2-selective inhibitors were further evaluated in dose response against both recombinant human TDP2 and TDP1 (hTDP2 and hTDP1) enzymes (Figure 1). Four TDP2-selective low micromolar inhibitors i.e. not inhibiting hTDP1 up to 111 µM, were checked by LC/MS (Figure S5) to assess for their presence in the vial and evaluated for their ability to inhibit endogenous hTDP2 within whole cell extracts from human TDP2-complemented DT40 chicken cells (hTDP2 WCE). The TDP2-selective inhibitors, NSC375976, NSC114532 and NSC3198 were found to inhibit hTDP2 with IC50 values of 3.5 ± 1.4, 4.1 ± 0.9 and 9.3 µM, respectively while all being inactive against recombinant hTDP1 up to 111 µM (Figure 2 and Table 1). All 3 compounds show robustness while being exposed to an increased complexity reaction mixture because they retain low micromolar activity against whole cell extracts (WCE) containing endogenous hTDP2 (Figure 2).
Figure 2.

Inhibition of human recombinant TDP2 (hTDP2) and endogenous human TDP2 from whole cell extracts (hTDP2 WCE) by NSC379576, NSC114532, and NSC3198. Concentrations of compounds are 0.5, 1.4, 4.1, 12.3, 37 and 111 µM. Concentrations of the positive control Compound 122 are 0.005, 0.017, 0.05, 0.15, 0.46, 1.4 µM.
Table 1.
IC50 values against human (H. sapiens, hTDP2), mouse (M. musculus, mTDP2) and zebrafish (D. rerio, zTDP2) TDP2 enzymes and against human TDP1 (hTDP1).
| IC50 (µM) | |||||
|---|---|---|---|---|---|
| Compound | Structure | hTDP2 | mTDP2 | zTDP2 | hTDP1 |
| NSC375976 | 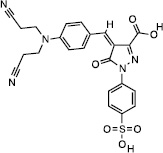 |
3.5 ± 1.4 (n=4) |
15 (n=1) |
5.2 (n=1) |
>111 |
| NSC114532 | 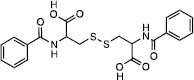 |
4.1 ± 0.9 (n=4) |
>111 | 15 (n=1) |
>111 |
| NSC3198 | 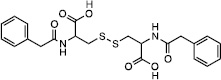 |
9.0, 9.5 (n=2) |
>111 | 20 (n=1) |
>111 |
The inhibition of TDP2 by these 3 compounds was then assessed across species by comparing their potencies against human (H. sapiens, hTDP2), mouse (M. musculus, mTDP2) and zebrafish (D. rerio, zTDP2) TDP2 enzymes (Figure 3). Only NSC379576 inhibited efficiently all three enzymes with IC50 values of 15 and 5.2 µM for mTdp2 and zTDP2, respectively (Figure 3 and Table 1). NSC114532 and NSC3198 both inhibited zTDP2 with IC50 values of 15 and 20 µM, respectively but interestingly, mTDP2 was totally resistant to both compounds up to 111 µM (Figure 3 and Table 1). These results suggest that the TDP2 binding site for both NSC114532 and NSC3198 is not conserved in the mouse enzyme similarly to what was recently observed for the first reported selective TDP2 inhibitor Compound 1 (Figure 2)22.
Figure 3.

Inhibition of human (H. sapiens, hTDP2), mouse (M. musculus, mTDP2) and zebrafish (D. rerio, zTDP2) TDP2 enzymes by NSC379576, NSC114532, and NSC3198. Concentrations of compounds are 0.5, 1.4, 4.1, 12.3, 37 and 111 µM.
A potential concern with the chemotype of NSC114532 and NSC3198 is the presence of a disulfide bond in the center of the structure (Table1). The presence of a high concentration of DTT (1mM) in the reaction buffer could therefore potentially cleave the molecule in two. To study the potential impact of the presence of a reducing agent on the stability of NSC114532 and NSC3198, we tested their ability to inhibit hTDP2 in the presence or absence of DTT. We did not observe any difference in the inhibition of hTDP2 by these two compounds under these conditions (Figure S6), suggesting that both compounds are not affected by the composition of the reaction buffer.
Molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) calculations23, as implemented in the AmberTools14 analysis suite24, were performed on NSC375976, NSC114532 and NSC3198. MM-PBSA is a thermodynamic cycle based method, whereby the solvation enthalpies of the complex (ΔHcomplex), receptor (ΔHreceptor) and ligand (ΔHligand) are estimated independently, then subtracted to obtain the binding enthalpy (ΔHbinding) via:
From MM-PBSA, NSC114532 and NSC3198 were determined to have binding enthalpies of −18±12 and −30±7.9 kcal/mol, respectively. NSC114532 and NSC3198 have essentially similar binding enthalpies, as expected from their structural similarity and similar IC50 values. NSC375976 has a higher binding enthalpy, 0.1±4.7 kcal/mol, than both NSC114532 and NSC3198, although it possesses the lowest IC50 of the group. This result is less surprising given the dissimilarity of the ligands. It is also possible that the IC50 values do not correlate perfectly with the binding enthalpies due to ligand interactions with other species in WCE. To compare the binding modes of the three inhibitors, we have performed MM-PBSA per-residue energy decomposition25. This method calculates the enthalpy of each residue’s non-bonded interactions with the ligand, highlighting residue-ligand contacts that are essential to ligand binding, as well as those that are detrimental to it.
NSC114532 and NSC3198 are symmetrical molecules around the disulfide bond. Because these two compounds are very close structural analogs, their modes of binding are strikingly similar, with the majority of the binding enthalpy coming from hydrogen bonding and electrostatic interactions between the ligands’ carboxy groups and R231 and R266 (Figure 4A and 4B).
Figure 4.

Binding poses and per-residue energy decomposition for residues important to binding. Per-residue energy decomposition results for (A) NSC114532, (B) NSC3198 and (C) NSC375976. Dominant binding poses of (D) NSC114532, (E) NSC3198 and (F) NSC375976.
NSC114532 and NSC3198 each also form favorable hydrophobic interactions with residues W297, L313 and F315 that lie in the DNA binding cleft (Figure 4D and 4E). NSC375976 does not share the basic chemical scaffold of NSC114532 and NSC3198, yet binds in nearly the same position (Figure 4C and 4F). In contrast to NSC114532 and NSC3198, NSC375976 binds predominantly to hydrophobic residues, with W297, L313 and F315 each contributing ~−1 kcal/mol in enthalpy. The NSC375976 sulfonate group is not able to form favorable electrostatic interactions with R231 and R266, with the adjacent phenyl group clashing slightly with R266. All three inhibitors form favorable van der Waals interactions with the T230 methyl group. The most unfavorable interaction between these inhibitors and hTDP2 is with D350. Each inhibitor places hydrophobic functional groups near the charged D350 side chain, incurring an enthalpic penalty relative to a solvent-exposed D350.
Residues R231, R266, W297, L313 and F315 have all been shown via mutational studies to interact favorably with the DNA substrate, with W297 and F315 having been proposed to form the basis of TDP2 selectivity towards 5’-tyrosine adducts7. Clearly, ligand binding in this region of the protein precludes substrate recognition by TDP2, thus inhibiting catalytic activity. Forming favorable contacts with the residues that select for 5’ adduct binding is likely to be the cause of these inhibitors’ selectivity towards TDP2, as 5’ adducts have been shown to be very poor substrates for TDP1. It should be noted that the two distinct molecular scaffolds represented by NSC114532, NSC3198 and NSC375976 both take advantage of the same structural characteristics of the DNA-binding groove, leaving open the possibility of the discovery of other unique scaffolds that have similar binding modes.
To further probe the basis of selectivity of NSC114532, NSC3198 and NSC375976, we docked each into hTDP1, using the same methodology as with hTDP2, again including both the active site and DNA-binding groove in the search area. The top docking poses from each compound (Supplementary Figure S4) identify the hTDP1 active site as the primary binding target. These results do not indicate that NSC114532, NSC3198 or NSC375976 bind hTDP1. Rather, they serve simply as an indication that, when forced to interact with hTDP1 in silico, these inhibitors preferentially interact with the active site. This provides another line of evidence supporting the hypothesis that DNA-binding cleft interactions dictate substrate specificity between hTDP1 and hTDP2 and that our inhibitors select for hTDP2 over hTDP1 based on many of the same interactions. The docking poses of NSC114532 and NSC3198 in hTDP1 are again similar, with both compounds forming hydrogen bonds with S399 and K495. NSC3198 stacks one phenyl ring against H263, while leaving approximately half of the molecule solvent-exposed with no notable favorable interactions with the enzyme. NSC114532 forms no notable favorable stacking interactions with either phenyl ring and forms an electrostatic clash between a carboxylate group and G538. NSC375976 remains largely solvent-exposed by packing against one wall of the active site while reaching slightly into the active site pocket. The only favorable hydrophobic interaction NSC375976 forms is between the phenylsulfonate ring and the aliphatic portion of the S459 sidechain, while the only hydrogen bond to the enzyme is made between the sulfonate group and H263. The combination of unfavorable interactions and inability to take advantage of nonspecific Van der Waals interactions explains these ligands’ inability to bind to hTDP1 and, therefore, their selectivity for hTDP2.
NSC114532, NSC3198 and NSC375976 can each be tailored to increase binding affinity to hTDP2 by improving electrostatic complementarity to the binding cleft to enhance binding enthalpy while leaving the Van der Waals contacts in place to maintain selectivity. It is important to note that NSC3198 and NSC114532 form very favorable electrostatic interactions with R231 and R266, residues that are important to the catalytic function of hTDP2. In the future development of these scaffolds, care must be taken to preserve these interactions, meaning that the carboxy groups, as well as the 4-atom linker that separates them, appear to be absolutely essential to effective binding. NSC375976 can likely form very favorable interactions between its sulfonate group and R231 and R266, if the sulfonate group is brought more proximal to the center of the compound. NSC375976 holds the lowest IC50 of the compounds studied, taking advantage of this potentially very favorable interaction may greatly improve binding characteristics.
Supplementary Material
Acknowledgments
This work was supported by a National Science Foundation CAREER award MCB-1149521 and National Institute of Health grant R01GM110387. This work was also supported in part by by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (Z01 BC 006161). Computational resources were provided in part by a National Science Foundation XSEDE allocation CHE110042 and through an allocation at NERSC supported by the U.S. Department of Energy Office of Science contract DE-AC02-05CH11231. The authors wish to thank Drs. Gary Pauly and Joel Schneider from the Chemical Biology Laboratory, CCR, NCI for their support in purity analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Supplementary data associated with this article can be found in the online version at
References and notes
- 1.Pommier Y, Huang SY, Gao R, Das BB, Murai J, Marchand C. DNA Repair (Amst) 2014;19:114–129. doi: 10.1016/j.dnarep.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu C, Pouliot JJ, Nash HA. Proc. Natl. Acad. Sci. U.S.A. 2002;99:14970–14975. doi: 10.1073/pnas.182557199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeng Z, Cortes-Ledesma F, El Khamisy SF, Caldecott KW. J. Biol. Chem. 2011;286:403–409. doi: 10.1074/jbc.M110.181016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caldecott KW. Nat. Struct. Mol. Biol. 2012;19:1212–1213. doi: 10.1038/nsmb.2455. [DOI] [PubMed] [Google Scholar]
- 5.Pommier Y, Leo E, Zhang H, Marchand C. Chem. Biol. 2010;17:421–433. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gomez-Herreros F, Romero-Granados R, Zeng Z, Alvarez-Quilon A, Quintero C, Ju L, Umans L, Vermeire L, Huylebroeck D, Caldecott KW, Cortes-Ledesma F. PLoS Genet. 2013;9:e1003226. doi: 10.1371/journal.pgen.1003226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schellenberg MJ, Appel CD, Adhikari S, Robertson PD, Ramsden DA, Williams RS. Nat. Struct. Mol. Biol. 2012;19:1363–1371. doi: 10.1038/nsmb.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi K, Kurahashi K, Gao R, Tsutakawa SE, Tainer JA, Pommier Y, Aihara H. Nat. Struct. Mol. Biol. 2012;19:1372–1377. doi: 10.1038/nsmb.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adhikari S, Karmahapatra SK, Elias H, Dhopeshwarkar P, Williams RS, Byers S, Uren A, Roy R. Anal Biochem. 2011;416:112–116. doi: 10.1016/j.ab.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raymond AC, Staker BL, Burgin AB., Jr J. Biol. Chem. 2005;280:22029–22035. doi: 10.1074/jbc.M502148200. [DOI] [PubMed] [Google Scholar]
- 11.Murai J, Huang SY, Das BB, Dexheimer TS, Takeda S, Pommier Y. J. Biol. Chem. 2012;287:12848–12857. doi: 10.1074/jbc.M111.333963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bahmed K, Nitiss KC, Nitiss JL. Proc. Natl. Acad. Sci. U.S.A. 2010;107:4057–4062. doi: 10.1073/pnas.0909917107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raoof A, Depledge P, Hamilton NM, Hamilton NS, Hitchin JR, Hopkins GV, Jordan AM, Maguire LA, McGonagle AE, Mould DP, Rushbrooke M, Small HF, Smith KM, Thomson GJ, Turlais F, Waddell ID, Waszkowycz B, Watson AJ, Ogilvie DJ. J. Med. Chem. 2013;56:6352–6370. doi: 10.1021/jm400568p. [DOI] [PubMed] [Google Scholar]
- 14.Ihlenfeldt WD, Voigt JH, Bienfait B, Oellien F, Nicklaus MC. J. Chem. Inf. Model. 2002;42:46–57. doi: 10.1021/ci010056s. [DOI] [PubMed] [Google Scholar]
- 15.Sali A, Blundell TL. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 16.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roe DR, Cheatham TE. J. Chem. Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 19.Hawkins PC, Skillman AG, Warren GL, Ellingson BA, Stahl MT. J. Chem. Inf. Model. 2010;50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hawkins PC, Skillman AG, Nicholls A. J. Med. Chem. 2007;50:74–82. doi: 10.1021/jm0603365. [DOI] [PubMed] [Google Scholar]
- 21.Tresadern G, Bemporad D, Howe T. J. Mol. Graph. Model. 2009;27:860–870. doi: 10.1016/j.jmgm.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 22.Marchand C, Abdelmalak M, Kankanala J, Huang SY, Kiselev E, Fesen K, Kurahashi K, Sasanuma H, Takeda S, Aihara H, Wang Z, Pommier Y. Chem. Biol. 2016 doi: 10.1021/acschembio.5b01047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller BR, McGee TD, Swails JM, Homeyer N, Gohlke H, Roitberg AE. J. Chem. Theory Comput. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 24.Wang J, Wang W, Kollman PA, Case DA. J. Mol. Graph. Model. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 25.Gohlke H, Kiel C, Case DA. J. Mol. Biol. 2003;330:891–913. doi: 10.1016/s0022-2836(03)00610-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.