

Abstract
Background
GM2 gangliosidosis-AB variants a rare autosomal recessive neurodegenerative disorder occurring due to deficiency of GM2 activator protein resulting from the mutation in GM2A gene. Only seven mutations in nine cases have been reported from different population except India.
Case presentation
Present case is a one year old male born to 3rd degree consanguineous Indian parents from Maharashtra. He was presented with global developmental delay, hypotonia and sensitive to hyperacusis. Horizontal nystagmus and cherry red spot was detected during ophthalmic examination. MRI of brain revealed putaminal hyperintensity and thalamic hypointensity with some unmyelinated white matter in T2/T1 weighted images. Initially he was suspected having Tay-Sachs disease and finally diagnosed as GM2 gangliosidosis, AB variant due to truncated protein caused by nonsense mutation c.472 G > T (p.E158X) in GM2Agene.
Conclusion
Children with phenotypic presentation as GM2 gangliosidosis (Tay-Sachs or Sandhoff disease) and normal enzyme activity of β-hexosaminidase-A and -B in leucocytes need to be investigated for GM2 activator protein deficiency.
Keywords: GM2 gangliosidosis, GM2A gene, GM2 activator protein, AB variant
Background
GM2 gangliosides are the glycosphingolipids present in the outer layer of mammalian cells that are enriched on the neuronic surfaces [1]. In normal condition, glycosphingolipids are catabolized by lysosomal exohydrolases and are unique as they require synthesis and interaction of three-gene products;α and β subunits of lysosomal glycosidase enzyme-β-hexosaminidase (EC 3.2.1.52) and presence of a small non-enzymatic lipid binding protein as an activator [1]. This tiny glycolipid transporter GM2 activator protein (GM2AP), acts as a substrate specific cofactor for the degradation of GM2 ganglioside by the enzyme β-hexosaminidase. Hence, deficiency of any of these protein that are encoded by the HEXA, HEXB and GM2A gene causes excessive intra lysosomal accumulation of GM2 and related glycolipids especially in neuronal cells resulting in GM2 gangliodosis [2].
Mutations in the HEXA gene encoding the α-chain of (β-hexosaminidase-A) Hex-A leads to Tay-Sachs disease or B variant, while mutations in the β-chain encompassing HEXB gene leads to deficiency of both Hex-A and Hex-B (total-Hex) causing Sandhoff disease or O variant. These disorders are fairly common in the Indian population [3]-[4]. The third variant of GM2 gangliosidosis, known as AB variant (OMIM-272750), is rarely encountered and only nine cases are reported till date world wide as described in Table 1 [2], [5]-[6]. This form of GM2 gangliosidosis is indistinguishable from infantile form of Tay-Sachs disease due to its phenotypic similarity. Here we report a novel case with a review of GM2A activator protein deficiency.
Table 1.
Review of molecularly proven cases of GM2 Gangliosidosis-AB variant
| Case | Mutation | Exon | Predicted protien change | Author | Ethnicity | Year |
|---|---|---|---|---|---|---|
| 1 | c.412T>C (p.C107R)a
(Homozygous) |
3 | Reduced interaction with Hex A | Schroder et al. [15] | US Black | 1991 |
| 2 | c.412T>C (p.C138R)a
(Homozygous) |
3 | Reduced interaction with Hex A | Xie et al. [16] | US Black | 1992 |
| 3 | c.506G>C (p.R169P) (Homozygous) |
4 | Pre-matured protein degradation | Schroder et al. [17] | Indian | 1993 |
| 4 | c.262_264delAAG(p.88Kdel) (Homozygous) |
3 | Absence of mature CRM | Schepers et al. [14] | Saudi Arabia | 1996 |
| 5 | c.410delA (p.H137PfsX34) (Homozygous) |
3 | Absence of mature CRM | Schepers et al. [14] | Spanish | 1996 |
| 6 | c.160G>T (p.E54X) (Homozygous) |
2 | Absence of mRNA or CRM | Chen et al. [9] | Laotian, Hmong |
1999 |
| 7 | c.522T>G (p.L174R) (Homozygous) |
4 | Pre-matured protein degradation | Kolodny et al. [18] | Indian | 2008 |
| 8 | c. 160G>T (p.E54X) (Homozygous) |
2 | Absence of mRNA or CRM | Renaud et al. [19] | Hmong | 2015 |
| 9 | c.164C>T (p.P55L) (Homozygous) |
2 | Reduced interaction with Hex A | Salih et al. [6] | Saudi Arabia | 2015 |
| 10 | c.472G>T (p.E158X) (Homozygous) |
4 | Absence of mRNA or CRM | Present case | Indian | 2015 |
a The mutations identified by Schroder et al. (1991) (CYS107ARG) and Xie et al. (1992) (CYS138ARG) are the same but derived from different amino acid numbering systems
Clinical presentation
Proband was a third child born to young third degree consanguineous parents from Maharashtra. One elder sister died at the age of 1½ years having similar complaints.
The case under report had a normal antenatal and perinatal history and was born with a birth weight of 3.05 kg at term. At the age of 12 months, global development delay with absence of social smile and poor head control was noticed. He was detected having hypotonia with a significantly poor tone in the limbs compared to the axial movements. The visual responses seemed quite poor. Head circumference was normal and no dysmorphism was observed. The child did not show any Mongolian patches on lumbosacral area which was detected, in our earlier case series with Tay-Sachs disease [7].
On clinical examination, there was no organomegaly. Significant hyperacusis was noted even to the most trivial sounds. Fundus examination showed bilateral cherry red spot in macula and horizontal nystagmus. Magnetic Resonance Imaging (MRI) scan of brain revealed putaminal hyperintensity and thalamic hypointensity with some unmyelinated white matter in T2/T1 weighted images (Fig 1(a)-(b)), which is commonly detected in cases with GM2 gangliosidosis.
Fig. 1.
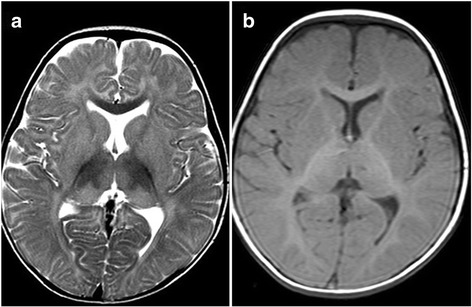
(a)-(b): Initial T2 weighted MRI pictures of brain revealed (a) putaminal hyperintensity and (b) thalamic hypointensity with some unmyelinated white matter in T2/T1 weighted images
After obtaining institutional ethics committee approval and informed written consent, further study was carried out. The blood samples of the proband and their parents were taken for the study. Clinical presentation was pointing towards GM2 gangliosidosis. Enzyme activity was determined by fluorimetric method using specific synthetic substrate. Hex-A was assayed with a sulphated substrate 4-methylumbelliferyl-N-acetyl-β-D-glucosamine-6-sulphate (MUGS) whereas total hexosaminidase (total-Hex) was measured from the hydrolysis of the synthetic substrate 4-methylumbelliferyl-Nacetyl-β-D-glucosamine (MUG) that releases fluorescent 4-methylumbelliferone when acted upon by β-hexosaminidase [8]. Enzyme activities for Hex-A [102 nmol/hr/mg protein; NR: 69.0 - 659.0 nmol/hr/mg protein] and total-Hex [414.9 nmol/hr/mg protein; NR: 288.4 – 1758.0 nmol/hr/mg protein] were found to be normal. This makes it highly unlikely for Tay-Sachs or B variant and Sandhoff disease or O variant. In presence of strong clinical presentation and normal lysosomal enzyme activity; GM2 gangliosidosis activator deficiency leading to AB variant was carried out by mutation analysis encompassing GM2 activator (GM2A) gene.
Molecular analysis was carried out by bi-directional sequencing of the coding region of GM2A gene together with intronic flanking region using the primer pairs as described earlier [9]. The amplification was carried out in a thermal cycler (ABI 2720) with 5-min denaturation at 94 °C followed by forty-three cycles each consisting of 30 s denaturation at 94 °C, 30 s of annealing at 54 °C, and 30 seconds extension at 72 °C. Final extension was carried out at 72°C for 10 min.
The GM2A gene analysis revealed a novel homozygous c.472G > T (p.E158X) nonsense mutation in exon-4confirming the diagnosis of GM2AP deficiency leading toGM2 gangliosidosis, AB variant (Fig. 2). Parents were found to be heterozygous for the same mutation. The deleterious effect of the mutation was further confirmed using bioinformatics tool; Mutation taster program.
Fig. 2.
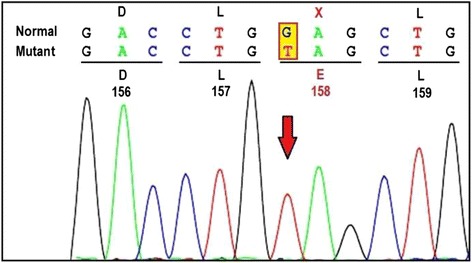
Bi-directional sequence chromatogram detected homozygous nonsense mutation viz. c.472 G > T (p.E158X) in exon-4 of GM2A gene
Discussion
GM2 activator deficiency as a cause of GM2 gangliosidosis is a rarely diagnosed sphingolipid disorder. Conzelmann and Sandhoff (1978) were the first one to present an evidence of defective activating factor necessary for degradation of GM2 ganglioside [10]. This was followed by demonstration of GM2 activator protein deficiency in liver cells in Non-Jewish child from England by Hechtmen et al. in 1982 [11]. Subsequently nine molecularly proven cases of GM2 activator deficiency from different parts of the world as summarized in Table 1 have been reported. Here we present tenth case having a novel mutation in GM2A gene. Clinically, proband was indistinguishable from classical infantile Tay-Sachs disease. However, due to the normal activity of Hex-A and total-hex enzymes in leucocytes, the c.DNA analysis of GM2A gene was carried out. The case under study showed homoallelic nonsense mutation (p.E158X) in GM2A gene, due to replacement of nucleotide Guanine (G) by Thymine (T) at c.DNA position 472 (c.472G > T). This is a novel pathogenic variant found to be responsible for a GM2AP deficiency in our patient with AB variant of GM2 gangliosidosis.
To the best of our knowledge, this mutation (p.E158X) is neither reported in any ethnic group, nor present in the dbSNP database (http://www.ncbi.nlm.nih.gov/SNP/). This variation has not been reported in the 1000 genome database and this region is conserved across species. In-silico analysis using the Mutation taster program suggests a probably damaging nature of the mutation. A subunit lacking the last 36 C-terminal residues is likely to make the protein non-functional. This premature stop codon is likely to have produced a truncated protein as the stop codon occurred near 3’end of the gene, which leads to unstable mRNAs. It is also likely that the proteins synthesized in the endothelium reticulum (ER) undergo a quality control check by the resident ER system that recognizes abnormal proteins and degrades them [12]. Though functional study has not been carried out in our case it is likely that nonsense point mutation in the present case seems to have produced a premature stop codon thus affecting the stability of the mutated protein or its Hex-A binding capabilities [13] or perhaps leading to early degradation in the ER or Golgi bodies [14], thus portraying the clinical presentation.
Only seven mutations in nine patients have been documented in the GM2A gene till date and all patients were found to be homozygous for individual mutant alleles associated with complete absence of GM2 ganglioside cleavage [2], [5, 6, 9, 14–19]. Earlier published reports have shown steady state levels of activator mRNA but none were found to have detectable activator cross reacting material (CRM) in patient’s cells except in a study reported by Chen et al. (1999) [9] where they have shown absence of detectable steady state mRNA nor any CRM.
The homoallelic nonsense mutation detected in the child was confirmed by the heterozygous state of both parents. This could be one of the reasons for the early death of the elder sister. Based on this, family was provided genetic counseling about the future recurrence risks of the disease and prevention by prenatal diagnosis.
Conclusion
It can be concluded from this case that children with global developmental delay and unmyelinated white matter with normal hexosaminidase study need to be further investigated for GM2 activator protein deficiency.
Abbreviations
CRM, Cross reacting material; ER, Endothelium reticulum; GM2AP, GM2 activator protein; Hex-A, β-hexosaminidase-A; Hex-T, β-hexosaminidase-total; mRNA, messenger RNA; MUG, 4-methylumbelliferyl-Nacetyl-β-D-glucosamine; MUGS, 4-methylumbelliferyl-N-acetyl-β-D-glucosamine-6-sulphate
Acknowledgements
We express our thanks to patient and his parents for their support.
Funding
This work is partly supported by Department of Health Research/Indian Council of Medical Research [grant no.: GIA/31(ii)/2014-DHR]. Funding agency was not involved in the study design, specimen collection, analysis, interpretation of the data and preparation of the manuscript.
Availability of data and materials
The dataset supporting the conclusion of this article is available in the ClinVar repository
Clinvar submission ID for GM2A gene
SUB1652067; c.472T→G (p.E158X)
https://submit.ncbi.nlm.nih.gov/subs/clinvar_wizard/SUB1652067/overview
Author’s contributions
JS and MM designed the experiment and standardized the protocols. MM and RB were involved in processing of the samples. CD was involved in collection of the clinical details. JS, CD, MM, FS and KS prepared the manuscript. All the authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests (financial or non-financial).
Consent for publication
Informed written consent was obtained from all the participants for publication of their clinical details and/or clinical images. A copy of the written consent is available for review by the editor of this journal.
Ethics approval and consent to participate
Present case under submission has been approved by the institutional ethics committee [FRIGE’s Institute of Human Genetics] wide approval number FRIGE/IEC/5/2010 dated 7th March, 2010. This process is in accordance with the Helsinki declaration.
An informed consent was obtained from the parents before enrolling for the investigations [This was in accordance with the requirement of the institutional ethics committee].
An informed consent for publication was also obtained from the individuals included in the submission [This was in accordance with the requirement of the institutional ethics committee].
Contributor Information
Jayesh Sheth, Phone: +91 79 26921414/65128444, Email: jshethad1@gmail.com.
Chaitanya Datar, Email: cd@sahyadrihospitals.com.
Mehul Mistri, Email: mistrimehul@yahoo.co.in.
Riddhi Bhavsar, Email: riddhib4@gmail.com.
Frenny Sheth, fshethad1@googlemail.com.
Krati Shah, Email: drkratishah@gmail.com.
References
- 1.Sandhoff K, Harzer K. Gangliosides and gangliosidoses: principles of molecular and metabolic pathogenesis. J Neurosci. 2013;33(25):10195–208. doi: 10.1523/JNEUROSCI.0822-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kolter T, Sandhoff K. Sphingolipid metabolism diseases. Biochim Biophys Acta. 2006;1758(12):2057–79. doi: 10.1016/j.bbamem.2006.05.027. [DOI] [PubMed] [Google Scholar]
- 3.Sheth J, Mistri M, Sheth F, Shah R, Bavdekar A, Godbole K. Burden of Lysosomal storage disorders in India: Experience of 387 affected children from a single diagnostic facility. JIMD Rep. 2014;12:51–63. doi: 10.1007/8904_2013_244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nalini A, Christopher R. Cerebral glycolipidoses: clinical characteristics of 41 pediatric patients. J Child Neurol. 2014;19(6):447–452. doi: 10.1177/088307380401900610. [DOI] [PubMed] [Google Scholar]
- 5.Mahuran D. Biochemical consequences of mutations causing the GM2 gangliosidoses. BiochimBiophys Acta. 1999;1455(2–3):105–38. doi: 10.1016/s0925-4439(99)00074-5. [DOI] [PubMed] [Google Scholar]
- 6.Salih MA, Seidahmed MZ, El Khashab HY, Hamad MH, Bosley TM, Burn S, et al. Mutation in GM2A Leads to a Progressive Chorea-dementia Syndrome. Tremor Other Hyper kinet Mov (NY) 2015;5:306. doi: 10.7916/D8D21WQ0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheth J, Mistri M, Datar C, Kalane U, Patil S, Kamate M. Expanding the spectrum of HEXA mutations in Indian patients with Tay–Sachs disease. Mol Genet Metab Rep. 2014;1:425–430. doi: 10.1016/j.ymgmr.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wendeler M, Sandhoff K. Hexosaminidase assays. Glycoconj J. 2009;26(8):945–52. doi: 10.1007/s10719-008-9137-5. [DOI] [PubMed] [Google Scholar]
- 9.Chen B, Rigat B, Curry C, Mahuran D. Structure of the GM2A Gene: Identification of an Exon 2 Nonsense Mutation and a Naturally Occurring Transcript with an In-Frame Deletion of Exon 2. Am J of Hum Genet. 1999;65(1):77–87. doi: 10.1086/302463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conzelmann E, Sandhoff K. AB variant of GM2 gangliosidosis: Deficiency of a father necessary for stimulation of hexosaminidase A-catalyzed degradation of gangliosides GM2 and glycolipid GA2. Proc Natl Acad Sci. 1978;75(8):3979–83. doi: 10.1073/pnas.75.8.3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hechtman P, Gordon BA, Ng Ying Kin NM. Deficiency of the Hexosaminidase A Activator protein in a case of GM2 Gangliosidosis. Variant AB. Pediatr Res. 1982;16:217–22. doi: 10.1203/00006450-198203000-00011. [DOI] [PubMed] [Google Scholar]
- 12.Henderson B, Nair SP, Coates ARM. Molecular Chaperones and Diseases. Inflamm Res. 1996;45(4):155–8. doi: 10.1007/BF02285154. [DOI] [PubMed] [Google Scholar]
- 13.Xie B, Rigat B, Smiljanic-Georgijev N, Deng H, Mahuran D. Biochemical Characterization of the Cys138Arg Substitution Associated with the AB Variant Form of GM2 Gangliosidosis: Evidence That Cys138Is Required for the Recognition of the GM2 Activator/GM2 Ganglioside Complex by β-Hexosaminidase A. Biochemistry. 1998;37(3):814–21. doi: 10.1021/bi971211s. [DOI] [PubMed] [Google Scholar]
- 14.Schepers U, Glombitza G, Lemm T, Hoffmann A, Chabas A, Ozand P. Molecular analysis of a GM2-activator deficiency in two patients with GM2-gangliosidosis AB variant. Am J of Hum Genet. 1996;59(5):1048–56. [PMC free article] [PubMed] [Google Scholar]
- 15.Shroder M, Schnabel D, Suzuki K, Sandhoff K. A mutation in the gene of a glycolipid-binding protein (GM2 activator) that causes GM2-gangliosidosis variant AB. FEBS Lett. 1991;290(1–2):1–3. doi: 10.1016/0014-5793(91)81211-P. [DOI] [PubMed] [Google Scholar]
- 16.Xie B, Wang W, Mahuran DJ. A cys138-to-arg substitution in the GM2 activator protein is associated with the AB variant form of GM2 gangliosidosis. Am J Hum Genet. 1992;50:1046–52. [PMC free article] [PubMed] [Google Scholar]
- 17.Schroder M, Schnabel D, Hurwitz R, Young E, Suzuki K, Sandhoff K. Molecular genetics of GM2-gangliosidosis AB variant: a novel mutation and expression in BHK cells. Hum Genet. 1993;92:437–440. doi: 10.1007/BF00216446. [DOI] [PubMed] [Google Scholar]
- 18.Kolodny E, Sathe S, Zeng BJ, Torres P, Alroy J, Pastores G. A novel GM2-activator deficiency mutation as a cause of AB variant GM2-Gangliosidosis. Mol Genet Metab. 2008;93(2):27–28. [Google Scholar]
- 19.Renaud D, Brodsky M. GM2-Gangliosidosis, AB Variant: Clinical, Opthalmological, MRI, and Molecular Findings. JIMD Rep. 2015 [Epub ahead of print] [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The dataset supporting the conclusion of this article is available in the ClinVar repository
Clinvar submission ID for GM2A gene
SUB1652067; c.472T→G (p.E158X)
https://submit.ncbi.nlm.nih.gov/subs/clinvar_wizard/SUB1652067/overview