

Abstract
Plasma cell leukemia (PCL) is an uncommon neoplasm of plasma cells, with an aggressive clinical course and poor outcome, even with current standard of care. It can occur either de novo (primary PCL) or as a progression of multiple myeloma (MM). This disease has unique diagnostic criteria but certain genetic markers and clinical features may overlap with MM. Due to the low prevalence of PCL, guidelines on its management are extrapolated from the management of MM and based on small retrospective studies and cases reports/series. We present an interesting case of PCL in a middle-aged African-American male, who was diagnosed incidentally after chest wall imaging for an unrelated complaint. The diagnostic approach, management and outcomes of PCL are discussed.
Keywords: Plasma cell leukemia, Multiple myeloma, Chest wall mass, Tumor lysis
Introduction
Multiple myeloma (MM) is a neoplasm of plasma cells accounting for 10–15% of hematopoietic neoplasms. It is more common in people of African descent, occurring twice as frequently compared to Caucasians [1]. Plasma cell leukemia (PCL) is a very rare type of MM characterized by high numbers (>20%) of circulating plasma cells in the peripheral blood. It comprises of an estimated 2–4% of cases of MM [2] and is an aggressive disease with poor prognosis [3]. The majority of cases occur de novo as primary PCL but it may also occur as leukemic transformation of underlying MM. Because of the rarity of this disease, knowledge of its presentation and clinical course has been largely dependent on small studies and case reports [4]. The clinical presentation typically involves symptoms attributed to end organ damage seen in MM (hypercalcemia, renal failure, anemia, and lytic bone lesions) or to leukemia (leukocytosis, thrombocytopenia and organomegaly). We report a case of PCL with atypical presentation as a chest wall mass and discuss the diagnostic approach as well as treatment options.
Case Report
A 65-year-old African-American male with a history of type 2 diabetes mellitus and inguinal and abdominal wall hernia presented with complaints of abdominal pain located in the left inguinal region, brought on by heavy lifting. Examination was significant for pallor, hepatomegaly and mild abdominal tenderness at the site of the inguinal hernia. CT scans of the abdomen and chest were notable for hepatosplenomegaly (fig. 1a) and a left chest wall mass with destruction of the underlying rib (fig. 1b). An ultrasound-guided core needle biopsy of the chest mass, showed a diffuse monotonous population of small to medium-sized lymphocytes with plasmacytoid features (fig. 2). The tumor cells were positive for CD138 and CD56 with monoclonal lambda restriction by in situ hybridization and showed a high proliferation index with Ki67 (60%). The cells were negative for CD20 and CD79a. A diagnosis of high-grade plasma cell neoplasm involving soft tissue was made. Laboratory investigation was significant for elevated serum calcium of 12.2 g/dl (reference 8.5–10.6) with normal parathyroid hormone level and evidence of acute renal failure [creatinine 1.9 mg/dl (reference 0.6–1.2)]. A complete blood count revealed mild leukocytosis [11.2 × 109/l (reference 3.2–10.6 × 109)], macrocytic anemia with a hemoglobin of 8.0 g/dl (reference 14.6–17.8) and mean corpuscular volume 101.9 fl (reference 77–94). The platelet count was normal. Serum protein electrophoresis and immunofixation showed a monoclonal spike with IgG lambda, and quantitative IgG levels were markedly elevated at 3,139 mg/dl (reference 791–1,643).
Fig. 1.
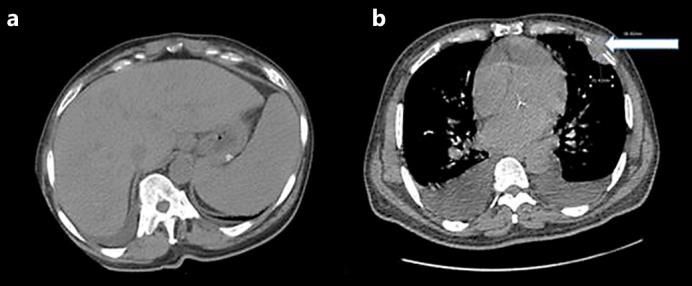
a CT scan of the abdomen showing marked hepatosplenomegaly. b CT scan of the chest showing a left chest wall mass with bony destruction of the left anterior fifth rib (arrow) and bilateral pleural effusions.
Fig. 2.
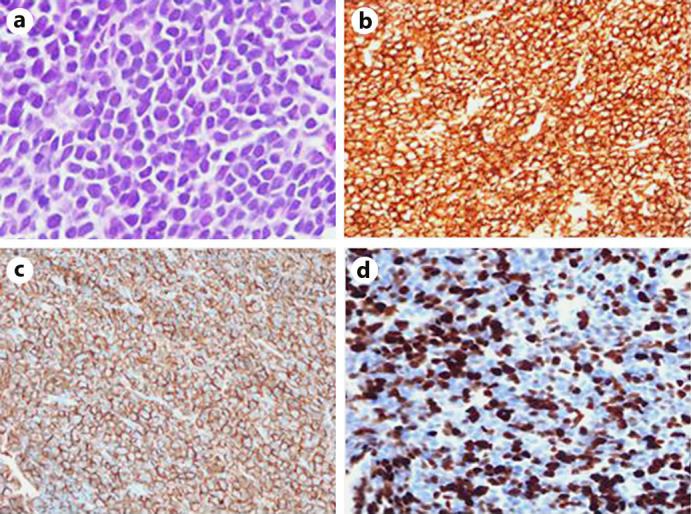
a Tissue morphology of chest wall mass demonstrating sheets of neoplastic cells. Cells were positive for CD138 (b) with aberrant CD56 expression (c) and a very high proliferative rate with Ki-67 (d).
A review of his peripheral blood smear showed 25–35% circulating atypical lymphocytes with plasmacytoid features (fig. 3), confirmed to be neoplastic plasma cells by flow cytometry and consistent with PCL. A bone marrow biopsy revealed marked infiltration with monoclonal plasma cells comprising >50% of total bone marrow cellularity. The diagnosis of PCL with extramedullary (chest wall) involvement was confirmed. Prior to the initiation of chemotherapy, the patient spontaneously developed hyperkalemia, worsening renal failure; creatinine 3.2 mg/dl and elevated uric acid to 8.9 mg/dl (reference 4–8.5), suggestive of tumor lysis syndrome. He was managed with i.v. hydration, allopurinol and rasburicase with good response. His course was further complicated by worsening leukocytosis, severe thrombocytopenia and hypercalcemia.
Fig. 3.
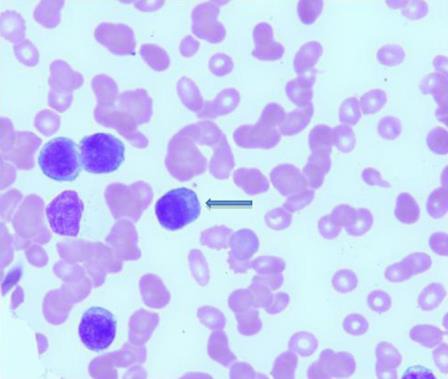
Peripheral blood smear. These cells with plasmacytoid morphology comprised 25–35% of the cells in the peripheral blood (arrow).
The patient was referred for clinical trial where he was given induction chemotherapy with CyBorD (cyclophosphamide, bortezomib, and dexamethasone) on days 1, 8, 15, and 22 of each 28-day cycle for a total of 4 cycles. Intrathecal chemotherapy was also administered for central nervous system prophylaxis. Patient was subsequently referred for stem cell transplant.
Discussion
PCL was first reported in 1906 in a patient who presented with bone pain, a palpable rib mass, rib fractures, anemia, and splenomegaly [5, 6]. Interestingly, this parallels our patient, in whom PCL was only diagnosed incidentally after presenting with abdominal pain, and then found to have hepatosplenomegaly and a neoplastic chest wall mass.
Although PCL lies in the spectrum of plasma cell dyscrasias (of which MM is most common), there are some distinguishing features of the disease. PCL tends to present with extramedullary involvement and very little bone involvement. A review of the literature revealed cases manifesting as lymphadenopathy, hepatomegaly, splenomegaly, leptomeningeal myelomatosis, and soft tissue tumors [6, 7]. In contrast, bone involvement with resultant lytic lesions, bone pain and pathological fractures is the predominant feature in MM. Patients with PCL are usually younger with a median age of 55, whereas that for MM patients is 65 [8].
The diagnosis of PCL is based on laboratory parameters. According to the consensus statement by the International Myeloma Working Group [4], PCL is defined by the presence of >20% circulating plasma cells and/or an absolute plasma cell count >2 × 109/l. As the diagnosis hinges on establishing an increased number of plasma cells in the peripheral blood, it is important to note that circulating plasma cells in PCL are often small with scant cytoplasm resembling plasmacytoid lymphocytes, and can easily be missed on automated differentials, especially when the white blood cell count is only minimally elevated. In the setting of organomegaly, soft tissue mass and hypercalcemia, serum and urine protein electrophoresis with immunofixation should be obtained to identify a monoclonal immunoglobulin. A careful review of the peripheral smear should then be performed with peripheral blood flow cytometry to confirm the diagnosis of PCL. Skeletal surveys can help in establishing bone involvement. A bone marrow biopsy with cytogenetics should be done in all patients diagnosed with PCL. Any soft tissue mass identified should be biopsied to evaluate for possible extramedullary involvement.
PCL is a highly aggressive tumor, with a high disease burden and proliferation index. As a result, a feared and potentially fatal complication is tumor lysis syndrome. This has been reported after commencing chemotherapy [9], but it may occur, as with our patient, even prior to chemotherapy. It is important to identify patients who are at risk and closely monitor for markers of cell lysis: elevated serum potassium, lactate dehydrogenase and phosphorus, and a fall in serum calcium. Elevated uric acid levels can cause uric acid nephropathy, a predominant cause of acute renal failure in tumor lysis syndrome. Due to the rare nature of PCL, there is no standard treatment as randomized studies are not available and treatment data are extrapolated from MM studies [4]. The clinical outcome following chemotherapy with conventional agents has been shown to be much worse in PCL compared to MM. New therapeutic strategies utilizing agents such as lenalidomide, bortezomib, and dexamethasone (RVD), melphalan, prednisone, bortezomib, and thalidomide (VMPT) and bortezomib, thalidomide, and dexamethasone (VTD) have shown encouraging outcomes [6].
The overall prognosis is poor with a median survival of 6–11 months [8, 10], but some improvements in outcomes have been shown with newer therapies and stem cell transplantation. In a study of 445 patients with primary PCL, using the SEER database and comparing overall survival in patients diagnosed between 1973 and 2005 to those diagnosed between 2006 and 2009, there was an increase in overall survival from 5 to 12 months. [11].
Since virtually all patients with PCL who attain a complete remission develop relapse, maintenance chemotherapy should be considered [12, 13]. Agents such as lenalidomide, bortezomib, and thalidomide have been used for maintenance therapy [6]. Moreover, because of the high rate of recurrence and poor outcome with chemotherapy alone, consolidation with hematopoietic stem cell transplant provides the best hope for remaining in complete remission [14]. Finally, patients with PCL, whenever possible, should be offered the opportunity to participate in clinical trials [6].
Conclusion
We report a case of PCL presenting in an unusual manner. Prompt diagnostic evaluation of a chest wall mass followed by peripheral smear evaluation led to timely diagnosis of PCL and early initiation of therapy. PCL has varied clinical presentation; as such, increased awareness and thorough evaluation with particular attention to the peripheral smear is paramount to establishing the diagnosis. Tumor lysis syndrome can complicate this aggressive disease; therefore, there should be close monitoring of renal function. Due to the low prevalence of PCL, the chemotherapy regimens used are largely based on those used for MM. It is important to report outcomes of therapies/regimens used to help establish guidelines for the management of PCL, pending a randomized clinical trial. Patients should also get early referral to a bone marrow transplant center, as there is a higher likelihood of achieving and maintaining complete remission with combined chemotherapy followed by hematopoietic stem cell transplantation.
Statement of Ethics
The authors have no ethical conflicts to disclose.
Disclosure Statement
The authors state no conflict of interests and have received no payment in the preparation of this paper.
References
- 1.Waxman AJ, Mink PJ, Devesa SS, Anderson WF, Weiss BM, et al. Racial disparities in incidence and outcome in multiple myeloma: a population based study. Blood. 2010;116:5501–5506. doi: 10.1182/blood-2010-07-298760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bladé J, Kyle RA. Nonsecretory myeloma, immunoglobulin D myeloma, and plasma cell leukemia. Hematol Oncol Clin North Am. 1999;13:1259–1272. doi: 10.1016/s0889-8588(05)70125-8. [DOI] [PubMed] [Google Scholar]
- 3.Ramsingh G, Mehan P, Luo J, Vij R, Morgensztern D. Primary plasma cell leukemia: a Surveillance, Epidemiology, and End Results database analysis between 1973 and 2004. Cancer. 2009;115:5734–5739. doi: 10.1002/cncr.24700. [DOI] [PubMed] [Google Scholar]
- 4.De Larrea CF, Kyle RA, Durie BG, Ludwig H, Usmani S, et al. Plasma cell leukemia: consensus statement on diagnostic requirements, response criteria, and treatment recommendations by the International Myeloma Working Group. Leukemia. 2013;27:780–791. doi: 10.1038/leu.2012.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gluzinski A, Reichentein M. Myeloma und leucaemia lymphatica plasmocellularis. Wien Klin Wochenschr. 1906;19:336. [Google Scholar]
- 6.Van de Donk NWCJ, Lokhorst HM, Anderson KC, Richardson PG. How I treat plasma cell leukemia. Blood. 2012;120:2376–2389. doi: 10.1182/blood-2012-05-408682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singla D, Rathod GB. Primary plasma cell leukemia in 32 years old male – a case report. IAIM. 2015;2:42–45. [Google Scholar]
- 8.Tiedemann RE, Gonzalez-Pazet N, Kyle RA, Santana-Davila R, Price-Troska T, et al. Genetic aberrations and survival in plasma cell leukemia. Leukemia. 2008;22:1044–1052. doi: 10.1038/leu.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaskiewicz AD, Herrington JD, Wong L. Tumor lysis syndrome after bortezomib therapy for plasma cell leukemia. Pharmacotherapy. 2005;25:1820–1825. doi: 10.1592/phco.2005.25.12.1820. [DOI] [PubMed] [Google Scholar]
- 10.Noel P, Kyle RA. Plasma cell leukemia: an evaluation of response to therapy. Am J Med. 1987;83:1062–1068. doi: 10.1016/0002-9343(87)90942-9. [DOI] [PubMed] [Google Scholar]
- 11.Gonsalves WI, Rajkumar SV, Go RS, Dispenzieri A, Gupta V, et al. Trends in survival of patients with primary plasma cell leukemia: a population-based analysis. Blood. 2014;124:907–912. doi: 10.1182/blood-2014-03-565051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D'Arena G, Valentini CG, Pietrantuono G, Guariglia R, Martorelli MC, et al. Frontline chemotherapy with bortezomib-containing combinations improves response rate and survival in primary plasma cell leukemia: a retrospective study from GIMEMA Multiple Myeloma Working Party. Ann Oncol. 2012;23:1499–1502. doi: 10.1093/annonc/mdr480. [DOI] [PubMed] [Google Scholar]
- 13.Beyer K, Rosner S, Woo KM, Devlin SM, Landau H, et al. Analysis of VDT-PACE Utilization in Multiple Myeloma Patients Treated at MSKCC for Relapsed Disease or Cytoreduction and Stem Cell Mobilization after Initial Induction Therapy. https://ash.confex.com/ash/2014/webprogram/Paper72932
- 14.International Myeloma Working Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003;121:749–757. [PubMed] [Google Scholar]