

Abstract
Background
Primary pleuropulmonary and mediastinal synovial sarcomas (PPMSSs) are extremely rare. The authors present the largest series in an Asian population.
Methods
Between 2000 and 2015, 26 genetically confirmed PPMSSs were included. The clinicopathologic features of all of the cases were reviewed. Immunohistochemical staining was carried out using the following antibodies: TLE1, cytokeratin (AE1/AE3), EMA, CD99, Bcl-2, CK7, CD34, S-100 protein, and Ki-67. The chromosomal translocation t(X;18)(p11.2;q11.2) was detected by fluorescence in situ hybridization (FISH) and reverse transcription polymerase chain reaction (RT-PCR). We compared the clinical, pathologic, immunohistochemical, and molecular features of this series with that of the previous series and soft tissue synovial sarcomas.
Results
This series included 17 males and nine females. The median age was 36.5 years (range, 16–72 years). The tumors involved the lung (76.9 %), pleura (15.4 %), and mediastinum (7.7 %). The median tumor size was 6 cm (range 2.3 ~ 24 cm). The majority of the tumors were well-circumscribed. The tumors were classified as monophasic (84.6 %), biphasic (3.8 %), and poorly differentiated (11.5 %) types. The tumors were graded as French Federation of Cancer Centers (FNCLCC) grade 2 (62.5 %) and FNCLCC 3 (37.5 %). Diffuse immunostaining for TLE1, BCL-2, and CD99 was identified in 91.7, 95.7, and 56.0 % of the tumors, respectively. Focal positivity was seen with EMA (84.6 %), CK7 (55.6 %), cytokeratin (AE1/AE3) (68.0 %), CD34 (5.0 %), and S-100 protein (21.7 %). A high Ki-67 index (≥10 %) was observed in 91.3 % of the tumors. The fusion transcripts included SS18-SSX1 (15/22, 68.2 %), SS18-SSX2 including variants (6/22, 27.3 %), and SS18-SSX4 (1/22, 4.5 %) fusions. The remaining four cases showed positivity for SS18 rearrangement by FISH. Surgical excision of tumors or lobectomy were performed in 20 patients, and seven of the patients underwent adjuvant therapy. Clinical follow-up was available in 73.1 % cases, with a median follow-up of 12.0 months. The median survival time was 14.5 months. Tumor resection (p = 0.024) and no residual tumor (p = 0.004) were associated with an improved overall survival time.
Conclusions
PPMSS is a highly aggressive neoplasm. Extensive surgical resection of the tumor and more effective adjuvant therapy should be advocated. PPMSS must be differentiated from similar diseases.
Keywords: Synovial sarcoma, Pleuropulmonary, Mediastinum, SS18-SSX fusion transcript, Prognosis
Background
Synovial sarcoma (SS) is a morphologically, clinically, and genetically well-defined soft-tissue neoplasm. SSs are characterized by the t(X;18)(p11.2;q11.2) translocation, which leads to SS18-SSX gene fusion [1, 2], and extremely rare neoplasms harbor SS18L1-SSX1, resulting from t(X;20) [3, 4]. Most tumors occur in the extremities near the joints, followed by the trunk and head and neck regions [1, 2]. Primary SSs have also been reported to arise from a variety of unusual locations, such as the mediastinum, retroperitoneum, and various viscera [1, 2].
Primary pleuropulmonary and mediastinal SSs (PPMSSs) are extremely unusual and have gradually been recognized as a clinicopathological entity [4, 5] since this type of peculiar tumor has been described in the literature over the past 26 years [6]. A diagnosis of PPMSS is difficult for clinicians and pathologists because of the rare nature of the tumor.
To the best of our knowledge, there have been 13 series consisting of five or more cases of PPMSSs in the English literature [7–19], and only seven of these studies were genetically confirmed. Among these t(X;18)(p11.2;q11.2)-positive series, five of them were confirmed by reverse transcription polymerase chain reaction (RT-PCR) [8, 12–14, 16], and the remaining two series were confirmed by fluorescence in situ hybridization (FISH) [15] and classical cytogenetics [11], respectively.
To better understand the clinicopathologic spectrum of these rare lesions, the authors describe a series of 26 genetically confirmed PPMSSs at one of the largest medical centers in China. This series is the third largest in the English literature and is the largest population-based analysis in an Asian population.
Methods
Patients
This study was approved by the West China Hospital Institutional Review Board. A SNOMED search of the hospital surgical pathology files from January 2000 to May 2015 identified 489 spindle cell tumors located in the pleuropulmonary and mediastinal area, whereas 24265 lung carcinomas were detected during this period. The tumors were reviewed by two pathologists with soft tissue tumor pathology expertise (H.Z. and H.C) and three general surgical pathologists (T.L., B.X., and T.Z). Two hundred and seventy-seven of the spindle cell tumors were sarcomas. Eighty-seven metastatic sarcomas (including 28 SSs) were excluded, and 190 of 277 spindle cells sarcomas were primary tumors. SS was the most common primary sarcoma (28/190, 14.7 %) in this location, followed by liposarcoma (14/190, 7.4 %), Ewing sarcoma (13/190, 6.8 %), leiomyosarcoma (13/190, 6.8 %), and chondrosarcoma (11/190, 5.8 %). Finally, 26 t(X;18)(p11.2;q11.2) -positive PPMSSs were included in this study.
Radiology methods
All the available images were reviewed by a radiologist with thoracic and oncologic imaging expertise (J.Y.). Several parameters were evaluated, including the margin, pleural effusion, mediastinal shift, tumor heterogeneity, enhancement, presence of calcification, necrosis/cystic change and lymphadenopathy.
Histologic evaluation
The tumors were classified into monophasic, biphasic, and poorly differentiated types according to the criteria proposed by the World Health Organization [1] and Enzinger and Weiss's Soft Tissue Tumors [2]. Grading was performed following the French Federation of Cancer Centers (FNCLCC) grading system [20].
Immunohistochemistry
Immunohistochemical staining was carried out on formalin-fixed paraffin-embedded (FFPE) tissue, using the EnVision Plus detection system (DAKO, Carpinteria, CA) with controls. Standard immunohistochemical studies were performed using the following antibodies: TLE1, cytokeratin (AE1/AE3), EMA, CD99, Bcl-2, CK7, CD34, S-100 protein, and Ki-67 (Table 1). The staining intensity of TLE1 was graded as “3+”, “2+”, “1+” or “-” (negative) according to Terry et al. [21]. The index of Ki-67 ≥ 10 % was considered high [22].
Table 1.
Antibodies performed and subcellular distribution
| Antibody | Laboratory | Clone | Dilution | Subcellular distribution |
|---|---|---|---|---|
| TLE1 | Santa Cruz Biotechnology, USA | polyclonal M-101 | 1:100 | Nucleus |
| cytokeratin | Dako, Carpintera CA | monoclonal AE1/AE3 | 1:200 | Cytoplasm/Membrane |
| EMA | Dako, Carpintera CA | monoclonal E29 | 1:100 | Cytoplasm/Membrane |
| CD99 | Dako, Carpintera CA | monoclonal 12E7 | 1:100 | Cytoplasm/Membrane |
| Bcl-2 | Dako, Carpintera CA | monoclonal 124 | 1:200 | Membrane |
| CK7 | Dako, Carpintera CA | monoclonal OV-TL 12/30 | 1:100 | Cytoplasm |
| CD34 | Buffalo Grove, IL | monoclonal QBEnd/10 | 1:50 | Cytoplasm |
| S-100 protein | Dako, Carpintera CA | polyclonal anti-S-100 | 1:400 | Cytoplasm |
| Ki-67 | Dako, Carpintera CA | monoclonal MIB-1 | 1:100 | Nucleus |
When staining for TLE1, 39 nonsynovial sarcomas were selected as the control samples, including ten solitary fibrous tumors (SFTs), six spindle cell carcinomas, ten type A thymomas, ten malignant mesotheliomas, and three type I pleuropulmonary blastomas.
Fluorescence in situ hybridization (FISH)
All the 26 tumors had material available for FISH studies. We used a commercially available Vysis LSI SS18 Dual Color Break Apart Probe (Abbott Molecular, Des Plaines, IL, USA) for SS18 on chromosome 18q11.2. The first probe was labeled in spectrum orange (telomeric, 5’ to SS18, 650 Kb), whereas the second probe was labeled in spectrum green (centromeric, 3’ to SS18, 1040 Kb). The FISH analyses were performed according to the manufacturer-provided protocol. FISH assays were carried out on 4-μm-thick FFPE tissue sections. The sections were deparaffinized in xylene twice for 30 min, and dehydrated in 100 % ethanol twice for 5 min. The sections were pretreated using the Paraffin Pretreatment Kit (Vysis). Tissue sections were then digested with Digest All-3 (Zymed, San Francisco, CA, USA) twice for 5 min. Tissue sections were denatured at 83 °C for 5 min and hybridized overnight at 42 °C in a humidified chamber. The slides were then washed with 0.3 % NP40/2 × SSC at 73 °C for 2 min, and again with 0.1 % NP40/2 × SSC at room temperature for 2 min. Slides were counterstained with 2.0 μg/ml of 4',6-diamidino-2-phenylindole (Abbott Molecular, Des Plaines, IL, USA). Tumor samples were scored by two investigators in 100 cells in a blind fashion in each case. A split signal pattern was considered positive for the gene rearrangement if the distance between the green and the red signals were greater than the diameter of any two signal. A case was considered positive for rearrangement when 10 % or more of the cells showed split apart signals.
Reverse transcription polymerase chain reaction (RT-PCR)
Using reverse transcriptase polymerase chain reaction (RT-PCR) analysis, 23 cases with enough material were analyzed for the presence of the fusion genes SS18-SSX1, SS18-SSX2, and SS18-SSX4. Total RNA was isolated from 10-μm sections of FFPE tissue material using the High Pure FFPE RNA Micro Kit (Qiagen, Valencia, CA), according to the manufacturer’s instructions. To eliminate the contamination of genomic DNA, RNA samples were treated with DNase I. RNA reverse-transcription into cDNA was performed using the Transcriptor First Strand cDNA Synthesis kit (Roche Applied Science, Indianapolis, IN) for 1 h at 42 °C and 5 min at 85 °C using SSX-1/2-b reverse primer: 5’CATTTTGTGGGCCAGATGC3’. All polymerase chain reactions (PCRs) were performed for 30 cycles using the GoTaq DNA polymerase with GoTaq green buffer (Promega, Madison, WI) with the following cycle conditions: denaturation at 94 °C for 7 minute, annealing at 58 °C for 30 s, and extension at 72 °C for 30 s. The primers were used in the following combinations: SS18-SSX consensus forward primer: 5'-agaccaacacagcctggaccac; SS18-SSX1–specific reverse primer: 5'-acactcccttcgaatcattttcg; SS18-SSX2–specific reverse primer: 5'-gcacttcctccgaatcatttc and SS18-SSX4–specific reverse primer: 5'-gcacttccttcaaaccattttct. Normal lung tissue was used as the negative control, and glyceraldehyde-3-phosphated ehydrogenase (GAPDH) as the reference gene. Products of classic fusion gene were 108 bp. The PCR products were gel purified by using the QIAquick Gel Extraction kit (Qiagen, Valencia, CA) and sequenced by using an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA).
Statistical analysis
Summary statistics were obtained using standard methods. Survival analyses were computed by the Kaplan-Meier method. Gender, age at presentation (<37 years old, ≥37 years old), tumor size (<5 cm, ≥5 cm), FNCLCC grade, mitotic rate (<10/10 high power filed, ≥10/10 high power filed), gene fusion type (SS18-SSX1, SS18-SSX2), tumor resection (yes, no) and residual tumor status (yes, no; assessed microscopically) (residual tumor was defined as treatment without tumor resection or resected tumors with positive margins) were considered to have prognostic value, and the log-rank test was used to compare the two aspects of the different groups. Differences were considered statistically significant when P < 0.05. SPSS 22.0 statistical software (IBM Corp, Armonk, NY, USA) was used for all statistical analysis.
Results
Clinical findings
The clinical findings are summarized in Table 2. This study included 17 males and nine females (ratios, 1.9:1.0) aged from 16 to 72 years (median, 36.5 years; mean, 37.8 years). The most common symptoms at presentation were cough, chest pain and dyspnea (24/26, 92.3 %), and the remaining two patients were found incidentally. The tumors involved the lung (20/26, 76.9 %), pleura (4/26, 15.4 %), and mediastinum (2/26, 7.7 %). The tumor size ranged from 2.3 to 24 cm (median, 6 cm; mean, 8.5 cm). None of the patients showed any evidence of primary malignancy elsewhere at the time of diagnosis. Smoking information was available for 18 patients, and 38.9 % (7/18) of the patients had a history of smoking.
Table 2.
Clinical and treatment features of 26 PPMSSs
| Case NO. | Age/Gender | Symptoms/preop course at initial Op | Smoking | Size (CM) | Location | Microscopically invasion | Microscopically margin | Lymph node metastasis | Treatment | Progression/interval | Outcome/Follow-up duration |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M/48 y | Cough, Dyspnea/24 m; Chest pain/1 m | NA | 15 | RLL | NA | NA | ND | Lobectomy | NA | NA |
| 2 | M/52 y | Cough, Expectoration, Blood in phlegm/4 d | NA | 3.5 | RLL | - | - | ND | Lobectomy | NA | NA |
| 3 | F/27 y | Cough, Expectoration/1 m | Never | 5 | RLL | pleura | - | - | Lobectomy, Lymph node dissection, Chemotherapy (EADM + IFO + DTIC) | NA | NA |
| 4 | F/42 y | Chest pain, Hemoptysis/6 m | Never | 13 | RMLL | pleura | - | - | Lobectomy, Lymph node dissection | NA | NA |
| 5 | M/30 y | Cough, Chest pain/15 d | 10 y | 24 | LP | ND | ND | ND | Chemotherapy (IFO + EADM + DDP) | NA | NA |
| 6 | M/27 y | Cough, Chest pain, Dyspnea/6 m; Blood in phlegm/1 w | Never | 15 | LLUL | ND | ND | ND | Biopsy only | Local lymph node metastasis/at presentation | DOD/11 m |
| 7 | F/53 y | Cough, Hemoptysis/6 m | Never | 6 | LUL | - | - | - | Lobectomy, Lymph node dissection, Chemotherapy (CTX + VCR + EADM) | Localized/45 m | DOD/51 m |
| 8 | M/20 y | Cough, Chest pain/NA | NA | 3 | RML | ND | ND | ND | Biopsy only | Intrapulmonary metastasis/at presentation | DOD/8 m |
| 9 | F/31 y | Cough, Dyspnea/2 m | NA | 15 | Anterior mediastinum | NA | NA | ND | Tumorectomy | Localized/6 m | DOD/12 m |
| 10 | F/72 y | Cough, Expectoration/3 d | NA | 5 | RLL | pleura | - | NA | Lobectomy, Lymph node dissection | Intrapulmonary metastasis/29 m | DOD/32 m |
| 11 | M/28 y | Cough/24 m; Hemoptysis/2 d | NA | 6 | LUL | - | - | NA | Lobectomy, Lymph node dissection | NA | NA |
| 12 | M/62 y | Cough, Blood in phlegm/3 m | 30 y | 11.6 | LL | ND | ND | ND | Bronchial artery infusion chemotherapy (FudR + DDP + NVB) | Intrapulmonary and thyriod metastasis/at presentation | DOD/15 m |
| 13 | F/34 y | Cough, Expectoration, Hemoptysis/1 m | Never | 3.5 | LLL | - | - | - | Lobectomy, Lymph node dissection, Chemotherapy (IFO + EADM) | Localized/26 m; Left chest wall/36 m;Left rib/39 m; Right lung and spine metastasis/51 m | AWD/52 m |
| 14 | M/26 y | Cough, Chest pain, Dyspnea/20 d | 7 y | 14 | LUL | pleura | + | ND | Lobectomy | Localized/8 m | DOD/11 m |
| 15 | F/38 y | Cough, Chest pain/NA | NA | 8 | LUL | NA | NA | ND | Lobectomy | NA | NA |
| 16 | F/46 y | Cough, Chest pain/24 m | Never | 2.3 | RUL | ND | ND | ND | Biopsy only | Brain metastasis/at presentation | DOD/10 m |
| 17 | F/39 y | Cough, Chest pain/1 m | Never | 10 | LUP | Lung; chest wall | - | - | Lobectomy, Lymph node dissection, Chemotherapy (DTIC + EADN) | Adrenal gland metastasis/2 m | DOD/15 m |
| 18 | M/46 y | Cough/NA | NA | 4.2 | LUL | - | - | ND | Lobectomy | Multiple organs metastasis/2 m | DOD/18 m |
| 19 | M/20 y | Asymptomatic | Never | 3.5 | RUP | - | - | ND | Tumorectomy, Chemotherapy (IFO + DDP + EADM), Radiotherapy | Left lung metastasis/24 m | AWD/24 m |
| 20 | M/36 y | Asymptomatic | Never | 4.5 | LLL | - | - | - | Lobectomy, Lymph node dissection | NO | NED/4 m |
| 21 | M/16 y | Cough, Hemoptysis/NA | Never | 6 | RL | pleura | + | ND | Partial tumorectomy | NO | DOD/18 m |
| 22 | M/22 y | Chest pain/2 w; Spontaneous hemopneumothorax/1+ w | 6 y | 5 | LP | lung | + | ND | Tumorectomy | NO | DOD/9 m |
| 23 | M/47 y | Chest pain, Dyspnea/5 m | 30 y | 18 | RL | ND | ND | ND | Chemotherapy (GT regimen) | NO | AWD/10 m |
| 24 | M/60 y | NA | 30 y | 7 | LUL | - | - | - | Lobectomy, Lymph node dissection, Postoperative | Localized/9 m | AWD/9 m |
| 25 | M/37 y | Cough, Expectoration,Chest pain, Dyspnea/1 m | 10 y | 4 | LUL | - | - | - | Lobectomy, Lymph node dissection | NO | NED/1 m |
| 26 | M/24 y | Dyspnea/3 m;Syncope/2 m | Never | 5.3 | Valvula bicuspidalis | myocardium | - | ND | Tumorectomy, Chemotherapy | Localized/2 m | DOD/26 m |
Abbreviation: NA not available, ND not done, “+” positive, “-” negative. W well-defined, I ill-defined, LLL left lower lobe, LUL left uper lobe, RLL right lower lobe, RUL right uper lobe, RML right middle lobe, RMLL right middle and lower lobe, LLUL left lower and uper lobe, LP left pleura, LUP left uper pleura, RUP right uper pleura, y year, m month, w week, d day, EADM Epirubicin, IFO Ifosfamide, DTIC Dacarbazine, DDP cis-Dichlorodiamineplatinum, CTX cyclophosphamide, VCR Vincristine, NVB vinorelbine, FudR 2'-deoxy-5-fluoro-uridin, MFSS monophasic fibrous synovial sarcoma, BSS biphasic synovial sarcoma, PDSS poorly differentiated synovial sarcoma
Surgical excision of the tumor or a lobectomy were performed in 20 patients (76.9 %). Ten (50 %) of these patients underwent local lymph node dissection, and seven (35 %) received postoperative chemotherapy or radiotherapy. Three of the six inoperable patients were treated with chemotherapy.
Radiologic findings
Radiology information were available for 23 patients, including ten conventional chest radiographs, 21 computed tomography (CT) images, and two magnetic resonance imaging (MRI) studies.
On the chest radiographs, all the masses were oval or round shaped with sharp or partly defined margins that were homogeneous without calcification or cavities. Ipsilateral pleural effusions were observed in six patients.
On CT (Fig. 1), the margins were well-defined in 14/21 (66.7 %) tumors, ill-defined in 1/21 (4.8 %), and infiltrative in 6/21 (28.6 %). Fifteen cases had contrast-enhanced CT data available, and those tumors showed heterogeneous (14/15, 93.3 %) or homogeneous enhancement (1/15, 6.7 %). Necrosis/cystic changes were observed in 11/21 tumors (52.4 %), and calcification was identified in 1/21 (4.8 %). Seven tumors (7/21, 33.3 %) showed lymphadenopathy. Enhancing tumor vessels were identified in two cases. A contralateral mediastinal shift was observed in 7/21 (33.3 %) cases, and ipsilateral pleural effusions were observed in 12/21 (57.1 %) patients.
Fig. 1.
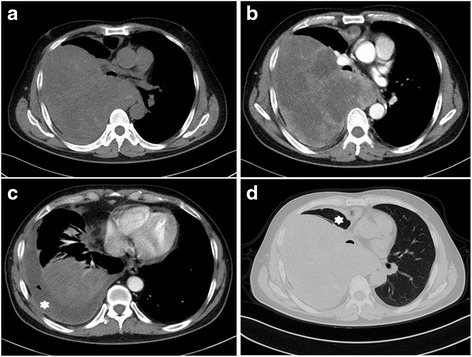
Primary synovial sarcoma of the lung in a 47-year-old male with chest pain and dyspnea. a Non-contrast CT (mediastinum window) scan demonstrating a large mass of the right lung, contiguous with the right lateral pleura and the mediastinum, note the mediastinal shift. b Contrast-enhanced CT scan (mediastinum window) showing the heterogeneous enhancement mass with predominantly cystic areas and a massive pleural effusion (white asterisk, c). d Contrast-enhanced CT scan (lung window) revealing obvious atelectasis on the right (white asterisk)
On MRI, the T1-weighted and T2-weighted images revealed lobulated lesions with sharp margins (2/2). One tumor showed a peripheral rim enhancement after the administration of gadopentetate dimeglumine. Cystic areas of hyperintensity suggestive of necrosis, hemorrhage, or myxoid material were observed in one tumor on T2-weighted images.
Gross and histologic findings
The macroscopic descriptions were available in 18 of the 20 resected specimens. Thirteen (72.2 %) tumors were well-circumscribed and surrounded by thin fibrous pseudocapsules or unencapsulated, and the remaining five (27.8 %) tumors were poorly circumscribed. On cut sections, the tumors were white-gray to tan and soft fleshy to rubbery in texture. Twelve tumors (66.7 %) contained focal necrosis and hemorrhage. Cystic changes were observed in four cases (22.2 %). No grossly visible calcification was observed.
The histologic findings are summarized in Table 3. The tumors were classified into monophasic SS (MSS) (22/26, 84.6 %), biphasic SS (BSS) (1/26, 3.8 %), and poorly differentiated SS (PDSS) (3/26, 11.5 %). The majority of the MSSs were composed of monomorphous spindle-shaped cells with moderate cytoplasm and hyperchromatic nuclei with inconspicuous, small nucleoli (Fig. 2a). The only case of BSS was predominantly composed of oval epithelial nests surrounded by malignant spindle cells (Fig. 2b). Among the three PDSSs, two cases were composed entirely of high-grade spindle-shaped cells (Fig. 2c) resembling malignant peripheral nerve sheath tumor (MPNST), and the remaining one tumor was predominantly composed of large epithelioid cells (Fig. 2d) and was reminiscent of carcinoma or mesothelioma. The mitotic rate ranged from two to 36 mitoses per ten high-power field (HPF)s. Other features included a hemangiopericytoma-like vasculature (76.9 %, 20/26), cystic changes (26.9 %, 7/26), myxoid degeneration (42.3 %, 11/26), necrosis (38.5 %, 10/26), and entrapped pneumocytes (3.8 %, 1/26) (Fig. 3a). Grading was performed on 24 samples with sufficient material. The tumors were classified as FNCLCC 2 (15/24, 62.5 %) and FNCLCC 3 (9/24, 37.5 %). The FNCLCC 3 tumors consisted of three PDSSs and six MSSs.
Table 3.
Histopathologic and immunohistochemical features of 26 PPMSSs
| Case NO. | Subtype | Grade (FNCLCC) | Mitotic Rate (/10HPF) | TLE1 | ki-67 | EMA | CD99 | CK7 | Bcl2 | cytokeratin | CD34 | S-100 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | MFSS | 2 | 7 | - | 50 % | - | - | - | + | + | - | - |
| 2 | MFSS | 3 | 36 | 2+ | 35 % | + | + | + | + | + | ND | - |
| 3 | MFSS | 2 | 2 | 3+ | 6 % | + | - | + | + | + | ND | - |
| 4 | PDSS | 3 | 12 | 2+ | ND | + | + | - | + | + | - | + |
| 5 | MFSS | NA | 2/3HPF | 2+ | ND | + | + | ND | ND | + | - | - |
| 6 | MFSS | 3 | 12 | 3+ | 4 % | + | - | - | + | + | ND | - |
| 7 | MFSS | 2 | 14 | 2+ | 35 % | + | + | + | + | + | - | + |
| 8 | MFSS | 2 | 2 | 3+ | 10 % | + | - | - | + | + | - | - |
| 9 | MFSS | 3 | 2 | 1+ | 10 % | + | - | + | + | - | + | - |
| 10 | MFSS | 2 | 9 | - | ND | + | + | - | + | - | - | - |
| 11 | MFSS | 2 | 8 | 2+ | 25 % | + | - | + | + | - | ND | + |
| 12 | MFSS | 2 | 6 | ND | 30 % | + | + | ND | - | + | - | - |
| 13 | MFSS | 2 | 4 | 1+ | 20 % | + | + | + | + | - | - | ND |
| 14 | MFSS | 2 | 5 | 2+ | 25 % | + | + | + | + | + | - | - |
| 15 | MFSS | 2 | 4 | 2+ | 60 % | + | - | + | + | + | - | ND |
| 16 | MFSS | NA | 2/8HPF | 3+ | 40 % | + | + | + | + | - | - | - |
| 17 | PDSS | 3 | 21 | 3+ | 40 % | + | + | - | + | - | - | - |
| 18 | MFSS | 2 | 17 | 1+ | 30 % | + | - | ND | ND | + | - | - |
| 19 | MFSS | 3 | 12 | 3+ | 75 % | + | + | - | + | - | - | - |
| 20 | MFSS | 2 | 8 | 3+ | 20 % | - | + | - | + | + | - | + |
| 21 | MFSS | 2 | 6 | 2+ | 35 % | - | ND | ND | + | ND | - | - |
| 22 | MFSS | 3 | 21 | 3+ | 45 % | + | - | ND | ND | + | - | - |
| 23 | MFSS | 2 | 2 | 2+ | 45 % | + | - | ND | + | + | - | - |
| 24 | MFSS | 3 | 11 | 3+ | 55 % | - | - | ND | + | - | - | - |
| 25 | BSS | 2 | 4 | 3+ | 20 % | + | + | + | + | + | ND | ND |
| 26 | PDSS | 3 | 21 | ND | 20 % | + | + | ND | + | + | ND | + |
Abbreviation: NA not available, ND not done, MFSS monophasic fibrous synovial sarcoma, BSS biphasic synovial sarcoma, PDSS poorly differentiated synovial sarcoma, HPF high power field, “+” positive, “-” negative
Fig. 2.
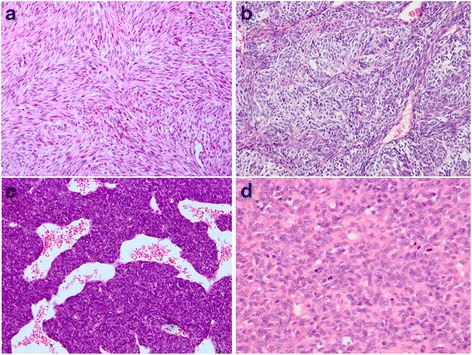
Microscopic features. a Monophasic synovial sarcoma, tumor consisting of uniform hyperchromatic spindled cells arranged in a prominent fascicular pattern (H&E, ×200). b Biphasic synovial sarcoma, composed of oval epithelial nests and surrounded by spindle cell components (H&E, ×200). c Poorly differentiated synovial sarcoma, showing high-grade spindle-shaped cells and a hemangiopericytoma-like vascular pattern, reminiscent of a malignant peripheral nerve sheath tumor (H&E, ×200). d Poorly differentiated synovial sarcoma, consisting of large epithelioid cells, indistinguishable from a poorly differentiated carcinoma or mesothelioma (H&E, ×400)
Fig. 3.
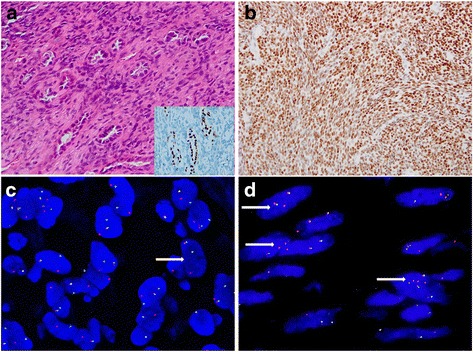
a Entrapped pneumocytes. The entrapped lung tissue could be observed in the peripheral areas of the monophasic synovial sarcoma, resembling a biphasic synovial sarcoma. The entrapped pneumocytes are positive for thyroid transcription factor 1 (TTF-1) (a inset, 400×). b Tumor cells with “3+” positively nuclear TLE1 expression (×200). c FISH demonstrating a balanced rearrangement of the SS18 locus observed in the majority of the neoplastic cells (×1000). d In Case 9, the majority of tumor cells show two paired signals and one or multiple extra red signals, loss of green signal (arrow) (×1000)
Detailed information regarding microscopic invasion and margins was available in 17 cases. Eight tumors infiltrated into the adjacent tissues, and the remaining nine were free of invasion. The surgical margins were negative in 14 tumors and positive in three tumors. Ten patients underwent lymph node dissections; the eight cases with available information were free of lymph node metastasis (Table 2).
Immunohistochemical findings
The immunohistochemical results are summarized in Table 3. TLE1 positivity was present in 91.7 % PPMSSs (22/24), with “≥2+” in 79.2 % (19/24) of the tumors (Fig. 3b). In the 39 nonsynovial sarcoma samples, 94.9 % (37/39) were completely negative for TLE1, whereas only two SFTs showed “1+” staining. None of the control tumors showed moderate or strong positivity (“≥2+”) for this marker. The sensitivity and specificity of TLE1 for PPMSS was 91.7 % and 94.9 %, respectively.
Diffuse staining for Bcl-2 and CD99 was present in 95.7 % (22/23) and 56.0 % (14/25) of PPMSSs, respectively. Focal positivity was seen with EMA (84.6 %, 22/26), CK7(55.6 %, 10/18), cytokeratin (AE1/AE3) (68.0 %, 17/25), CD34 (5.0 %,1/20) and S-100 protein (21.7 %, 5/23). A high Ki-67 index (≥10 %) was observed in 91.3 % (21/23) of the tumors.
FISH and RT-PCR
The molecular results are described in Table 4. By FISH, 25 cases showed SS18 rearrangement (Fig. 3c). In one of the FISH-positive cases (case 9), at least 60 % of the nuclei contained two paired signals with multiple extra red signals, and loss of green signal (Fig. 3d). One case failed FISH based on absent fluorescent signals; this case (case 2) showed a SS18-SSX1 fusion on RT-PCR.
Table 4.
FISH and RT-PCR features of 26 PPMSSs
| Case NO. | FISH | RT-PCR | Fusion site of exon | Fusion site of codon |
|---|---|---|---|---|
| 1 | + | SS18-SSX1 | SS18:exon 10 SSX1:exon 6 | SS18:codon 410 SSX1:codon 111 |
| 2 | failed | SS18-SSX1 | SS18:exon 10 SSX1:exon 6 | SS18:codon 410 SSX1:codon 111 |
| 3 | + | SS18-SSX1 | SS18:exon 10 SSX1:exon 6 | SS18:codon 410 SSX1:codon 111 |
| 4 | + | SS18-SSX1 | SS18:exon 10 SSX1:exon 6 | SS18:codon 410 SSX1:codon 111 |
| 5 | + | ND | ND | ND |
| 6 | + | SS18-SSX2 | SS18:exon 10 SSX2:exon 6 | SS18:codon 410 SSX2:codon 111 |
| 7 | + | SS18-SSX1 | SS18:exon 10 SSX1:exon 6 | SS18:codon 410 SSX1:codon 111 |
| 8 | + | SS18-SSX1 | SS18:exon 10 SSX1:exon 6 | SS18:codon 410 SSX1:codon 111 |
| 9 | + | SS18-SSX1 | SS18:exon 10 SSX1:exon 6 | SS18:codon 410 SSX1:codon 111 |
| 10 | + | ND | ND | ND |
| 11 | + | SS18-SSX1 | SS18:exon 10 SSX1:exon 6 | SS18:codon 410 SSX1:codon 111 |
| 12 | + | SS18-SSX1 | SS18:exon 10 SSX1:exon 6 | SS18:codon 410 SSX1:codon 111 |
| 13 | + | SS18-SSX1 | SS18:exon 10 SSX1:exon 6 | SS18:codon 410 SSX1:codon 111 |
| 14 | + | SS18-SSX1 | SS18:exon 10 SSX1:exon 6 | SS18:codon 410 SSX1:codon 111 |
| 15 | + | SS18-SSX2 | SS18:exon 10 SSX2:exon 6 | SS18:codon 410 SSX2:codon 111 |
| 16 | + | SS18-SSX1 | SS18:exon 10 SSX1:exon 6 | SS18:codon 410 SSX1:codon 111 |
| 17 | + | - | - | - |
| 18 | + | SS18-SSX2, variant | SS18:exon 9 SSX2:exon 5 | SS18:codon 366 SSX2:codon 94 |
| 19 | + | SS18-SSX2 | SS18:exon 10 SSX2:exon 6 | SS18:codon 410 SSX2:codon 111 |
| 20 | + | SS18-SSX2, variant | SS18:exon 9 SSX2:exon 5 | SS18:codon 366 SSX2:codon 94 |
| 21 | + | SS18-SSX2 | SS18:exon 10 SSX2:exon 6 | SS18:codon 410 SSX2:codon 111 |
| 22 | + | SS18-SSX1 | SS18:exon 10 SSX1:exon 6 | SS18:codon 410 SSX1:codon 111 |
| 23 | + | ND | ND | ND |
| 24 | + | SS18-SSX4 | failed | failed |
| 25 | + | SS18-SSX1 | SS18:exon 10 SSX1:exon 6 | SS18:codon 410 SSX1:codon 111 |
| 26 | + | SS18-SSX1 | SS18:exon 10 SSX1:exon 6 | SS18:codon 410 SSX1:codon 111 |
ND not done, “+” positive, “-” negative
Of the 23 cases subjected to RT-PCR, 22 were positive for SS18-SSX gene fusion, whereas the remaining one was negative (case 17, positive by FISH). The fusion transcripts included SS18-SSX1 (15/22, 68.2 %), SS18-SSX2 (6/22, 27.3 %), and SS18-SSX4 (1/22, 4.5 %) fusions. DNA sequencing showed that the fusion sites of all SS18-SSX1 and the four classical SS18-SSX2 tumors were involved in exon 10 of the SS18 gene (codon 410) and exon 6 of the SSX1 or SSX2 genes (codon 111), which is typical of the ordinary fusion site of synovial sarcoma [23]. The remaining two SS18-SSX2 tumors (cases 18 and 20) showed involvement of exon 9 of the SS18 gene (codon 366) and exon 5 of the SSX2 gene (codon 94) (Fig. 4a and b), which has not been described in the literatures. SS18-SSX4 is shown in the gel-electrophoresis (Fig. 4c), whereas the lanes of SS18-SSX1 and SS18-SSX2 were clear. However, sequencing of this rare sample failed.
Fig. 4.
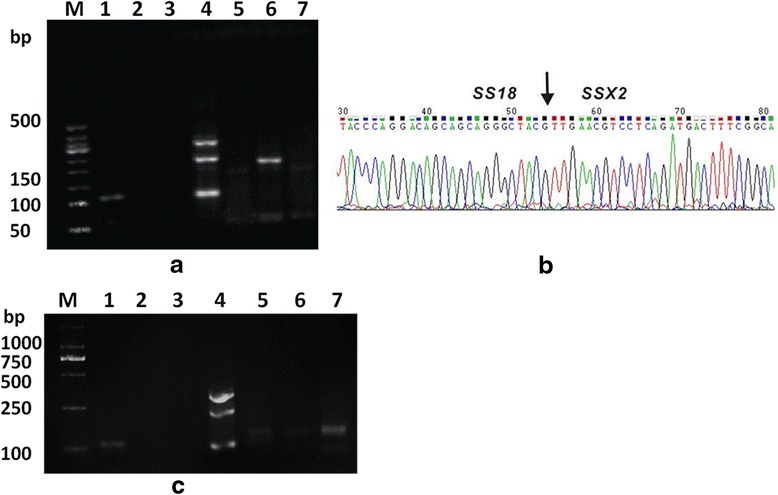
RT-PCR for SS18-SSX gene fusions. Rare fusion site for SS18-SSX2 fusion transcript (case 18) (a and b). a Gel electrophoresis of the RT-PCR product. Lane M, size marker; Lane 1, positive control; Lane 2, negative control; Lane 3, blank control; Lane 4, internal control (GAPDH); Lane 5, SS18-SSX1; Lane 6, SS18-SSX2; Lane 7, SS18-SSX4. b Partial nucleotide sequence of the SS18-SSX2 fusion transcript; the fusion sites (arrow) are involved in exon 9 of the SS18 gene (codon 366) and exon 5 of the SSX2 gene (codon 94). c SS18-SSX4 (case 24). The gel-electrophoresis demonstrates obvious SS18-SSX4, with clean lanes of SS18-SSX1 and SS18-SSX2
Survival analysis
Relapse and survival information are summarized in Table 2. Follow-up data were available for 73.1 % of cases (19/26) with a median follow-up time of 12.0 months; these data included 15 patients with localized disease and four patients with tumor metastasis at presentation. Among the 15 patients with localized disease, ten (66.7 %) suffered disease progression two to 51 months after diagnosis (median, 8.5 months; mean, 15.9 months), including six patients with local recurrence and five patients with metastasis. Thirteen patients (68.4 %) died of the disease within 51 months. Four patients (21.1 %) were alive with the disease. Two (10.5 %) were disease free with a short follow-up time. No patients lived without evidence of disease for more than 24 months. In the entire cohort, the median survival time (MST) was 14.5 months (range 1–52 months). The 2-year disease-specific survival was 27.7 %.
Log-rank analyses on the prognostic parameters were as follows: gender (χ2 = 1.25, p = 0.246 > 0.05), age (≥37 vs. <37; χ2 = 0.064, p = 0.800 > 0.05), tumor size (≥5 cm vs. <5 cm; χ2 = 0.56, p = 0.454 > 0.05), FNCLCC grade (χ2 = 0.17, p = 0.685 > 0.05), fusion gene types (SS18-SSX1 vs. SS18-SSX2; χ2 = 0.10, p = 0.756 > 0.05), mitotic rate (≥10/10 HPFs vs. <10/10 HPFs; χ2 = 0.01, p = 0.925 > 0.05), tumor resection (yes vs. no; χ2 = 5.13, p = 0.024 < 0.05) and tumor residual status (yes vs. no; χ2 = 8.55, p = 0.004 < 0.05). Surgical resection and no residual tumor were associated with significantly improved overall survival (OS). No significant difference was observed between two sides of the other survival factors.
Discussion
PPMSSs are extremely rare; however, according to our series, SS accounted for 14.7 % of all primary sarcoma in pleuropulmonary and mediastinal area and seems to be the most common primary sarcoma type in this region. A preference for location at the lung (76.9 %) was observed, which was similar to the findings of prior studies [11, 14, 15]. Additionally, lung appears to be the most common organ-based location for SS [24]. Therefore, more attention should be paid to this peculiar entity.
In this study, the tumors displayed a male predominance (17 males and nine females; ratio, 1.9:1.0), which is in agreement with a previous series (ratio, 2.0:1.0) [9]. In contrast, a study from Japan showed a female dominance (ratio, 0.6:1.0) [12]. However, there was no obvious sex predominance in the majority of previous studies [10, 11, 13–17]. Our findings demonstrated that PPMSSs often develop in young to middle-aged adults (median, 36.5 years; mean, 37.8 years), older than that of the soft tissue counterpart (median, 34.0 years [25]). Historical series showed similar findings. Chan et al. [26] observed that SS in older people were more likely to occur in unusual regions. The mean age of patients with PPMSS in this study was somewhat lower than that reported in the largest series [14] (37.8 years vs. 42 years) and much lower than that of the Japanese series [12] (37.8 years vs. 58 years). Cough, chest pain and dyspnea were the common presenting signs and symptoms in this study, whereas a few cases were detected incidentally. In our study, only a single patient developed pneumothorax; however, in the series of Cummings et al. [27], pneumothorax appeared to be a frequent event in cystic pulmonary SSs, partly because of the superficial location of the masses.
Radiographically, PPMSSs are typically homogenous and well-defined, and ipsilateral pleural effusions could be observed in most cases. On CT, the tumors in this study could present with sharp (66.7 %) or ill-defined (33.3 %) margins. The tumors were manifested by heterogeneous enhancement and rarely by homogeneous enhancement, which was similar to the findings of earlier studies [16–19] and their soft tissue counterparts [28]. Calcification was absent in our series and in most previous series although it is a common feature of soft tissue SSs [28]. A mediastinal shift was usually observed in the patients with very large masses, and ipsilateral pleural effusions are also a common sign. Generally, the “triple signal” pattern (hypointense, isointense, and hyperintense) was a frequent event on T2-weighted magnetic resonance imaging [17].
Grossly, the majority of tumors (72.2 %) in this study were well circumscribed, and the tumors exhibited a variety of hemorrhage, necrosis, and cystic changes, which were similar to those seen in previous reported SSs [14, 17].
Histologically, MSS (84.6 %) was the main subtype, followed by PDSS (11.5 %) and BSS (3.8 %); these features are similar to the prior PPMSS series [11–16] and soft series [25, 28]. In this series, microscopic calcification was absent, and some previous series also showed that calcification appears to be much less common than in soft tissue lesions (15 % vs. 30 %) [9, 11, 13, 14, 28].
The proportions of FNCLCC 2 (62.5 %)) and FNCLCC 3 (37.5 %) in our series were similar to the largest series (75.8 % and 24.2 %, respectively) [14] and most soft tissue SSs (69.0 ~ 74.0 % and 26.0 ~ 31.0 %, respectively) [29, 30]. However, the proportion of FNCLCC 3 in Begueret’s study [13] was as high as 70 %. The explanation for the difference between the results of Begueret et al. [13] and other authors requires further studies.
TLE1 has recently emerged as a new marker for the diagnosis of SS [15, 21]. However, the diagnostic value of this antibody remains controversial [31]. In this series, TLE1 demonstrated high sensitivity (91.7 %) and specificity (94.9 %) for PPMSSs. Although some SFTs showed weak positivity for TLE1, none of the nonsynovial sarcomas were moderately or strongly reactive for this marker. Therefore, TLE1 could be used as a screening marker prior to the detection of t(X;18)(p11.2;q11.2) [32].
A high Ki-67 index (≥10 %) in our study occurred in 91.3 %, which was higher than in the prior Japanese series [12] (45.5 %) and in most soft tissue counterparts [22] (26.5 ~ 86.0 %). Ki-67 has been suggested as a prognostic factor in synovial sarcoma [22], and the poor prognosis might be partly explained by this high Ki-67 staining in our series.
In this series, the fusion transcripts included SS18-SSX1 (68.2 %), SS18-SSX2 (27.3 %), and SS18-SSX4 (4.5 %) fusions, and the proportion of SS18-SSX1 and SS18-SSX2 fusions were similar to that in previous studies [12–14]. However, some different findings were as follows. First, we found a single case harboring a SS18-SSX4 fusion transcript. This fusion presents in only 1–2 % of SSs [25], and to the best of our knowledge, this extremely rare fusion has not been previously reported in PPMSS before. This case was classified as MSS (FNCLCC 3). Local recurrence occurred 9 months after the tumor excision and postoperative radiotherapy. Second, two cases harboring rare fusion site of SS18-SSX2 fusion transcript were identified and this fusion site has not been previously reported in SSs. Further studies are needed to determine whether the variant fusion site is specific to PPMSS. Third, one case was found to harbor a complex pattern by FISH analysis. This case exhibited two paired signals with multiple extra red signals and loss of green signal, which are suggestive of a cryptic rearrangement of the 5’ region of the SS18 gene; this case was proven to be SS18-SSX1 by subsequent RT-PCR. This finding has not been described in PPMSSs, although it has been reported in soft tissue SSs [33, 34], and the presence of aneuploidy or amplificon of chromosome 18q might account for this event. This case was MSS (FNCLCC 3) and was the only case with massive necrosis in our series. This patient developed a local recurrence only 6 months after the tumor excision and died 12 months after diagnosis. However, more samples are needed to further analyze the correlation of prognosis and the special genetic changes.
The differential diagnosis of PPMSS is broad and might be very challenging, especially in small biopsy samples. This neoplasm must be differentiated from secondary SS and other primary spindle cell neoplasms in these locations.
The intrathoracic area is an extraordinarily rare location for SS. For example, in our Medical Centre, among the 56 cases of SS in this location, 50 % were metastatic tumors. Therefore, the possibility of a secondary SS should always be excluded. In this series, detail clinical information ruled out the possibility of metastatic lesions arising from other sites.
Sarcomatoid carcinoma could have a conspicuous spindle cell component and show focal positivity for only epithelial markers, simulating SS. SS could also display epithelioid features and might be confused with carcinomas or mesothelioma. Therefore, sarcomatoid carcinoma and mesothelioma should always be the primary consideration in the differential diagnosis of SS. However, carcinoma and mesothelioma could be distinguished from SS in several aspects. First, the radiologic findings usually show an infiltrative border for carcinoma and a diffuse growth pattern for mesothelioma, whereas PPMSS typically appears as a well-circumscribed mass with uniform opacity [17]. Second, sarcomatoid carcinoma and mesothelioma are usually more pleomorphic than SS, and extensive sampling of the lesion could help identify an underlying histologic subtype of lung carcinoma. Third, neither sarcomatoid carcinoma nor mesothelioma exhibit strong reactivity for TLE1 in this study and Lino-Silva et al. [15] although Matsuyama et al. [35] found TLE1 staining in 69 % of mesotheliomas. Fourth, identification of t(X;18)(p11.2;q11.2) could be invaluable for extremely challenging cases.
Type A thymomas and type I pleuropulmonary blastomas could share clinical and morphological features with PPMSS. However, type I pleuropulmonary blastomas typically develop in infants and during early childhood. In contrast, PPMSSs typically occur in older patients [13, 14]. Careful morphologic inspection and ancillary immunohistochemical stains and molecular studies are helpful in distinguishing between these lesions.
Intrathoracic SFTs might share some morphologic features with PPMSSs. SSs always do not exhibit diffuse reactivity for CD34 [2], whereas SFT usually do not show strong positivity for TLE1, as seen in the this study. SFTs are negative for t(X;18)(p11.2;q11.2) and have been shown to harbor a characteristic NAB2-STAT6 fusion gene [36]. Very recently, STAT6 has proven to be a highly sensitive and specific marker for the diagnosis of SFT [37].
MPNST could closely resembles SS and appears to be the most challenging tumor type in the differential diagnosis of SS in any location. A full panel of markers including TLE1 and molecular genetic testing for t(X;18)(p11.2;q11.2) could be useful for distinguishing between the two tumors.
Poorly differentiated SS might be almost entirely poorly differentiated with uniform small round cell morphology, resembling Ewing sarcoma. Extensive sampling and careful inspection are required to identify any accompanying more typical areas of synovial sarcoma, which play an important role in establishing the diagnosis. Immunostaining for TLE1 and identification of SS18-SSX fusion instead of EWSR1-FLI-1/EWSR1-ERG [38] could help to confirm the diagnosis.
The preferred treatment of SSs is radical resection combined with radiotherapy and or chemotherapy, which could remarkably or partly decrease the recurrent or metastatic rate [39, 40]. In our series, 20 patients (76.9 %) underwent lobectomy or tumor resection, and 82.4 % of these patients obtained negative margins. Seven of these surgical patients (35 %) underwent adjuvant radiotherapy or/and chemotherapy. Several previous PPMSSs series have described the treatment for PPMSS [9–13, 16]; however, few previous studies analyzed the correlation between treatment and survival. Our results indicated that effective excision of the tumors with negative margins could significantly improve survival (p = 0.004), and tumor resection is important when available (p = 0.024).
According to the current study, PPMSSs appear to be more aggressive than the soft tissue counterparts (MST, 14.5 months vs. 52 months [41]); these results are similar to the results of previous studies. The prognosis of PPMSSs in this study appear more unfavorable than in prior series of PPMSSs (MST, 14.5 months vs. 30 ~ 50 months) [11–13, 15, 16]. None of the subjects in our series survived for more than 5 years, and none of the subjects lived with no evidence of disease for more than 2 years; the 5-year survival rate in prior series was approximately 30 % [13], and it could reach 50 % in soft tissue SSs [11, 42]. The dismal prognosis of PPMSSs might partly be due to later presentation and difficulties in obtaining negative surgical margins.
In terms of prognostic factors, age (better in young patients), tumor size (<5 cm), mitotic rate (<10/10HPF), FNCLCC (low grade), status of the surgical margins (negative), and SS18-SSX type (SS18-SSX2) were reported to be associated with better survival in soft tissue SSs [25, 30]. We found tumor resection (p = 0.024) and no residual tumor (p = 0.004) were significantly associated with better survival. The other factors had no effect on survival.
Conclusions
Here, we present the clinicopathological and molecular features of 26 t(X;18)(p11.2;q11.2)-positive PPMSSs. These tumors seem to be more aggressive than the soft tissue counterparts and more aggressive than prior PPMSSs series. Extensive surgical resection of the tumor and more effective adjuvant therapy should be advocated. The diagnosis of PPMSS might be challenging. A combination of clinical studies, careful morphologic analysis, and a full panel of immunomarkers including TLE1 and genetic studies is helpful in confirming the diagnosis.
Abbreviations
BSS, biphasic synovial sarcoma; CT, computed tomography; FFPE, formalin-fixed paraffin-embedded; FISH, fluorescence in situ hybridization; FNCLCC, French Federation of Cancer Centers; HPF, high-power field; MPNST, malignant peripheral nerve sheath tumor; MSS, monophasic synovial sarcoma; MST, median survival time; OS, overall survival.; PDSS, poorly differentiated synovial sarcoma; PPMSS, primary pleuropulmonary and mediastinal synovial sarcoma; RT-PCR, reverse transcription polymerase chain reaction; SFT, solitary fibrous tumor; SS, synovial sarcoma
Acknowledgements
No acknowledgements to make.
Funding
This work was supported by National Natural Science Foundation of China (no. 81472510, 81272944, and 30971148).
Availability of data and materials
All measurements were collected and recorded in Microsoft Excel. The detail clinicopathologic and genetic data are stored in the Surgical Pathology archives at our institution. Both Dr. Lan and Dr. Zhang have copies of these documents. All material is available upon request.
Authors’ contributions
TL carried out the genetic and immunohistochemical studies, data analysis and drafted the manuscript. HC performed the histopathological examinations, data analysis, and helped to draft the manuscript. HC contributed equally to this work with TL, and is the co-first author for this paper. BX carried out the genetic and immunohistochemical studies, and prepared the manuscript. TZ helped to perform the histopathological examinations. RP carried out the RT-PCR studies. MC carried out the FISH studies. FY participated in the sequence alignment and primers design. JY reviewed the radiologic images. XH helped to perform the immunohistochemical studies. YW helped to collect the clinical data. HZ conceived of the study, and participated in its design and coordination, performed histopathological examinations and revised the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
This study was approved by the West China Hospital Institutional Review Board (no. 20150975WCH). Formal informed consent of the individual patients was not necessary.
Contributor Information
Ting Lan, Email: tinglanxt@163.com.
Huijiao Chen, Email: joan-ch@sohu.com.
Bo Xiong, Email: 541623443@qq.com.
Tingqing Zhou, Email: whf-ztq@163.com.
Ran Peng, Email: 2015224020003@stu.scu.edu.cn.
Min Chen, Email: Mindy5555@163.com.
Feng Ye, Email: fengye@scu.edu.cn.
Jin Yao, Email: shelleyyao@163.com.
Xin He, Email: cd120hexin@126.com.
Yaqin Wang, Email: silver19@163.com.
Hongying Zhang, Phone: +86 28 85423847, Email: hy_zhang@scu.edu.cn, Email: hy_zh@263.net.
References
- 1.Suurmeijer AJH, de Bruijn D, Geurts van Kessel A, Miettinen MM. Synovial sarcoma. In: Fletcher CDM, Bridge JA, Hogendoor P, Mertens F, editors. WHO classification of tumours of soft tissue and bone. 4th ed. Lyon: IARC; 2013. p. 213–5.
- 2.Goldblum JR, Folpe AL, Weiss SW. Enzinger and Weiss's Soft Tissue Tumors. 6. Philadelphia: Saunders; 2013. Malignant soft tissue tumors of uncertain type; pp. 1052–70. [Google Scholar]
- 3.Storlazzi CT, Mertens F, Mandahl N, Gisselsson D, Isaksson M, Gustafson P, et al. A novel fusion gene, SS18L1/SSX1, in synovial sarcoma. Genes, chromosomes & cancer. 2003;37(2):195–200. doi: 10.1002/gcc.10210. [DOI] [PubMed] [Google Scholar]
- 4.Dacic S, Franks TJ, Ladanyi M. Synovial sarcoma. In: Travis WD, Brambilla E, Burke AP, Marx A, Nicholson AG, editors. WHO classification of tumours of the lung, pleura, thymus and heart. 4. Lyon, France: IARC; 2015. pp. 127–8. [DOI] [PubMed] [Google Scholar]
- 5.Mirzoyan M, Muslimani A, Setrakian S, Swedeh M, Daw HA. Primary pleuropulmonary synovial sarcoma. Clinical lung cancer. 2008;9(5):257–61. doi: 10.3816/CLC.2008.n.040. [DOI] [PubMed] [Google Scholar]
- 6.Witkin GB, Miettinen M, Rosai J. A biphasic tumor of the mediastinum with features of synovial sarcoma. A report of four cases. Am J Surg Pathol. 1989;13(6):490–9. doi: 10.1097/00000478-198906000-00005. [DOI] [PubMed] [Google Scholar]
- 7.Gaertner E, Zeren EH, Fleming MV, Colby TV, Travis WD. Biphasic synovial sarcomas arising in the pleural cavity. A clinicopathologic study of five cases. Am J Surg Pathol. 1996;20(1):36–45. doi: 10.1097/00000478-199601000-00004. [DOI] [PubMed] [Google Scholar]
- 8.Aubry MC, Bridge JA, Wickert R, Tazelaar HD. Primary monophasic synovial sarcoma of the pleura: five cases confirmed by the presence of SYT-SSX fusion transcript. Am J Surg Pathol. 2001;25(6):776–81. doi: 10.1097/00000478-200106000-00009. [DOI] [PubMed] [Google Scholar]
- 9.Suster S, Moran CA. Primary synovial sarcomas of the mediastinum: a clinicopathologic, immunohistochemical, and ultrastructural study of 15 cases. Am J Surg Pathol. 2005;29(5):569–78. doi: 10.1097/01.pas.0000157934.50936.3e. [DOI] [PubMed] [Google Scholar]
- 10.Zeren H, Moran CA, Suster S, Fishback NF, Koss MN. Primary pulmonary sarcomas with features of monophasic synovial sarcoma: a clinicopathological, immunohistochemical, and ultrastructural study of 25 cases. Hum Pathol. 1995;26(5):474–80. doi: 10.1016/0046-8177(95)90242-2. [DOI] [PubMed] [Google Scholar]
- 11.Essary LR, Vargas SO, Fletcher CD. Primary pleuropulmonary synovial sarcoma: reappraisal of a recently described anatomic subset. Cancer. 2002;94(2):459–69. doi: 10.1002/cncr.10188. [DOI] [PubMed] [Google Scholar]
- 12.Okamoto S, Hisaoka M, Daa T, Hatakeyama K, Iwamasa T, Hashimoto H. Primary pulmonary synovial sarcoma: a clinicopathologic, immunohistochemical, and molecular study of 11 cases. Hum Pathol. 2004;35(7):850–6. doi: 10.1016/j.humpath.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 13.Begueret H, Galateau-Salle F, Guillou L, Chetaille B, Brambilla E, Vignaud JM, et al. Primary intrathoracic synovial sarcoma: a clinicopathologic study of 40 t(X;18)-positive cases from the French Sarcoma Group and the Mesopath Group. Am J Surg Pathol. 2005;29(3):339–46. doi: 10.1097/01.pas.0000147401.95391.9a. [DOI] [PubMed] [Google Scholar]
- 14.Hartel PH, Fanburg-Smith JC, Frazier AA, Galvin JR, Lichy JH, Shilo K, et al. Primary pulmonary and mediastinal synovial sarcoma: a clinicopathologic study of 60 cases and comparison with five prior series. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2007;20(7):760–9. doi: 10.1038/modpathol.3800795. [DOI] [PubMed] [Google Scholar]
- 15.Lino-Silva LS, Flores-Gutierrez JP, Vilches-Cisneros N, Dominguez-Malagon HR. TLE1 is expressed in the majority of primary pleuropulmonary synovial sarcomas. Virchows Archiv : an international journal of pathology. 2011;459(6):615–21. doi: 10.1007/s00428-011-1160-4. [DOI] [PubMed] [Google Scholar]
- 16.Kim GH, Kim MY, Koo HJ, Song JS, Choi CM. Primary Pulmonary Synovial Sarcoma in a Tertiary Referral Center: Clinical Characteristics, CT, and 18F-FDG PET Findings, With Pathologic Correlations. Medicine. 2015;94(34) doi: 10.1097/MD.0000000000001392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frazier AA, Franks TJ, Pugatch RD, Galvin JR. From the archives of the AFIP: Pleuropulmonary synovial sarcoma. Radiographics : a review publication of the Radiological Society of North America, Inc. 2006;26(3):923–40. doi: 10.1148/rg.263055211. [DOI] [PubMed] [Google Scholar]
- 18.Baheti AD, Sewatkar R, Hornick JL, Saboo SS, Jagannathan JP, Ramaiya NH, et al. Imaging features of primary and recurrent intrathoracic synovial sarcoma: a single-institute experience. Clin Imaging. 2015;39(5):803–8. doi: 10.1016/j.clinimag.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 19.Zhang WD, Guan YB, Chen YF, Li CX. CT imaging of primary pleuropulmonary synovial sarcoma. Clin Radiol. 2012;67(9):884–8. doi: 10.1016/j.crad.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 20.Coindre JM. Grading of soft tissue sarcomas: review and update. Archives of pathology & laboratory medicine. 2006;130(10):1448–53. doi: 10.5858/2006-130-1448-GOSTSR. [DOI] [PubMed] [Google Scholar]
- 21.Terry J, Saito T, Subramanian S, Ruttan C, Antonescu CR, Goldblum JR, et al. TLE1 as a diagnostic immunohistochemical marker for synovial sarcoma emerging from gene expression profiling studies. Am J Surg Pathol. 2007;31(2):240–6. doi: 10.1097/01.pas.0000213330.71745.39. [DOI] [PubMed] [Google Scholar]
- 22.Skytting BT, Bauer HC, Perfekt R, Nilsson G, Larsson O. Ki-67 is strongly prognostic in synovial sarcoma: analysis based on 86 patients from the Scandinavian Sarcoma group register. Br J Cancer. 1999;80(11):1809–14. doi: 10.1038/sj.bjc.6690602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei Y, Sun M, Nilsson G, Dwight T, Xie Y, Wang J, et al. Characteristic sequence motifs located at the genomic breakpoints of the translocation t(X;18) in synovial sarcomas. Oncogene. 2003;22(14):2215–22. doi: 10.1038/sj.onc.1206343. [DOI] [PubMed] [Google Scholar]
- 24.Coindre JM, Pelmus M, Hostein I, Lussan C, Bui BN, Guillou L. Should molecular testing be required for diagnosing synovial sarcoma? A prospective study of 204 cases. Cancer. 2003;98(12):2700–7. doi: 10.1002/cncr.11840. [DOI] [PubMed] [Google Scholar]
- 25.Ladanyi M, Antonescu CR, Leung DH, Woodruff JM, Kawai A, Healey JH, et al. Impact of SYT-SSX fusion type on the clinical behavior of synovial sarcoma: a multi-institutional retrospective study of 243 patients. Cancer Res. 2002;62(1):135–40. [PubMed] [Google Scholar]
- 26.Chan JA, McMenamin ME, Fletcher CD. Synovial sarcoma in older patients: clinicopathological analysis of 32 cases with emphasis on unusual histological features. Histopathology. 2003;43(1):72–83. doi: 10.1046/j.1365-2559.2003.01643.x. [DOI] [PubMed] [Google Scholar]
- 27.Cummings NM, Desai S, Thway K, Stewart S, Hill DA, Priest JR, et al. Cystic primary pulmonary synovial sarcoma presenting as recurrent pneumothorax: report of 4 cases. Am J Surg Pathol. 2010;34(8):1176–9. doi: 10.1097/PAS.0b013e3181e85c87. [DOI] [PubMed] [Google Scholar]
- 28.Murphey MD, Gibson MS, Jennings BT, Crespo-Rodriguez AM, Fanburg-Smith J, Gajewski DA. From the archives of the AFIP: Imaging of synovial sarcoma with radiologic-pathologic correlation. Radiographics : a review publication of the Radiological Society of North America, Inc. 2006;26(5):1543–65. doi: 10.1148/rg.265065084. [DOI] [PubMed] [Google Scholar]
- 29.Krieg AH, Hefti F, Speth BM, Jundt G, Guillou L, Exner UG, et al. Synovial sarcomas usually metastasize after >5 years: a multicenter retrospective analysis with minimum follow-up of 10 years for survivors. Annals of oncology : official journal of the European Society for Medical Oncology/ESMO. 2011;22(2):458–67. doi: 10.1093/annonc/mdq394. [DOI] [PubMed] [Google Scholar]
- 30.ten Heuvel SE, Hoekstra HJ, Bastiaannet E, Suurmeijer AJ. The classic prognostic factors tumor stage, tumor size, and tumor grade are the strongest predictors of outcome in synovial sarcoma: no role for SSX fusion type or ezrin expression. Applied immunohistochemistry & molecular morphology : AIMM/official publication of the Society for Applied Immunohistochemistry. 2009;17(3):189–95. doi: 10.1097/PAI.0b013e31818a6f5c. [DOI] [PubMed] [Google Scholar]
- 31.Kosemehmetoglu K, Vrana JA, Folpe AL. TLE1 expression is not specific for synovial sarcoma: a whole section study of 163 soft tissue and bone neoplasms. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2009;22(7):872–8. doi: 10.1038/modpathol.2009.47. [DOI] [PubMed] [Google Scholar]
- 32.Knosel T, Heretsch S, Altendorf-Hofmann A, Richter P, Katenkamp K, Katenkamp D, et al. TLE1 is a robust diagnostic biomarker for synovial sarcomas and correlates with t(X;18): analysis of 319 cases. European journal of cancer (Oxford, England : 1990) 2010;46(6):1170–6. doi: 10.1016/j.ejca.2010.01.032. [DOI] [PubMed] [Google Scholar]
- 33.Amary MF, Berisha F, Bernardi Fdel C, Herbert A, James M, Reis-Filho JS, et al. Detection of SS18-SSX fusion transcripts in formalin-fixed paraffin-embedded neoplasms: analysis of conventional RT-PCR, qRT-PCR and dual color FISH as diagnostic tools for synovial sarcoma. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2007;20(4):482–96. doi: 10.1038/modpathol.3800761. [DOI] [PubMed] [Google Scholar]
- 34.Torres L, Lisboa S, Cerveira N, Lopes JM, Lopes C, Teixeira MR. Cryptic chromosome rearrangement resulting in SYT-SSX2 fusion gene in a monophasic synovial sarcoma. Cancer Genet Cytogenet. 2008;187(1):45–9. doi: 10.1016/j.cancergencyto.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 35.Matsuyama A, Hisaoka M, Iwasaki M, Iwashita M, Hisanaga S, Hashimoto H. TLE1 expression in malignant mesothelioma. Virchows Archiv : an international journal of pathology. 2010;457(5):577–83. doi: 10.1007/s00428-010-0975-8. [DOI] [PubMed] [Google Scholar]
- 36.Robinson DR, Wu YM, Kalyana-Sundaram S, Cao X, Lonigro RJ, Sung YS, et al. Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat Genet. 2013;45(2):180–5. doi: 10.1038/ng.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ouladan S, Trautmann M, Orouji E, Hartmann W, Huss S, Buttner R, et al. Differential diagnosis of solitary fibrous tumors: A study of 454 soft tissue tumors indicating the diagnostic value of nuclear STAT6 relocation and ALDH1 expression combined with in situ proximity ligation assay. Int J Oncol. 2015;46(6):2595–605. doi: 10.3892/ijo.2015.2975. [DOI] [PubMed] [Google Scholar]
- 38.Im YH, Kim HT, Lee C, Poulin D, Welford S, Sorensen PH, et al. EWS-FLI1, EWS-ERG, and EWS-ETV1 oncoproteins of Ewing tumor family all suppress transcription of transforming growth factor beta type II receptor gene. Cancer Res. 2000;60(6):1536–40. [PubMed] [Google Scholar]
- 39.Menendez LR, Brien E, Brien WW. Synovial sarcoma. A clinicopathologic study. Orthop Rev. 1992;21(4):465–71. [PubMed] [Google Scholar]
- 40.Spurrell EL, Fisher C, Thomas JM, Judson IR. Prognostic factors in advanced synovial sarcoma: an analysis of 104 patients treated at the Royal Marsden Hospital. Annals of oncology : official journal of the European Society for Medical Oncology/ESMO. 2005;16(3):437–44. doi: 10.1093/annonc/mdi082. [DOI] [PubMed] [Google Scholar]
- 41.Spillane AJ, A'Hern R, Judson IR, Fisher C, Thomas JM. Synovial sarcoma: a clinicopathologic, staging, and prognostic assessment. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2000;18(22):3794–803. doi: 10.1200/JCO.2000.18.22.3794. [DOI] [PubMed] [Google Scholar]
- 42.Sultan I, Rodriguez-Galindo C, Saab R, Yasir S, Casanova M, Ferrari A. Comparing children and adults with synovial sarcoma in the Surveillance, Epidemiology, and End Results program, 1983 to 2005: an analysis of 1268 patients. Cancer. 2009;115(15):3537–47. doi: 10.1002/cncr.24424. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All measurements were collected and recorded in Microsoft Excel. The detail clinicopathologic and genetic data are stored in the Surgical Pathology archives at our institution. Both Dr. Lan and Dr. Zhang have copies of these documents. All material is available upon request.