

Abstract
The prolactin (PRL) family of hormones and cytokines participates in the regulation of optimal reproductive performance in the mouse and rat. Members of the PRL family are expressed in the anterior pituitary, uterus, and/or placenta. In the present study, we investigated the ontogeny of PRL family 7, subfamily b, member 1 (PRL7B1; also called PRL-like protein-N, PLP-N) expression in the developing mouse placenta and established a mouse model for investigating the biological function of PRL7B1. Transcripts for Prl7b1 were first detected on Gestation Day (d) 8.5. From gestation d8.5 through d14.5, Prl7b1 was expressed in trophoblast cells residing at the interface between maternal mesometrial decidua and the developing placenta. On gestation d17.5, the predominant cellular source of Prl7b1 mRNA was migratory trophoblast cells invading into the uterine mesometrial decidua. The Prl7b1 null mutant allele was generated via replacement of the endogenous Prl7b1 coding sequence with beta-galactosidase (LacZ) reporter and neomycin cassettes. The mutant Prl7b1 allele was successfully passed through the germline. Homozygous Prl7b1 mutant mice were viable and fertile. Under standard animal housing conditions, Prl7b1 had undetectable effects on placentation and pregnancy. Hypoxia exposure during pregnancy evoked adaptations in the organization of the wild-type placenta that were not observed in Prl7b1 null placentation sites. In summary, PRL7B1 is viewed as a part of a pathway regulating placental adaptations to physiological stressors.
Keywords: hypoxia, placentation, pregnancy, prolactin family
INTRODUCTION
The prolactin (PRL) locus in the mouse and rat encodes a family of proteins with hormone and cytokine activities contributing to the regulation of reproduction [1–3]. Members of the PRL family signaling through the PRL receptor (PRLR; defined as classical actions) have been shown to contribute to pregnancy-dependent regulation of ovarian function, mammary gland development, and islet beta cell expansion [4–6]. Most members of the PRL family do not activate their targets through the PRLR and instead possess other modes of action (defined as nonclassical actions) [1, 3]. These nonclassical PRL family members possess a broad range of pregnancy-associated cellular targets and actions, influencing vascular remodeling [7, 8], hematopoiesis [9–13], immune cell function [14, 15], and adaptations to physiological stressors [16, 17].
PRL family 7, subfamily b, member 1 (PRL7B1; also called PRL-like protein-N, PLP-N) [18] is expressed by invasive trophoblast cells (both endovascular and interstitial invasive trophoblast) at the placentation site [19, 20]. Intrauterine trophoblast cell invasion and remodeling of the uterine spiral arterioles at the maternal-fetal interface are prominent features of rat, mouse, as well as human placentation [21–24]. Invasive trophoblast cells are proposed to play key roles in uterine spiral arteriole remodeling [23, 25–27]. Current evidence indicates that invasive trophoblast cells modulate the uterine vasculature by replacing the endothelium of the targeted uterine spiral arterioles and by their production of angiogenic factors [28, 29]. Thus, Prl7b1 is situated at a key cellular site pivotal to the establishment of the hemochorial placenta; however, the physiological role of trophoblast-derived PRL7B1 is unknown. In the present study, the ontogeny of Prl7b1 expression in the developing mouse placenta was examined, a Prl7b1 null mouse model for investigating the biology of PLP-N was established, and adaptive responses to maternal hypoxia at placentation sites of wild-type and Prl7b1 null mice assessed.
MATERIALS AND METHODS
Animals and Tissue Collection
C57BL/6 mice were purchased from The Jackson Laboratory. Animals were housed in an environmentally controlled facility, with lights on from 0600 to 2000 h and were allowed free access to food and water. Timed pregnancies were generated by cohabitating male and female mice. The presence of a seminal plug in the vagina was designated as Day (d) 0.5 of gestation.
Prl7b1 mutant mouse embryonic stem cells were obtained from the National Institutes of Health Knock-Out Mouse Project (KOMP) repository (www.komp.org; VG10354) [30]. The University of Kansas Medical Center Transgenic and Gene-Targeting Facility injected the mutant embryonic stem cells into albino C57BL/6 blastocysts to generate germline competent chimeras. Male chimeras were mated to C57BL/6 females to establish a germline stock of the mutant strain. Genotyping was performed using genomic DNA isolated from tail biopsies and polymerase chain reaction (PCR) with forward primers specific for the wild-type allele (5′ cttcaacgtgacttaa 3′) and mutant allele (5′ ttgattcccactttgtggttc 3′) and a common reverse primer (5′ cccaggacaggcaagataaa 3′). PCR amplicons for the wild-type and mutant alleles were 795 and 458 bp, respectively.
Maternal hypoxia exposure was achieved by placing gestation d7.5 mice in hypoxia chambers connected to an oxygen sensor/controller Pro-OX P110 (BioSpherix). Chambers were briefly opened each day (2–3 min) to monitor the health of the animals and replenish food and water. Tissue samples for histological analysis, including in situ hybridization and immunohistochemistry, were collected at indicated gestation days and immediately frozen in dry ice-cooled heptane and stored at −80°C until processed.
Trophoblast tissues were dissected from placentation sites from gestation d9.5 to d17.5 as previously described [31]. Briefly, trophoblast tissues were recovered from placentation sites with the aid of fine forceps and a dissecting microscope (10–20×). Isolated tissues represent enrichments and each contain some contaminating decidua. The dissected tissues were snap frozen in liquid nitrogen and stored at −80°C until processed for RNA extraction.
All experimentation with animals was performed in accordance with guidelines recommended by the National Institutes of Health. The University of Kansas Medical Center Animal Care and Use Committee approved the protocols for the care and use of animals.
RNA Analysis
RNA was extracted from tissue using TRI Reagent (Sigma-Aldrich) according to the manufacturer's instructions. RT-PCR was performed as previously described [32]. Primer sequences used for RT-PCR, included: Prl7b1 (forward: 5′ attggcagtggatcaggtgtt 3′; reverse: 5′ ttcatgatgcgatccagaag 3′; amplicon: 425 bp; NM_029355) and Gapdh (forward: 5′ accacagtccatgccatcac 3′; reverse: 5′ tccaccaccctgttgctgta 3′; amplicon: 452 bp; NM_001289726).
Tissue Analyses
All histochemical staining was performed on 10 μm cryosections, which were prepared with the aid of a cryostat and stored at −80°C until use. Frozen sections were air-dried and fixed in cold phosphate-buffered saline (PBS) containing 4% paraformaldehyde except for the β-galactosidase (LacZ) histochemical staining (see below).
In situ hybridization was performed as previously described [21]. Plasmids containing cDNAs for Prl7b1, Tpbpa, and Prl2a1 were used as templates to synthesize sense and antisense digoxigenin-labeled RNA probes according to the manufacturer's instructions (Roche Molecular Biochemicals). Prehybridization, hybridization, and detection of alkaline phosphatase-conjugated anti-digoxigenin were performed as previously reported [21].
Isolectin B4 histochemical staining was performed as previously described [17]. Endogenous peroxidase activity was quenched by incubation in methanol containing 0.3% H2O2. Sections were then incubated with PBS containing 0.1% Triton X-100 and 5 μg/ml biotinylated isolectin B4 (B-1205; Vector Laboratories) for 30 min. Detection of isolectin B4 binding was achieved by using ABC Kit (PK-4000; Vector Laboratories) and AEC peroxidase substrate kit (SK-4200; Vector Laboratories) according to the manufacturer's instructions.
Immunohistochemical staining was performed as previously described [32]. Sections were incubated overnight at 4°C with antibodies against pan-cytokeratin (TROMA-1; Developmental Studies Hybridoma Repository; 1:20 dilution) to detect trophoblast cells or PECAM1 (CD31, 14-0311-81; eBioscience; 1:50 dilution) to detect endothelial cells. Sections for LacZ histochemical staining were air-dried and incubated in fixative solution (PBS, containing 1% paraformaldehyde, 2 mM MgCl2, 0.2% glutaraldehyde, 5 mM ethylene glycol tetraacetic acid pH8.0 and 0.2% NP-40) for 10 min on ice, followed by rinsing with PBS containing 2 mM MgCl2. Sections were then incubated in PBS containing 2 mM MgCl2, 0.01% sodium deoxycholate, and 0.02% NP-40 for 10 min on ice. LacZ enzymatic activity was detected by incubating sections with staining solution (PBS containing 2 mM MgCl2, 0.01% sodium deoxycholate, 0.02% NP-40, 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 20 mM Tris HCl, pH 7.5, and 1 mg/ml X-gal) at 37°C for 4–8 h. Images were captured using a Leica MZFIII stereomicroscope (Leica Microsystems GmbH) or a Nikon Eclipse 55i microscope (Nikon Instruments Inc.), both equipped with Leica charge-coupled device cameras (Leica).
Morphological Measurements
Morphological measurements of the sizes of placental compartments were performed with National Institutes of Health Image J software as previously described [31–34]. The chorioallantoic placenta consists of the junctional zone and labyrinth zone. Definitions of the junctional zone and labyrinth zone compartments within the placentation site have been described [31]. The junctional zone consists of an area bordered by the uterine mesometrial deciduum and the labyrinth zone. It contains trophoblast giant cells, spongiotrophoblast cells, and glycogen cells. The labyrinth zone is defined as the region of the chorioallantoic placenta vascularized by the allantois. This compartment contains syncytial trophoblast, cytotrophoblast, trophoblast giant cells, and fetal mesenchyme as well as its associated vasculature.
The thickness of the junctional zone was estimated from cross-sectional area measurements of isolectin B4 histochemical-stained placentation sites. Measurements were expressed as the ratio of each zone to the total cross-sectional area of the chorioallantoic placenta (junctional + labyrinth zones) and as the ratio of the labyrinth zone cross-sectional area to the junctional zone cross-sectional area. Chorioallantoic zone measurements were made from a histological plane at the center of each placentation site perpendicular to the flat fetal surface of the placenta. Sample sizes for the analyses were at least five placentation sites from at least five different pregnancies per treatment group.
Statistical Analysis
Data are presented as the mean ± standard error of the mean. Differences between two groups were assessed by Student t test. Comparisons between more than two groups were made using analysis of variance and multiple comparisons were performed using Tukey post hoc test.
RESULTS
Ontogeny of Prl7b1 Expression and the Invasive Trophoblast Cell Lineage
The invasive trophoblast cell population arises early during mouse placental development. Prl7b1 expression can be effectively used to track the ontogeny of the invasive trophoblast cell lineage (Fig. 1A). The lineage first appears in a subpopulation of cells within the ectoplacental cone and was first evident at gestation d8.5 and continued through the course of pregnancy (Fig. 1, A and B). Prl7b1 transcripts were found in both endovascular and interstitial invasive trophoblast cell populations.
FIG. 1.
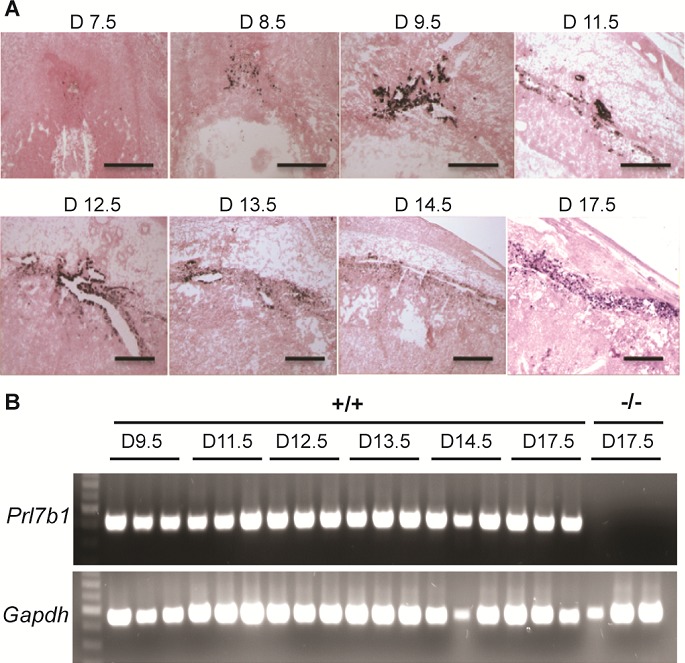
Ontogeny of Prl7b1 in the mouse placenta. A) In situ hybridization for Prl7b1 transcripts on frozen sections of placentation sites from indicated gestation days in the mouse. Bar = 1 mm. B) RT-PCR analysis of Prl7b1 transcripts in trophoblast dissected from placentation sites on indicated gestation days in the mouse. Three samples per gestation day were analyzed.
Establishment of Prl7b1 Null Mouse
The restricted expression of Prl7b1 in the invasive trophoblast cell lineage (Fig. 1) [19, 20] prompted an investigation of its biological role during placentation and pregnancy. Prl7b1 null mice were generated by gene-targeting strategies. The entire Prl7b1-coding region was replaced with a LacZ cassette (inserted at the ATG codon in the first exon) followed by a neomycin-resistance cassette (Fig. 2A). The mutation was successfully transmitted through the germline (Fig. 2B). Breeding of mice heterozygous for the Prl7b1 null mutation resulted in offspring genotypes that did not significantly deviate from the expected Mendelian ratio (number of litters: 29; number of pups: 201; Mendelian ratio: wild type, 45; heterozygotes, 106; Prl7b1 nulls, 50; mean litter size: 6.93). Wild-type and Prl7b1 null mutant mice exhibited similar fertility. The number of placentation sites and fetal and placental weights on gestation d17.5 were similar between wild-type and Prl7b1 null mice (number of placentation sites: wild type, 7.88 ± 1.0, n = 8 vs. Prl7b1 null, 8.56 ± 0.53, n = 9; fetal weights (g): wild type, 0.88 ± 0.04, n = 8 vs. Prl7b1 null, 0.79 ± 0.04, n = 9; placental weights (g): wild type, 0.095 ± 0.007, n = 8 vs. Prl7b1 null, 0.095 ± 0.002, n = 9). Furthermore, litter size was comparable for wild-type and Prl7b1 null mice at the first and second pregnancies (wild type: first pregnancy, 8.1 ± 0.9, n = 10; second pregnancy, 9.0 ± 1.2, n = 7; Prl7b1 null: first pregnancy, 8.4 ± 1.2, n = 18; second pregnancy, 8.1 ± 1.9, n = 7). Mouse placentas homozygous for the mutant allele did not express Prl7b1 (Fig. 2C) but instead exhibited LacZ activity in the invasive trophoblast cell population in a pattern similar to Prl7b1 expression in wild-type mice and confirming the integrity of the targeted mutagenesis (Fig. 3). Overall Prl7b1 null placentation sites exhibit structural and gene expression patterns (cytokeratin, Tpbpa, Prl2a1) on gestation d17.5 closely resembling wild-type placentation sites (Fig. 4). In summary, under standard laboratory housing conditions, wild-type and Prl7b1 null mice exhibited similar reproductive performance and placental structure.
FIG. 2.

Targeted disruption of the Prl7b1 gene in the mouse. A) Schematic representation of the mouse Prl7b1 gene (top) and the targeting construct (bottom). B) PCR analysis of genomic Prl7b1 locus from wild-type (+/+), heterozygous (+/−), and null (−/−) animals. C) RT-PCR analysis of Prl7b1 transcripts in gestation d17.5 placentas from wild-type (+/+) and Prl7b1 null (−/−) mice. Three samples per genotype were examined.
FIG. 3.
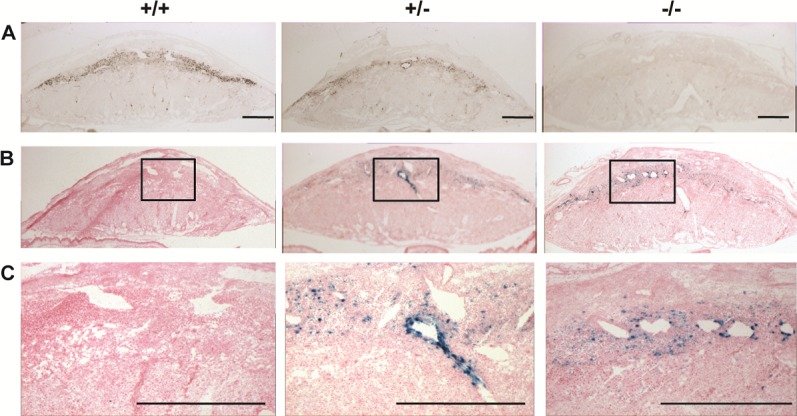
Examination of Prl7b1 expression at the maternal-fetal interface in wild-type and Prl7b1 mutant mice. A) In situ hybridization of Prl7b1 transcripts on tissue sections of gestation d17.5 placentation sites from wild-type (+/+), heterozygous (+/−), and Prl7b1 null (−/−) animals. B and C) LacZ reporter activity in gestation d17.5 placentation site tissue sections from wild-type (+/+), heterozygous (+/−), and Prl7b1 null (−/−) animals. Panel C represents high magnification images corresponding to the boxed areas in B. Bar = 1 mm.
FIG. 4.
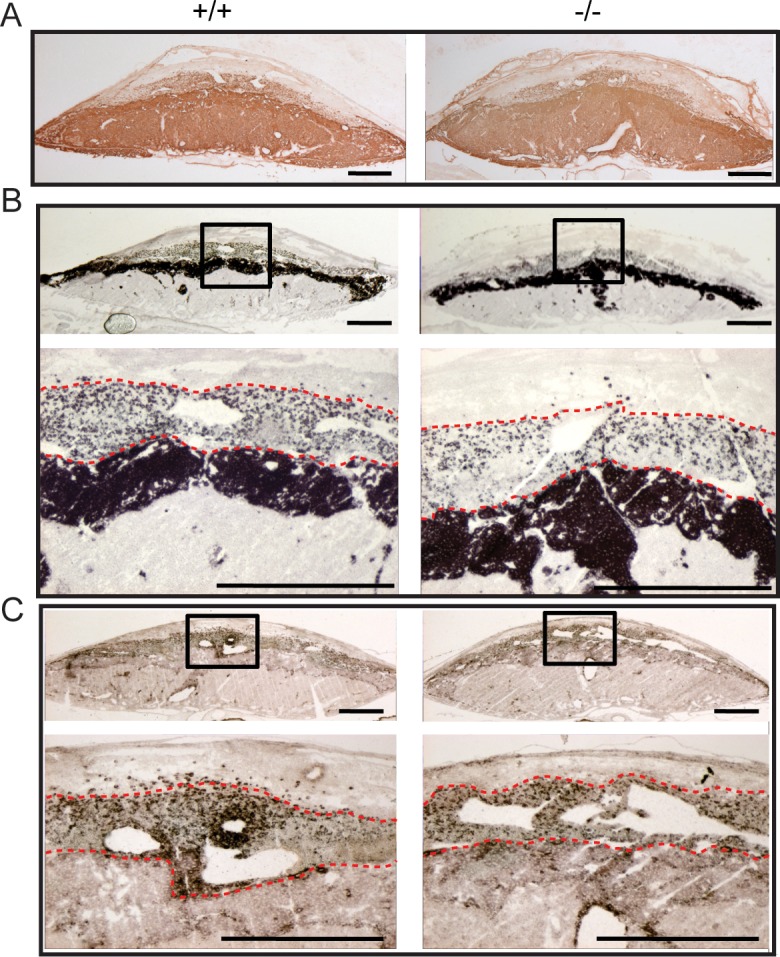
Wild-type and Prl7b1 null placentation sites exhibit similar organization and gene expression patterns on gestation d17.5. A) Trophoblast cell distributions in wild-type (+/+) and Prl7b1 null (−/−) placentation identified by cytokeratin immunohistochemical staining. In situ hybridization for Tpbpa (B) and Prl2a1 (C) transcripts in wild-type (+/+) and Prl7b1 null (−/−) placentation sites. Panels B and C contain low magnification (upper images) and high magnification images corresponding to the boxed areas (lower images). The red dashed lines form the boundaries of the invasive trophoblast cell infiltration into the decidual compartment. Bar = 1 mm.
Prl7b1 and Pregnancy-Dependent Adaptations to Maternal Hypoxia
The absence of a pronounced reproductive phenotype associated with the Prl7b1 null mutation was reminiscent of earlier observations of mice possessing null mutations for other members of the PRL gene family [3, 16, 17]. In these instances, null mutations at Prl4a1 and Prl8a2 loci did not significantly impact reproductive fitness under standard laboratory husbandry conditions but were each associated with compromised adaptations to maternal hypoxia. Maternal hypoxia evokes adaptive responses that alter the development of the placentation site [33, 34]. Groups of pregnant wild-type females mated with wild-type males and Prl7b1 null females mated with Prl7b1 null male were exposed to a range of oxygen tensions (9.5%, 10.5%, 11.5%, or 21% [atmospheric] O2) from gestation d7.5 to d17.5 (Fig. 5A), which spans the time period for placental Prl7b1 expression (Fig. 1). Pregnancies from wild-type mice were more vulnerable to maternal hypoxia (9.5% O2) and were associated with increased resorption rates, decreased fetal weights, and decreased fetal/placental weight ratios (Fig. 5, B–E). Uterine arteries were enlarged with prominent radial branches directed to each placentation site in Prl7b1 null mice exposed to low oxygen in comparison to similarly treated wild-type controls (Fig. 5F). In response to maternal hypoxia, wild-type placentas exhibited a striking reorganization, which included expansion of the junctional zone (Fig. 6). This is a response previously observed in pregnant rats exposed to maternal hypoxia [33, 34]. Most interestingly, these maternal hypoxia-dependent responses were not observed in placentation sites from Prl7b1 null mice (Fig. 6).
FIG. 5.

Effects of maternal hypoxia on pregnancy outcome at gestation d17.5 in wild-type and Prl7b1 null mice. A) Schematic diagram describing the maternal hypoxia exposure is shown. B) Conceptus reabsorption rate in wild-type (21% O2, n = 10; 11.5% O2, n = 4; 10.5% O2, n = 6; 9.5% O2, n = 9) and Prl7b1 null (21% O2, n = 14; 11.5% O2, n = 10; 10.5% O2, n = 9; 9.5% O2, n = 8) dams exposed to various oxygen tensions (mean ± SEM). Fetal weights (C) and fetal/placental weight ratios (D) from wild-type (21% O2, n = 10; 11.5% O2, n = 4; 10.5% O2, n = 6; 9.5% O2, n = 18) and Prl7b1 null (21% O2, n = 14; 11.5% O2, n = 10; 10.5% O2, n = 9; 9.5% O2, n = 24) dams exposed to various oxygen tensions (samples correspond to pregnancies; mean ± SEM). Asterisks in B, C, and D correspond to significant differences, *P < 0.05; **P < 0.01. E) Representative images of fetuses and placentas from wild-type and Prl7b1 null pregnancies exposed to maternal hypoxia (9.5% O2). F) Representative images of uteri containing conceptuses, fetuses, and placentas from wild-type and Prl7b1 null dams exposed to maternal hypoxia (10.5% or 9.5% O2).
FIG. 6.

Effects of maternal hypoxia on organization of placentation sites at gestation d17.5 in wild-type and Prl7b1 null mice. Isolectin B4 histochemical staining (A) and PECAM1 immunohistochemical staining (B) of gestation d17.5 placentation sites from wild-type and Prl7b1 null animals exposed to atmospheric oxygen (21% O2) or maternal hypoxia (10.5% or 9.5% O2). C) In situ hybridization for Tpbpa transcripts on gestation d17.5 placentation sites from wild-type and Prl7b1 null animals exposed to maternal hypoxia (9.5% O2). Panel C contains low magnification (upper images) and high magnification images corresponding to the boxed areas (lower images). Bar = 1 mm. Quantitative analysis of placentation sites of wild-type and Prl7b1 null dams exposed to various oxygen tensions (wild type: 21% O2, n = 6; 10.5% O2, n = 5; 9.5% O2, n = 5; Prl7b1 null: 21% O2, n = 5; 10.5% O2, n = 5; 9.5% O2, n = 5). Each sample corresponds to placental measurements from a different pregnancy. D) Junctional zone/placenta ratio. E) Labyrinth zone/placenta ratio. F) Labyrinth zone/junctional zone ratio. Values are presented as mean ± SEM. Letters above each mean value possessing a different identity reflect significant differences among the relevant groups (P < 0.05).
Collectively, Prl7b1 null pregnant mice failed to exhibit placental adaptations to maternal hypoxia. The absence of pregnancy-dependent adaptations to maternal hypoxia in Prl7b1 null mice was associated with enhanced fetal survival and fetal/placental growth.
DISCUSSION
Expansion of the PRL family is linked to pregnancy and uteroplacental function [1, 3]. We have some knowledge of the biology of classical members of the PRL family, which influence cell signaling through activation the PRLR. Insights into the biology of nonclassical PRL members is more limited. In this report, we investigate PRL7B1, a PRL family paralog expressed in migratory trophoblast cells situated at the uterine-trophoblast interface and embedded within the differentiated uterine stroma ([19, 20] and the present study). PRL7B1 deficient mice were successfully generated using standard embryonic stem cell-mediated mouse mutagenesis. Under standard housing conditions, pregnancy outcome did not appear to differ between wild-type and PRL7B1-deficient mice. Differences associated with PRL7B1 emerged when pregnant mice were exposed to hypoxia. The nature of these responses were somewhat complex. Placentas from wild-type animals exhibited a characteristic adaptation to hypoxia whereupon the junctional zone of the chorioallantoic placenta is preferentially expanded in size. This adaptive expansion was not observed in PRL7B1-deficient placentas. The outcome of the hypoxia-exposed pregnancies differed according to genotype. At gestation d17.5, uterine vascularity and fetal outcomes were superior in hypoxia-exposed PRL7B1 deficient than in hypoxia-exposed wild-type pregnancies. The results indicate that PRL7B1 contributes to the regulation of placental-associated adaptations to physiological stressors.
The absence of a pregnancy-related phenotype in mice with a deficiency of a PRL family member may seem perplexing but is not new. PRL4A1 and PRL8A2 are pregnancy-associated cytokines/hormones [35, 36]. Similar to the Prl7b1 null mutant mouse, genetic disruptions of Prl4a1 and Prl8a2 yield modest pregnancy-related phenotypes when examined under standard housing conditions [16, 17]. Functional redundancies are certainly possible, especially from such a large family of related proteins; however, such observations challenge the evolutionary drive for retaining these genes within the mouse genome. Instead their preservation may relate to other aspects of the reproductive success of the species, especially in less than favorable habitats. To this end, PRL4A1 and PRL8A2 have been shown to facilitate pregnancy-dependent adaptations to physiological stressors and improve pregnancy outcomes [16, 17]. Maternal hypoxia exposures led to a discrimination between wild-type and PRL7B1-deficient pregnancies but in an unexpected direction.
Maternal hypoxia exposure has been effectively used as an experimental tool to dissect pregnancy-dependent adaptations [33, 34]. Among these adaptations are structural changes in the organization of the placentation site. In the rat, maternal hypoxia exposure evokes an accelerated and increased depth of endovascular trophoblast invasion and a preferential expansion of the junctional zone relative to the labyrinth zone of the chorioallantoic placenta [33, 34]. In the mouse, structural adaptations to maternal hypoxia are restricted to expansion of the junctional zone (present study). The junctional zone is the origin of progenitors for invasive trophoblast cell development and the major placental site for maternally directed endocrine activities and energy storage [24, 37]. Hypoxia-stimulated junctional zone growth came at a cost to the development of the labyrinth zone, which is responsible for bidirectional transport of nutrients and wastes between maternal and fetal compartments. Failed pregnancies at 9.5% oxygen in wild-type females may relate to a maladaptation associated with an inappropriate balance between junctional zone and labyrinth zone development. The presence of PRL7B1 was required for hypoxia-dependent expansion of the junctional zone. PRL7B1 may possess autocrine and/or paracrine actions within the context of hypoxia-driven expansion of trophoblast cells within the junctional zone. Interestingly, another feature of the PRL7B1-positive migratory trophoblast is their accumulation of glycogen [38, 39], which may provide a linkage to PRL7B1 actions and their involvement in adaptations to hypoxia. Relative junctional zone size has also been associated with nutrient availability. Dietary protein restriction leads to an expansion of the junctional zone [40], whereas a high fat diet is associated with attenuated junctional zone development [41]. These diet-mediated effects on junctional zone development may be secondary to the regulation of PRL7B1 expression [42]. Alternatively, PRL7B1 actions may be indirect through systemic effects on nutrient availability, which is consistent with known roles for other PRL family paralogs on maternal metabolic adaptations [43–45].
Species-specific PRL family expansions have been viewed as a strategy for improving pregnancy-dependent adaptations to environmental challenges [3]. Several PRL family members (PRL, PRL4A1, PRL8A2) improve adaptations to physiological stressors, including pregnancy outcomes [15, 16, 46–48]. Our findings with the PRL7B1-deficient mouse challenges this concept. Fetal viability at gestation d17.5 in hypoxia-exposed pregnancies was superior in females devoid of PRL7B1 in comparison to wild-type females. These observations are seemingly paradoxical and infer a level of unexpected complexity. A few possibilities are evident. First, perhaps we are not viewing the PRL7B1 phenotype along the appropriate timeline. The evolutionary drive for retention of Prl7b1 in the mouse and rat genomes may serve as a sensor of pregnancy quality and actually a successful adaptation to a stressor. Termination of pregnancies may be adaptive, preventing impending maternal demise or the production of offspring with pathologies affecting their survival. Second, it is possible that there is specificity associated with the PRL family and the nature of the physiological stressor. Maybe the Prl7b1 gene has been retained in the genome because it provides adaptive benefits to other types of physiological stressors (dietary, thermal, etc.) but not hypoxia and could represent an evolutionary prioritization of stressors. It is also conceivable that Prl7b1 is an evolutionary relic or path to a future mode of adaptation to an undetermined stressor. Third, we should also consider that the contributions of PRL7B1 to the regulation of adaptations to physiological stressors may not be essential for survival of the species. Some of these issues should clarify as we learn more about the targets and biological actions of PRL7B1.
ACKNOWLEDGMENT
We thank Stacy McClure for administrative assistance. We acknowledge the technical assistance of the University of Kansas Medical Center Transgenic and Gene Targeting Institutional Facility for generating the mutant mice. This facility is supported by a Center of Biomedical Research Excellence (GM104936).
Footnotes
Current address: Departments of Biological Sciences and Pharmaceutical Sciences, St. John's University, Queens, NY 11439.
Current address: Department of Biochemistry, Bangabandhu Sheikh Mujib Medical University, Dhaka, Bangladesh.
This work was supported by the National Institutes of Health, HD020676.
REFERENCES
- Soares MJ. The prolactin and growth hormone families: pregnancy-specific hormones/cytokines at the maternal-fetal interface. Reprod Biol Endocrinol. 2004;2:51. doi: 10.1186/1477-7827-2-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam SMK, Ain R, Konno T, Ho-Chen JK, Soares MJ. The rat prolactin gene family locus: species-specific gene family expansion. Mamm Genome. 2006;17:858–877. doi: 10.1007/s00335-006-0010-1. [DOI] [PubMed] [Google Scholar]
- Soares MJ, Konno T, Alam SMK. The prolactin family: effectors of pregnancy-specific adaptations. Trends Endocrinol Metab. 2007;18:114–121. doi: 10.1016/j.tem.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Horseman ND, Zhao W, Montecino-Rodriguez E, Tanaka M, Nakashima K, Engle SJ, Smith F, Markoff E, Dorshkind K. Defective mammopoiesis, but normal hematopoiesis, in mice with a targeted disruption of the prolactin gene. EMBO J. 1997;16:6926–6935. doi: 10.1093/emboj/16.23.6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormandy CJ, Camus A, Barra J, Damotte D, Lucas B, Buteau H, Edery M, Brousse N, Babinet C, Binart N, Kelly PA. Null mutation of the prolactin receptor gene produces multiple reproductive defects in the mouse. Genes Dev. 1997;11:167–178. doi: 10.1101/gad.11.2.167. [DOI] [PubMed] [Google Scholar]
- Huang C, Snider F, Cross JC. Prolactin receptor is required for normal glucose homeostasis and modulation of beta-cell mass during pregnancy. Endocrinology. 2009;150:1618–1626. doi: 10.1210/en.2008-1003. [DOI] [PubMed] [Google Scholar]
- Jackson D, Volpert OV, Bouck N, Linzer DI. Stimulation and inhibition of angiogenesis by placental proliferin and proliferin-related protein. Science. 1994;266:1581–1584. doi: 10.1126/science.7527157. [DOI] [PubMed] [Google Scholar]
- Volpert O, Jackson D, Bouck N, Linzer DI. The insulin-like growth factor II/mannose 6-phosphate receptor is required for proliferin-induced angiogenesis. Endocrinology. 1996;137:3871–3876. doi: 10.1210/endo.137.9.8756559. [DOI] [PubMed] [Google Scholar]
- Lin J, Linzer DI. Induction of megakaryocyte differentiation by a novel pregnancy-specific hormone. J Biol Chem. 1999;274:21485–21489. doi: 10.1074/jbc.274.30.21485. [DOI] [PubMed] [Google Scholar]
- Bittorf T, Jaster R, Soares MJ, Seiler J, Brock J, Friese K, Müller H. Induction of erythroid proliferation and differentiation by a trophoblast-specific cytokine involves activation of the JAK/STAT pathway. J Mol Endocrinol. 2000;25:253–262. doi: 10.1677/jme.0.0250253. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Lin J, Linzer DI. Reactivation of a hematopoietic endocrine program contributes to recovery from thrombocytopenia. Mol Endocrinol. 2002;16:1386–1393. doi: 10.1210/mend.16.6.0847. [DOI] [PubMed] [Google Scholar]
- Zhou B, Kong X, Linzer DI. Enhanced recovery from thrombocytopenia and neutropenia in mice constitutively expressing a placental hematopoietic cytokine. Endocrinology. 2005;146:64–70. doi: 10.1210/en.2004-1011. [DOI] [PubMed] [Google Scholar]
- Zhou B, Lum HE, Lin J, Linzer DI. Two placental hormones are agonists in stimulating megakaryocyte growth and differentiation. Endocrinology. 2002;143:4281–4286. doi: 10.1210/en.2002-220447. [DOI] [PubMed] [Google Scholar]
- Müller H, Liu B, Croy BA, Head JR, Hunt JS, Dai G, Soares MJ. Uterine natural killer cells are targets for a trophoblast cell-specific cytokine, prolactin-like protein A. Endocrinology. 1999;140:2711–2720. doi: 10.1210/endo.140.6.6828. [DOI] [PubMed] [Google Scholar]
- Ain R, Tash JS, Soares MJ. Prolactin-like protein-A is a functional modulator of natural killer cells at the maternal-fetal interface. Mol Cell Endocrinol. 2003;204:65–74. doi: 10.1016/s0303-7207(03)00125-4. [DOI] [PubMed] [Google Scholar]
- Ain R, Dai G, Dunmore JH, Godwin AR, Soares MJ. A prolactin family paralog regulates reproductive adaptations to a physiological stressor. Proc Natl Acad Sci U S A. 2004;101:16543–16548. doi: 10.1073/pnas.0406185101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam SMK, Konno T, Dai G, Lu L, Wang D, Dunmore JH, Godwin AR, Soares MJ. A uterine decidual cell cytokine ensures pregnancy-dependent adaptations to a physiological stressor. Development. 2007;134:407–415. doi: 10.1242/dev.02743. [DOI] [PubMed] [Google Scholar]
- Soares MJ, Alam SM, Duckworth ML, Horseman ND, Konno T, Linzer DI, Maltais LJ, Nilsen-Hamilton M, Shiota K, Smith JR, Wallis M. A standardized nomenclature for the mouse and rat prolactin superfamilies. Mamm Genom. 2007;18:154–156. doi: 10.1007/s00335-007-9003-y. [DOI] [PubMed] [Google Scholar]
- Wiemers DO, Ain R, Ohboshi S, Soares MJ. Migratory trophoblast cells express a newly identified member of the prolactin gene family. J Endocrinol. 2003;179:335–346. doi: 10.1677/joe.0.1790335. [DOI] [PubMed] [Google Scholar]
- Wiemers DO, Shao L-J, Ain R, Dai G, Soares MJ. The mouse prolactin gene family locus. Endocrinology. 2003;144:313–325. doi: 10.1210/en.2002-220724. [DOI] [PubMed] [Google Scholar]
- Ain R, Canham LN, Soares MJ. Gestational stage-dependent intrauterine trophoblast cell invasion in the rat and mouse: novel endocrine phenotype and regulation. Dev Biol. 2003;260:176–190. doi: 10.1016/s0012-1606(03)00210-0. [DOI] [PubMed] [Google Scholar]
- Pijnenborg R, Vercruysse L. Animal models of deep trophoblast invasion In Pijnenborg R, Brosens I, Romero R. (eds.), Placental Bed Disorders Cambridge: Cambridge University Press; 2010. 127 139 [Google Scholar]
- Pijnenborg R, Vercruysse L, Hanssens M. The uterine spiral arteries in human pregnancy: facts and controversies. Placenta. 2006;27:939–958. doi: 10.1016/j.placenta.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Soares MJ, Chakraborty D, Rumi MAK, Konno T, Renaud SJ. Rat placentation: an experimental model for investigating the hemochorial maternal-fetal interface. Placenta. 2012;33:233–243. doi: 10.1016/j.placenta.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Red-Horse K, Zhou Y, Genbacev O, Prakobphol A, Foulk R, McMaster M, Fisher SJ. Trophoblast differentiation during embryo implantation and formation of the maternal-fetal interface. J Clin Invest. 2004;114:744–754. doi: 10.1172/JCI22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace AE, Fraser R, Cartwright JE. Extravillous trophoblast and decidual natural killer cells: a remodelling partnership. Hum Reprod Update. 2012;18:458–471. doi: 10.1093/humupd/dms015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares MJ, Chakraborty D, Kubota K, Renaud SJ, Rumi MA. Adaptive mechanisms controlling uterine spiral artery remodeling during the establishment of pregnancy. Int J Dev Biol. 2014;58:247–259. doi: 10.1387/ijdb.140083ms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris LK. Trophoblast-vascular cell interactions in early pregnancy: how to remodel a vessel Placenta 2010. 31 (Suppl): S93 S98 [DOI] [PubMed] [Google Scholar]
- Harris LK. Transformation of the spiral arteries in human pregnancy: key events in the remodelling timeline Placenta 2011. 32 (Suppl 2): S154 S158 [DOI] [PubMed] [Google Scholar]
- Valenzuela DM, Murphy AJ, Frendewey D, Gale NW, Economides AN, Auerbach W, Poueymirou WT, Adams NC, Rojas J, Yasenchak J, Chernomorsky R, Boucher M, et al. High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat Biotechnol. 2003;21:652–659. doi: 10.1038/nbt822. [DOI] [PubMed] [Google Scholar]
- Ain R, Konno T, Canham LN, Soares MJ. Phenotypic analysis of the placenta in the rat. Methods Mol Med. 2006;121:295–313. doi: 10.1385/1-59259-983-4:293. [DOI] [PubMed] [Google Scholar]
- Konno T, Graham AR, Rempel LA, Ho-Chen JK, Alam SMK, Bu P, Rumi MAK, Soares MJ. Subfertility linked to combined luteal insufficiency and uterine progesterone resistance. Endocrinology. 2010;151:4537–4550. doi: 10.1210/en.2010-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario GX, Konno T, Soares MJ. Maternal hypoxia-activated endovascular trophoblast cell invasion. Dev Biol. 2008;314:362–375. doi: 10.1016/j.ydbio.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty D, Rumi MAK, Konno T, Soares MJ. Natural killer cells direct hemochorial placentation by regulating HIF-dependent trophoblast lineage decisions. Proc Natl Acad Sci U S A. 2011;108:16295–16300. doi: 10.1073/pnas.1109478108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller H, Ishimura R, Orwig KE, Liu B, Soares MJ. Homologues for prolactin-like proteins A and B are present in the mouse. Biol Reprod. 1998;58:45–51. doi: 10.1095/biolreprod58.1.45. [DOI] [PubMed] [Google Scholar]
- Orwig KE, Ishimura R, Müller H, Liu B, Soares MJ. Identification and characterization of a mouse homolog for decidual/trophoblast prolactin-related protein. Endocrinology. 1997;138:5511–5517. doi: 10.1210/endo.138.12.5628. [DOI] [PubMed] [Google Scholar]
- Soares MJ, Chapman BM, Rasmussen CA, Dai G, Kamei T, Orwig KE. Differentiation of trophoblast endocrine cells. Placenta. 1996;17:277–289. doi: 10.1016/s0143-4004(96)90051-x. [DOI] [PubMed] [Google Scholar]
- Adamson SL, Yong L, Whiteley KJ, Holmyard D, Hemberger M, Pfarrer C, Cross JC. Interactions between trophoblast cells and the maternal and fetal circulation in the mouse placenta. Dev Biol. 2002;250:358–371. doi: 10.1016/s0012-1606(02)90773-6. [DOI] [PubMed] [Google Scholar]
- Coan PM, Conroy N, Burton GJ, Ferguson-Smith AC. Origin and characteristics of glycogen cells in the developing murine placenta. Dev Dyn. 2006;235:3280–3294. doi: 10.1002/dvdy.20981. [DOI] [PubMed] [Google Scholar]
- Doherty CB, Lewis RM, Sharkey A, Burton GJ. Placental composition and surface area but not vascularization are altered by maternal protein restriction in the rat. Placenta. 2003;24:34–38. doi: 10.1053/plac.2002.0858. [DOI] [PubMed] [Google Scholar]
- Mark PJ, Sisala C, Connor K, Patel R, Lewis JL, Vickers MH, Waddell BJ, Sloboda DM. A maternal high-fat diet in rat pregnancy reduces growth of the fetus and the placental junctional zone, but not labyrinth zone growth. J Dev Origins Health Dis. 2011;2:63–70. [Google Scholar]
- Broad KD, Keverne EB. Placental protection of the fetal brain during short-term food deprivation. Proc Natl Acad Sci U S A. 2011;108:15237–15241. doi: 10.1073/pnas.1106022108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freemark M. Regulation of maternal metabolism by pituitary and placental hormones: roles in fetal development and metabolic programming Horm Res 2006. 65 (Suppl 3): 41 49 [DOI] [PubMed] [Google Scholar]
- Newbern D, Freemark M. Placental hormones and the control of maternal metabolism and fetal growth. Curr Opin Endocrinol Diabetes Obes. 2011;18:409–416. doi: 10.1097/MED.0b013e32834c800d. [DOI] [PubMed] [Google Scholar]
- John RM. Epigenetic regulation of placental endocrine lineages and complications of pregnancy. Biochem Soc Trans. 2013;41:701–709. doi: 10.1042/BST20130002. [DOI] [PubMed] [Google Scholar]
- Dorshkind K, Horseman ND. Anterior pituitary hormones, stress, and immune system homeostasis. Bioessays. 2001;23:288–294. doi: 10.1002/1521-1878(200103)23:3<288::AID-BIES1039>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Dugan AL, Thellin O, Buckley DJ, Buckley AR, Ogle CK, Horseman ND. Effects of prolactin deficiency on myelopoiesis and splenic T lymphocyte proliferation in thermally injured mice. Endocrinology. 2002;143:4147–4151. doi: 10.1210/en.2002-220515. [DOI] [PubMed] [Google Scholar]
- Dugan AL, Schwemberger S, Babcock GF, Buckley D, Buckley AR, Ogle CK, Horseman ND. Effects of prolactin level on burn-induced aberrations in myelopoiesis. Shock. 2004;21:151–159. doi: 10.1097/01.shk.0000108401.56565.e2. [DOI] [PubMed] [Google Scholar]