

Abstract
Poly(vinyl alcohol) hydrogels have a long and successful history of applications in biomedicine. Historically, these matrices were developed to be nondegradable—limiting their utility to applications as permanent implants. For tissue engineering and drug delivery, herein we develop spontaneously eroding physical hydrogels based on PVA. We characterize in detail a mild, noncryogenic method of producing PVA physical hydrogels using poly(ethylene glycol) as a gelating agent, and investigate PVA molar mass as a means to define the kinetics of erosion of these biomaterials. PVA hydrogels are characterized for associated inflammatory response in adhering macrophages, antiproliferative effects mediated through delivery of cytotoxic drugs to myoblasts, and pro-proliferative activity achieved via presentation of conjugated growth factors to endothelial cells. Together, these data present a multiangle characterization of these novel multifunctional matrices for applications in tissue engineering and drug delivery mediated by implantable biomaterials.
Introduction
Hydrogel biomaterials offer unique opportunities in diverse biomedical applications, specifically due to their biocompatibility, soft human tissue-like mechanical properties, and well-developed means for controlled delivery of therapeutic cargo to the surrounding environment.1−3 However, while academic developments are impressive and widely adopted, progression from laboratory to clinic has been slow.4 In large part, this is due to the limited choice of suitable hydrogel matrices that combine the sought-after material characteristics and drug delivery properties with due biocompatibility.5,6 Indeed, virtually any water-soluble macromolecule can be used to make up a hydrogel,1,6,7 yet only a small number of hydrogels have regulatory approval for use in human patients. One potentially fruitful approach toward broadening the scope and utility of hydrogels in clinical applications could be based on engineering and fine-tuning of the materials already approved for the use in humans into novel applications.
One hydrogel biomaterial with a long and successful history of use in humans is based on poly(vinyl alcohol) (PVA).8−12 A specific example of successful use of these hydrogels is for production of embolic bodies13 and implants for prevention of postoperative tissue collapse.9 Historically, these matrices were designed to be nonbiodegradable, permanent implants. Another area of use of PVA hydrogels—specifically physical hydrogels—is immobilization of enzymes and cells for biomass conversion.10−12 These hydrogels are typically prepared via “cryo-gelation” employing repetitive freeze–thaw cycles to make up highly porous matrices, a feature highly beneficial for diffusion and fast exchange of solutes between the hydrogel and solution bulk. Cryogel matrices too are designed as highly stable materials poised to retain their stability over extended periods of time, and are typically comprised of high molar mass chains of PVA (100 kDa and above). These examples highlight the utility of PVA in biomedicine but reveal that, in their well-established form, these hydrogels lack means for biodegradation and are rather ill-suited for controlled retention and slow release of drugs.
Over the past few years, we devoted significant attention to re-engineering of PVA physical hydrogels to make these materials suited in a variety of biomedical applications.14−16 Specifically, physical hydrogels based on PVA were successfully engineered to undergo spontaneous erosion under physiological conditions.15,17 Biodegradation or erosion is pivotal for applications such as controlled drug release mediated by implantable biomaterials. It is also important for tissue engineering applications whereby de novo assembled tissue should be able to fully replace the biomaterial.18 The cornerstone to our design of degradable PVA hydrogels was the synthesis of polymer samples with defined molar mass and narrow dispersity, that is, high uniformity of chains by length. Toward this end, PVA was prepared from its polymer precursor, poly(vinyl acetate), obtained via the controlled radical polymerization technique (RAFT).19 The latter allows controlling polymer molar mass through the ratio of concentrations of the monomer and the chain transfer reagent and can be varied in a broad range. In the studied range of PVA molar mass (up to 35 kDa), physical hydrogels prepared thereof underwent spontaneous erosion under physiological conditions and hydrogels comprised of shorter polymer chains underwent significantly faster erosion.15
From a different perspective, synthesis of PVA through RAFT polymerization also afforded polymer chains with terminal groups amenable for bioconjugation.14 Indeed, RAFT polymerization as such proceeds as “insertion” of monomer units between the so-called “R” and “Z” functionalities of the RAFT agent with an end result that most of the synthesized chains are equipped with the chosen R and Z groups.20 In our work, the R group contained a phthalimide-protected amine. Upon removal of the phthalimide, end-localized amine groups were converted into protected thiol functionality, such that upon a reaction with dithiothreitol (DTT) the chains release 2-nitro-5-thiobenzoate, a chromophore with a high extinction coefficient, for solution based facile quantification of chains (Figure 1).15 A terminal disulfide group is also highly attractive for bioconversions and conjugations through thiol–disulfide exchange chemistry most attractive due to fast kinetics, high specificity of reactions, and possibility to conduct conjugation under physiological conditions (phosphate buffered saline, pH 7.4). Proof of concept conjugation was demonstrated toward diverse cargo, from oligopeptides14 to globular proteins and growth factors17 as well as cholesterol21 for “branding” liposomes within PVA hydrogels.16
Figure 1.

Chemical formula of PVA (top) and schematic illustration of chemical reactions of PVA via terminal groups activated toward thiol–disulfide exchange. Cleavage of the terminal disulfide (be it in solution or within the gel phase) liberates a chromophore—allowing to quantify the polymer chains via a solution-based UV–vis readout; reaction with thiol-modified growth factors creates hydrogel matrices for localized stimulation of proliferation of adhering cells; conjugation to cholesterol is used toward anchoring PVA chains into liposomes and “branding” the latter within the structure of PVA hydrogels—for delivery of hydrophobic drugs.
We also invested much effort into designing physical hydrogels through a noncryogenic manufacturing route, specifically to avoid macro-porosity and maximize retention of polymer chains in the hydrogel matrix.22,23 In the majority of our previous studies, hydrogels were obtained through “salting out”, gradual drawing of water from the hydrogel using concentrated solutions of a kosmotropic salt (sodium sulfate). Water extraction and ensuing hydrogelation of PVA chains could also be achieved using liquid, oligomeric samples of PEG.17 The latter approach is potentially more friendly toward immobilized biological cargo such as proteins. However, detailed investigation of this method of PVA hydrogelation is missing. One goal of the present study was therefore to quantitatively characterize the PEG-assisted hydrogelation of PVA with regards to the hydrogel composition, that is, retention of chains within the hydrogel phase during assembly and incubation under physiological conditions.
Another aim of this study considers the fate of polymer chains released from the hydrogel and the possible effects these may elicit on the adhering cells or adjacent tissues. On a systemic level, PVA chains with a molar mass as high as >100 000 were shown to be eliminated from the body via a renal pathway.24 This observation is encouraging and means that upon implantation, spontaneously eroding matrices release polymer chains amenable for excretion. In this work, we take a closer look and analyze the fate of PVA chains with regard to the uptake by the cells adhering to the hydrogel matrix and most importantly, with regard to ensuing inflammatory response, depending on the molar mass of chains constituting the hydrogel.
Further aspects which we investigate in this work pertain to the interplay between controlled matrix dissolution, opportunities in bioconjugation, and therapeutic effects elicited by the hydrogels. Indeed, polymer molar mass may have a dual influence on the biomaterials’ properties in that chain length defines both the stability of the hydrogel to dissolution and the content of available sites for conjugation (through polymer terminal groups). Both parameters are important to control the use of biomaterials in biomedicine and are analyzed below in detail. With regard to bioconjugation and drug delivery, we specifically investigate the suitability of PVA chains with varied molar mass as extensions toward “branding” of incorporated liposomes in the structure of the hydrogel for delivery of hydrophobic drugs16 and functionalization of hydrogels with growth factors for accelerated proliferation of adhering endothelial cells.17 Taken together, this work presents a multiangle analysis of PVA physical hydrogels as functional biomaterials with properties defined by the rational choice of chain lengths of the constituting polymer chains.
Materials and Methods
All chemicals were purchased from Sigma-Aldrich and used without any further purification unless stated otherwise. Poly(dimethyl silicone), PDMS elastomer (Sylgard 184), and curing agent were obtained from Dow Corning, USA. The secondary antibody, Alexa Fluor 488 F(ab′)2 fragment of goat antimouse IgG, HUVECs, M200 medium, LSGS, fetal bovine serum (FBS < 5 EU/mL), and PrestoBlue cell viability kit were obtained from Invitrogen. Fluorescent lipids 1-oleoyl-2-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl]-sn-glycero-3-phosphocholine (NBD-PC) were purchased from Avanti Polar Lipids, USA. All buffer solutions were prepared using ultrapure water (Milli-Q gradient A 10 system, 18.2 MΩ/cm resistivity).
General Considerations
PVA Synthesis
The synthesis of PVA via RAFT polymerization to produce amine terminal groups was performed as described elsewhere.15,19 In short, vinyl acetate was polymerized using a phthalimide-containing RAFT agent. The resulting polymer was first treated with aqueous hydrazine to remove the terminal phthalimide functionality and further saponified (i.e., treated to remove the acetate groups) to produce PVA chains with terminal amine groups. Further end group modification was carried out by dissolving amine terminated PVA in carbonate buffer (pH 8.3). To this was added first a solution of Ellman’s reagent and then a freshly prepared solution of 2-iminothiolane. The mixture gradually became deep orange (illustrating progression of reaction) and was allowed to stir for 16 h. The target polymer, PVA-ER, was recovered via precipitation into excess methanol and filtration followed by trituration with copious amounts of methanol.15 In order to prevent degradation of the disulfide linkage, the dissolution of PVA-ER was carried out by gentle and short heating in MQ water. For fluorescent labeling of the amine terminal groups, PVA was dissolved in NaHCO3 buffer (pH 8.3) and mixed with FITC dissolved in dimethyl sulfoxide (DMSO). The reaction was incubated overnight after which the polymer was purified by gel filtration on a PD-10 Sephadex G-25 M column and freeze-dried.22
Master and PDMS Stamp Preparation
Micropatterns were created by standard photolithography and following the manufacturer’s manual of the photoresist (SU-8 3025, MicroChem, USA).22 To produce elastomeric molds, PDMS elastomer and curing agent were mixed in a 10:1 volume ratio, respectively, degassed, poured over the photoresist structured micropatterns, and cured for 3 h at 80 °C.
Microtransfer Molding (μTM)
Surface adhered hydrogels were prepared via μTM. In a typical procedure, 1.5 μL of PVA solution was placed on the PDMS stamp, covered with a 9 mm cover glass slide, and placed in a clamping system fastened at finger tight pressure for 24 h. Upon disassembly, the surface adhered PVA films were stabilized in PEG400 (MW 400 Da) for 1 h at 37 °C. Once stabilized, samples were washed three times in 4 °C PBS to remove any residual PEG400.
Visualization
Visualization of the samples was performed using CLSM (CLSM, Axiovert microscope coupled to an LSM 700 confocal laser scanning module, Zeiss, Germany) as well as an inverted microscope (Axio Observer Z1, Zeiss, Germany).
Cell Work
Primary human umbilical vein endothelial cells, pooled (Gibco) and RAW 264.7 mouse leukemic monocyte macrophage (European Collection of Cell Cultures), and C2C12 mouse myoblast (American Type Culture Collection) cell lines were used. HUVECs were cultured in M200 medium supplemented with 1% penicillin/streptomycin (P/S) and low serum growth supplement according to vendor recommendations. Macrophages were cultured in Dulbecco’s modified Eagle’s Medium (DMEM) supplemented with 10% FBS and 1% P/S and passaged with a cell scraper. Myoblast cells were cultured in DMEM supplemented with 10% FBS, 1% P/S, and 1 mM sodium pyruvate. All cells were cultured in 75 cm2 culture flasks at 37 °C and 5% CO2. Trypsinization was used to passage HUVEC and C2C12 cells. All samples were UV-sterilized for 15 min prior to cell seeding.
Cell Viability
Cell viability was determined by removing the medium and adding 200 μL of fresh medium supplemented with 20 μL of PrestoBlue and incubated at 37 °C and 5% CO2 for 30 min (60 min for HUVEC). The samples were transferred to a black 96-well plate for fluorescence reading using a plate reader (Enspire PerkinElmer) (ex/em λ = 560/590 nm).
Hydrogel Degradation
To quantitatively analyze the polymer content, hydrogels prepared using PVA-ER (4, 8, 12, or 16 wt % with molecular weights ranging from 6, 17, and 31 kDa) were stabilized as described above and subsequently incubated in PBS at 4 or 37 °C for 1 h, 24 h, 3 days, or 7 days. After any time point, the PBS was collected and hydrogels were replenished with fresh PBS. To quantify the amount of released polymer from the hydrogel and the amount left in the matrix, DTT was added to a final concentration of 5 g/L. The release of the NTB chromophore is directly proportional to the polymer chains present in the solution and hydrogel matrix. The absorbance (λ = 412 nm) of aspirated supernatants was measured using a plate reader after 10 min of incubation at 37 °C. For comparison, the total amount of polymer loaded was individually analyzed in nonstabilized hydrogels.
Macrophages and Inflammatory Response
Hydrogel samples were prepared as described above using PVA samples of 6, 17, and 31 kDa and 12 wt % solutions. To promote cell adhesion, PLL was added to the polymer solution yielding a final concentration of 1 g/L. For flow cytometry, PVA-FITC was used for the hydrogel assembly. For visualization of cell adhesion, RAW 264.7 cells were seeded onto the substrates (60 000 cells/well in 2 mL of medium in 12 well plates) and allowed to adhere at 37 °C and 5% CO2 for 24 h. The cells were washed twice with 2 mL of PBS and fixed using 4% PFA solution for 20 min and subsequently washed two times in PBS. The nuclei were stained with DAPI (1 μg/mL) for 1 h, subsequently washed with PBS twice, and stored in PBS at 4 °C until mounted on a glass cover slide using mounting media (Eukitt, Sigma) for visualization.
Polymer Uptake
To quantify polymer uptake, RAW 264.7 cells were seeded directly onto the substrates (30 000 cells/well in 200 μL of media in 48 well plates). The cells were allowed to adhere at 37 °C and 5% CO2 for 24 h. For flow cytometry analysis, the samples were rinsed twice with 500 μL of PBS and harvested using 100 μL of trypsin. The samples were further washed twice with 200 μL of cold PBS and collected for analysis. The cells were analyzed using a C6 Flow Cytometer (Accuri Cytometers, Inc.) using an excitation wavelength of λ = 488 nm. At least 1000 cells were analyzed. The autofluorescence of cells grown on glass slides has been subtracted in all the presented results.
Reagents for Quantification of Nitric Oxide via the Griess Assay
Reagent A was prepared by dissolving 100 mg of N-(1-napthyl)ethylenediamine dihydrochloride in 100 mL of MQ water, yielding a final concentration of 1 g/L. Reagent B was prepared by dissolution of 1 g of sulfanilic acid in 94.2 mL of MQ water supplemented with 5.77 mL of phosphoric acid (86.6%), giving a final concentration of 10 g/L. For the Griess nitrite standard (0.1 M), 6.9 mg of sodium nitrite was dissolved in 1 mL of MQ water. All reagents were stored at 4 °C and protected from light.
Analyses of the Inflammatory Response
RAW 264.7 cells in DMEM media (10% FBS, 1% P/S) were seeded onto the hydrogel substrates (30 000 cells/well in 200 μL of medium) and placed in the incubator at 37 °C and 5% CO2 for 24 h. For the controls, cells were seeded on untreated coverslips. After 24 h, media was removed and replenished with 180 μL of DMEM (phenol-red free, 10%, 1% P/S, 1% l-glutamine, 20 μL of PBS) containing the reagents of interest. NO production was stimulated at this point through the addition of 1 μg/mL LPS (Escherichia coli 026:B6). Samples with unstimulated cells (20 μL of PBS) were included as a reference. The cells were incubated for an additional 24 h, and 50 μL of media was transferred to a new multiplate for NO quantification via Griess assay (50 μL sample + 50 μL reagent B, 5 min wait, 50 μL reagent A, 5 min wait, readout of absorbance at 548 nm) using a plate reader (Enspire PerkinElmer). NO levels were quantified from a standard curve of a serially diluted sodium nitrite standard prepared on the same plate. Cell viability was performed after removal of medium for the Griess assay.
Lipogels
Liposome Assembly
Unilamellar liposome solutions were prepared by evaporation of chloroform solution of 5 mg of DOPC lipids and 0.8 mg of thiocholesterol, followed by further drying under a vacuum for 1 h and subsequent hydration using 50 μL of carbonate buffer containing PVA-ER (40 g/L) and 200 μL MQ water. The solution was extruded through 100 nm filters. For all PVA molecular weights employed, the liposome-polymer and polymers making up the matrix were identical. For fluorescently labeled liposomes, 1 wt % of NBD-PC was added to the lipid mixture. For encapsulation of paclitaxel, 200 μL of 4 g/L paclitaxel dissolved in chloroform was added to the lipid and thiocholesterol mixture. The extruded liposome solution was dialyzed against HEPES buffer (pH, 7.4, 10 mM HEPES, 150 mM NaCl) for 1 h, exchanging the buffer after 30 min. DLS (Malvern Instruments Zetasizer nano S90) was employed to analyze the liposomes with hydrodynamic diameters and polydispersity indices.
μTM
Surface adhered lipogels were prepared by heating PVA (24 wt %, 6, 17, and 31 kDa) to 90 °C for 5 min to homogenize the solution. Once brought to room temperature, PVA and liposome solution were mixed in a 1:1 volume ratio, resulting in a 12 wt % polymer solution. For cell experiments, PLL was blended in to give a final concentration of 1 g/L. PDMS molds with 2 μm deep and 10 μm wide wells with centers spaced by 20 μm were utilized. Upon disassembly, the surface adhered PVA films were stabilized in PEG400 (MW 400 Da) for 1 h at 37 °C. Once stabilized, samples were washed three times in 4 °C PBS to remove any residual PEG400.
Cell Viability
The amount of encapsulated paclitaxel in the liposomes was quantified by mixing 200 μL of paclitaxel encapsulated liposomes with 200 μL of chloroform. The aqueous and organic phases were allowed to settle before extraction of the organic phase and measurement of the UV absorbance (λ = 246 nm, NanoDrop 2000 Thermo Scientific). The concentration of the encapsulated paclitaxel was calculated using a calibration curve. Liposome solution was blended with PVA, giving a final concentration of 65 nM paclitaxel. Myoblast cells were seeded in 200 μL of medium in 48 well plates with a cell density of 12 000 cells/well and incubated at 37 °C and 5% CO2 for 48 h.
Growth Factor Immobilization
Surface adhered PVA hydrogels were prepared using PVA solutions (12 wt %, 6, 17, and 31 kDa) with either aminated or Ellman’s modified terminal groups via μTM as described above using PDMS molds with 2 μm deep and 10 μm wide wells with centers spaced by 20 μm. For improved cell adhesion, PVA hydrogels were surface coated with 2 g/L dopamine hydrochloride dissolved in Tris buffer (10 mM, pH 8.5) for 1 h at 4 °C and washed with PBS before conjugation of growth factors. For growth factor immobilization, a solution of LSGS (Invitrogen) was mixed at a 1:1 volume ratio with 9 × 10–5 M 2-iminothiolane (dissolved in PBS containing 1 mM EDTA; pH 8) and stirred for 1 h at 25 °C, and subsequently purified via size exclusion chromatography using a NAP column. For conjugation, 150 μL of purified thiolated growth factor solution was added to PVA-ER and PVA-NH2 hydrogels, the latter for control. The samples were incubated for 3 h and then washed three times in PBS to remove any unconjugated LSGS components. Cells were cultured on PDA coated PVA-NH2 or PVA-ER hydrogels preincubated with thiolated LSGS in LSGS free medium supplemented with 2% FBS. For negative and positive controls, cells were cultured on PDA coated PVA-NH2 hydrogels in LSGS free medium with 2% FBS or LSGS supplemented medium, respectively. To quantify cell viability, cells were seeded directly on top of the substrates (8400 cells per well in 200 μL of medium in 48 well plates) and incubated for 72 h at 37 °C and 5% CO2. Viability was assessed using Presto Blue (Invitrogen) following the manufacturer’s protocols. For a direct cell count, cells were seeded directly on top of the substrates (8400 cells per well in 200 μL of medium in 48 well plates) and incubated for 72 h at 37 °C and 5% CO2. For cell counting, the cells were washed two times in HBSS, fixed using 4% PFA solution for 10 min, and then rinsed three times with PBS. The nuclei were stained with DAPI (1 μg/mL) for 20 min and subsequently rinsed three times with PBS. Cell counts of cells adhering to the hydrogel were obtained by visualization using a Zeiss Axio Observer Z1 microscope.
Cell Staining for Visualization
For visualization of hydrogels and adhered cells, PVA-ER was supplemented with PVA-FITC, yielding a final concentration of 1 g/L. Cells were seeded directly on top of the substrates (80 000 cells per well in 1 mL of medium in 12 well plates) and incubated for 48 h at 37 °C and 5% CO2. The cells were washed two times in HBSS, fixed using 4% PFA solution for 10 min, and then rinsed three times with PBS. The cells were permeabilized for 15 min using 0.1% T-PBS, followed by blocking using 2% BSA in T-PBS for 30 min and rinsing three times with T-PBS. The nuclei and actin filaments were stained with DAPI (1 μg/mL) and phalliodin (0.1 μg/mL), respectively, for 20 min and subsequently rinsed two times with T-PBS and one time with PBS. The samples were mounted on a glass slide using mounting media (Vectashield) and visualized.
Data Analysis
For all data points, at least three independent experiments with a minimum of three replicates for each sample were performed and reported as mean ± standard deviation. Data was analyzed in Microsoft Excel and plotted in OriginPro (Origin Lab, v. 8.5). Statistical significance was determined through Student’s t test or Tukey’s test in conjunction with ANOVA and reported as significant if P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***). ImageJ was used for image analysis.
Results and Discussion
Materials’ characterization and analyses were carried out using surface-adhered microstructured hydrogels.16,22,23 We have established this methodology as a convenient platform to assemble hydrogels with chosen microscale topography as well as to quantify polymer gelation as well as incorporation and release of diverse cargo. The surface-adhered format allows using a broad range of characterization methods including microscopy observation. Finally, the prepared specimen can be directly used as substrates for adhesion and proliferation of mammalian cells toward characterization of hydrogels as biomaterials.17 Microstructured hydrogels were prepared using elastomeric poly(dimethylsiloxane) stamps obtained from silicon wafers with a nominated surface topography (in this work, 10 μm wide circles with a 20 μm intercenter spacing). Solutions of PVA (varied polymer molar mass and solids content) were placed between the stamp and a glass coverslip to fill the stamp cavities and clamped at finger-tight pressure for 24 h. During this time, PVA undergoes partial dehydration, and upon removal of clamps, PVA microstructures remain on the glass coverslip. To make hydrogels, these dehydrated PVA were immersed and incubated in liquid, oligomeric PEG and then washed with PBS and visualized using confocal laser scanning microscopy, Figure 2A. Fluorescently labeled PVA was used to obtain three-dimensional images of hydrogels prepared using polymer samples with a molar mass of 6, 17, and 31 kDa. In each case, well-defined, robust hydrogel structures were produced, illustrating that PEG treatment can be used to make physical hydrogels of PVA with a polymer molar mass as low as 6 kDa.
Figure 2.
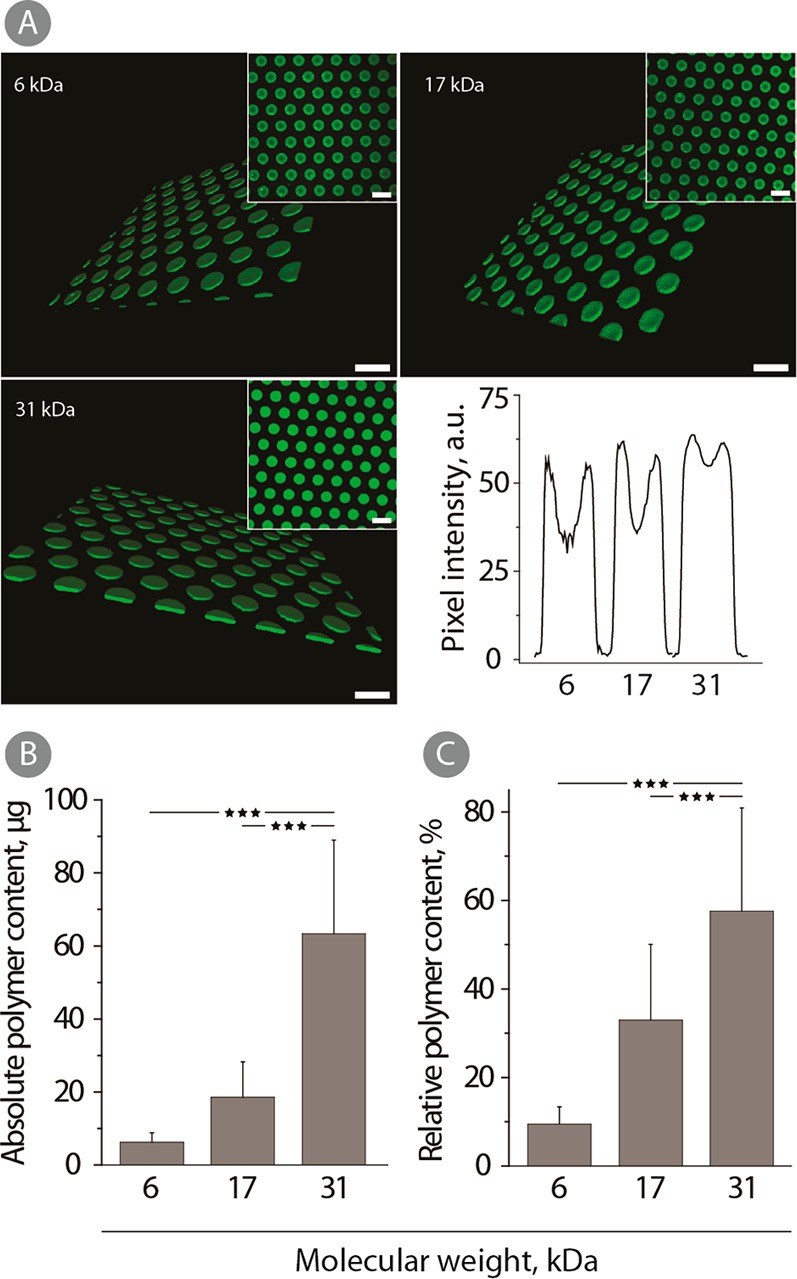
(A) 3D CLSM images (insets: 2D images) and pixel intensity profiles of physical hydrogels prepared using 12 wt % solutions of fluorescently labeled polymer samples of 6, 27, and 31 kDa. All samples were stabilized in PEG400 for 1 h at 37 °C and stored in PBS at 4 °C until visualization. Scale bars: 20 μm. (B) Absolute and (C) relative polymer content in hydrogels prepared from 12 wt % PVA-ER using 6, 17, and 31 kDa. All samples were stabilized for 1 h in PEG400 at 37 °C and incubated in PBS for 1 h at 4 °C. The values correspond to the absorbance measured after the direct addition of DTT to the hydrogels (n = 3; ***p < 0.001).
For quantitative analysis of polymer content in the hydrogel structure, we used the terminal group quantification method. PVA chains containing terminal mixed disulfide linkages with 2-nitro-5-thiobenzoate readily undergo thiol–disulfide exchange with DTT and release a chromophore which is then quantified using solution based UV–vis spectroscopy.25 For hydrogels assembled using PVA with an average molar mass of 6, 17, and 31 kDa, PEG treatment, and a single equilibration step in ice-cold PBS, polymer content was progressively higher for the polymers with increased chain length, Figure 2B,C. Absolute polymer content for hydrogels comprised of 31 kDa PVA was nearly 10-fold higher than that for 6 kDa PVA hydrogels (Figure 2B). Furthermore, the relative polymer content was also increased with the PVA molar mass. In other words, a greater fraction of PVA was retained within the hydrogel for polymer samples with increased chain length. These results are quite similar to the observations made for the hydrogels prepared via the “salting out” technique.25
At a constant polymer average molar mass, the solids content in the hydrogel phase can be fine-tuned through the choice of the polymer concentration in the feed solution, Figure 3A. Indeed, with increasing PVA concentration from 8 to 12 and further to 16 wt % (6 kDa PVA), the polymer content in the hydrogel phase exhibited a corresponding linear increase. While this observation is rather expected, it illustrates a facile means to tune the PVA content in the gel phase and thus set the properties of the hydrogel such as the concentration of terminal groups for bioconjugation. Quantitative measurements are well supported by the microscopy observations, Figure 3B. Higher polymer feed affords more robust hydrogels with a better developed contrast in the digital interference contrast mode (DIC) of bright field microscopy observation.
Figure 3.

(A) Absolute polymer content in hydrogels prepared from 6 kDa PVA-ER (8, 12, or 16 wt %) after 1 h in PEG400 at 37 °C and incubation in PBS for 1 h at 37 °C. The values correspond to the absorbance measured after the direct addition of DTT to the hydrogels (n = 3; *p < 0.05). (B) DIC images of corresponding hydrogels in the dry state and after 1 h of PBS incubation. Scale bars: 10 μm.
Spontaneous degradation of PVA physical hydrogels was considered to characterize the lifetime of these biomaterials under physiological conditions. For comparison, dissolution of these matrices was also studied in ice-cold PBS—conditions poised to largely suppress the degradation of interpolymer hydrogen bonds and thus maintain the stability of the hydrogels. The latter conditions would be favorable for, e.g., bioconjugation purposes such that chemical modification of the hydrogel does not compromise its integrity. Polymer content within hydrogels was quantified over a period of 7 days and presented as a function of the polymer molar mass and incubation temperature, Figure 4. Quantitative measurements were accompanied by visual observation of the hydrogels using DIC microscopy, Figure 5.
Figure 4.

Quantitative analysis of polymer content within the PVA hydrogels prepared using 12 wt % solutions of polymer samples of 6, 17, and 31 kDa and PEG-induced hydrogelation. Hydrogels were incubated in PBS at 4 or 37 °C for 7 days with periodic quantification of polymer chains within the hydrogel through the terminal group quantification method (n = 3; */#p < 0.05, **/##p < 0.01, and ***/###p < 0.001).
Figure 5.

Microscopy images of the hydrogel samples prepared using PDMS molds with 10 μm wide circles and 20 μm intercenter spacing, 12 wt % solutions of polymer samples of 6, 17, and 31 kDa, and PEG-induced hydrogelation. Hydrogels were incubated in PBS at 4 or 37 °C for 7 days. Images correspond to the quantitative data presented in Figure 4.
Relative polymer content within the hydrogel exhibited a large drop within the first 24 h of incubation in PBS at 37 °C to a level under 50% by weight, Figure 4A–C. Rather surprisingly, this observation was true for each polymer molar mass and dissolution profiles expressed in relative polymer content were in fact near identical for samples of 6, 17, and 31 kDa (Figure 4, panels A, B, and C, respectively). However, absolute polymer content expressed in units of mass was drastically lower for the 6 kDa sample than for 17 kDa and for the latter, considerably lower than for the 31 kDa counterpart (cf. Y axis scale for panels D–F). Visual observations suggest that, for the 6 kDa sample, the hydrogel dissolution was near complete within the first 24 h of incubation at physiological temperature (Figure 5). For the 17 kDa sample and more so for the 31 kDa polymer, hydrogels are well-defined and robust by appearance throughout the 7 day incubation time period. Along with expectations, incubation of hydrogels at 4 °C significantly suppressed the matrix dissolution—paving the way to facile chemical modification of the hydrogels at low temperatures. Even for the lowest molar mass polymer sample, at 4 °C, as much as 75% of the polymer chains were still found within the hydrogel phase after a week-long incubation. This result is also well supported by the visual observation of hydrogels in which case the 6 kDa polymer hydrogels are still visible after 3 days of incubation in a hydrated state in PBS. Taken together, the results presented in Figure 2–5 illustrate a fine level of control over incorporation of PVA into the hydrogel matrix and kinetics of erosion in physiological buffer—specifically as a function of polymer molar mass.
For applications as resorbable tissue sealants or other implantable biomaterials, PVA physical hydrogels need to be characterized in terms of the fate of polymer chains being released from the hydrogel matrix. On a systemic level, chains of molar mass in the 100–200 kDa mass range were shown to be translocated from the abdomen into the blood and then removed from the body by renal secretion without noticeable nephrotoxicity.24 Herein, we aimed to investigate a localized fate of polymer chains, specifically their possible uptake by the cells making up tissues in the immediate vicinity of the biomaterial. More specifically, we aimed to investigate the interaction of gradually resorbing PVA matrices with macrophages, a human cell type orchestrating the foreign body response to implants.26 To this end, PVA hydrogels were assembled using fluorescently labeled polymer samples. Macrophages (RAW 264.7) were cultured on the hydrogels, and upon cell detachment, fluorescence of cells was monitored using flow cytometry, Figure 6A. For each PVA average molar mass (6, 17, and 31 kDa), the histogram corresponding to fluorescence of cells exhibited a full population shift to higher levels of fluorescence, indicating a pronounced level of polymer interaction with cells. Confocal laser scanning microscopy images (Figure 6B) illustrate the presence of polymer-associated fluorescence inside the cells, thus revealing that much of the acquired fluorescent signal is due to polymer uptake, not surface absorption. Quantitatively, nearly 100% of cells tested positive for the polymer-associated fluorescence, and this observation was true for each PVA average molar mass (Figure 6C). Interestingly, the absolute level of cell fluorescence was highest for the 17 kDa sample than 6 or 31 kDa counterparts (Figure 6D). A plausible explanation could be that, for 6 kDa samples, the total polymer content in the hydrogel phase is significantly lower than that for the 17 kDa counterpart. In turn, the 31 kDa sample is characterized with an increased total polymer content yet in this case, degradation of the hydrogel is also significantly slower than for the 17 kDa.
Figure 6.
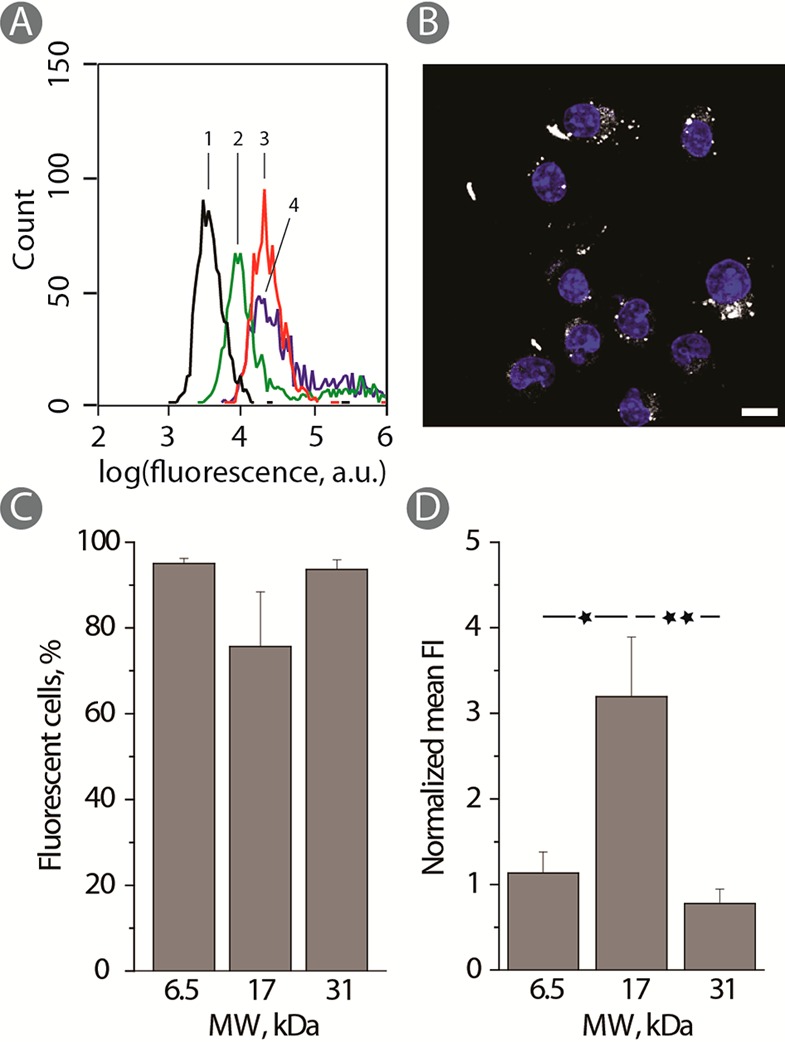
(A) Typical flow cytometry histogram for RAW 264.7 cells cultured on PVA-FITC hydrogels. (1 - black; control) PVA-NH2; (2 - green) 17 kDa; (3 - red): 31 kDa; (4 - blue): 6 kDa. (B) CLSM image illustrating polymer uptake of RAW 264.7 cells cultured on 17 kDa PVA-FITC physical hydrogels. Scale bars: 10 μm (blue: DAPI). (C) Percentage fluorescent cells and (D) normalized mean fluorescence of cells (D) adhering for 24 h to PVA-FITC hydrogels with different molecular weights and using PLL for rendering the hydrogels cell adhesive (n = 3; *p < 0.05 and **p < 0.01).
Inflammatory processes mediated by macrophages are highly important in the context of foreign body response.26 Materials degradation and the accompanying uptake of polymer chains released from the hydrogel may possibly affect the adhering cells through intracellular effects of polymer chains. To investigate this for the physical hydrogels based on PVA, the inflammatory response of macrophages was quantified through measuring secreted nitric oxide (inflammatory marker) via the Griess assay. Cultured on PVA hydrogel matrices of 6, 17, or 31 kDa, macrophages revealed a decrease in their metabolic activity to ∼80% possibly implying an effect elicited by the substrate on proliferation or metabolism of cells27—in this case the effect being minor. However, there was no measurable associated inflammatory response and levels of released nitric oxide were below the detection limit. Upon addition of lipopolysaccharide (LPS), a potent bacterial pro-inflammatory stimulant, macrophages showed a pronounced inflammatory response which measured 60–80% of control (cells cultured on glass coverslips and stimulated with LPS). The levels of nitric oxide coincide well with the levels of metabolic activity, and this implies that materials elicit no pro- or anti-inflammatory response added to activity of LPS. Finally, addition of a commercial inhibitor of inducible nitric oxide synthase (L-NAME) resulted in expected inhibition in synthesis of nitric oxide—illustating that intracellular biochemisty of regulation of synthesis of this marker of inflammation is not altered by the hydrogels or the products of its degradation (PVA chains). Together, the data in Figure 7 illustrate that PVA hydrogels are fully benign in the context of their interaction with macrophages.
Figure 7.

Cell viability and nitric oxide produced by RAW 264.7 cells cultured on PVA hydrogels prepared using 6, 17, and 31 kDa as well as glass coverslips (n = 3; *p < 0.05). For nitric oxide, statistical significance is given compared to LPS stimulated cells cultured on glass.
Hydrogel biomaterials are uniqe tissue-like matrices, yet their highly hydrated state makes controlled drug retention and release rather challenging.28 One approach to circumnavigate this makes use of composite hydrogels whereby the polymer network is embedded with dedicated reservoirs of drug molecules. Such reservoirs can be nanodispersions, microparticles, polymersomes, etc.28,29 In our previous work,16 as drug reservoirs we used liposomes—the latter being a powerful tool of nanotechnology, specifically in drug delivery.30 The key to our method of production of lipogels (liposomes incorporated in hydrogels) was the technique of “branding” whereby cholesterol-modified PVA is anchored into the lipid bilayer of the liposome and polymer extensions are incorporated into the structure of the hydrogel. Herein, we investigate lipogels prepared using PVA samples of 6, 17, and 31 kDa whereby liposome branding is accomplished using polymers of matched molar mass. Visualization of lipogels via CLSM and DIC illustrates that, in each case, robust hydrogels were formed and liposomes were distributed in the hydrogel, Figure 8A. This result is important in that it suggests that lipogels with vastly different rates of matrix dissolution can be successfully assembled to suit diverse applications. In an effort to quantify therapeutic effects mediated by lipogels, liposomes were loaded with a hydrophobic drug, paclitaxel, and used to deliver their toxic payload to adhering myoblasts. The relevance of these experiments to biomedicine lies in that PVA matrices are investigated toward production of vascular grafts31,32 and controlled delivery of cytotoxic drugs is the mainstream of design of cadivascular implants.33,34 Lipogels mediated efficient killing of cells cultured on these matrices, and in each case, over 48 h of incubation cell viability was decreased to 60%. Antiproliferative activity of lipogels revealed no trend with regard to the molar mass of the polymer making up the hydrogel matrix, suggesting that, in this particular case, the rate of hydrogel dissolution is not decisive in the effects mediated on the adhering cells.
Figure 8.
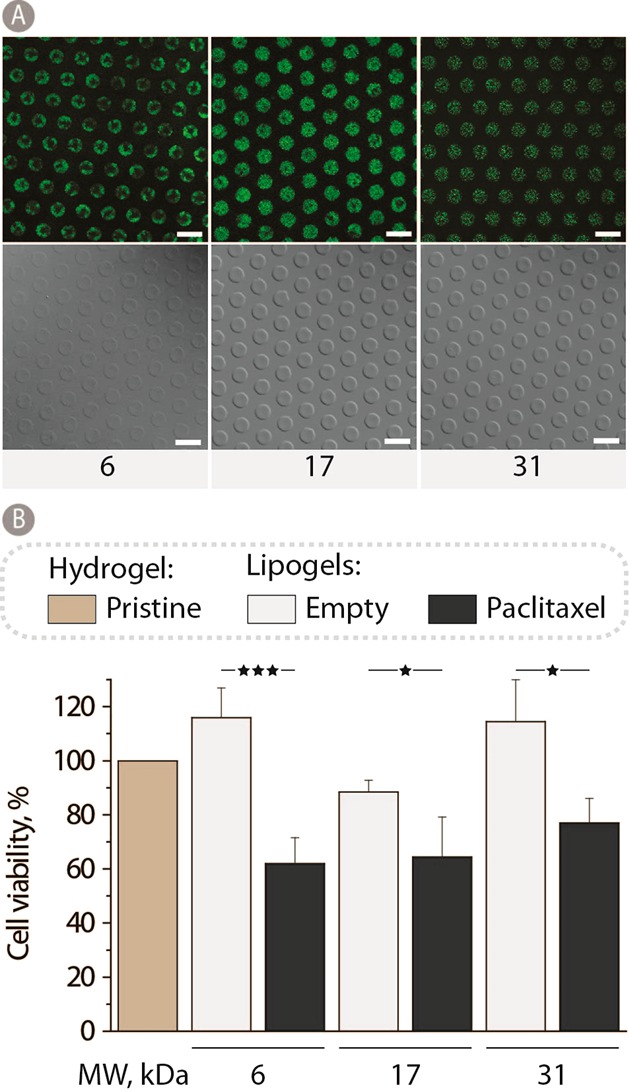
(A) Fluorescence and DIC images of 12 wt % PVA lipogels using 6, 17, and 31 kDa PVA. All samples were stabilized in PEG400 for 1 h at 37 °C and stored in PBS at 5 °C until visualization. Scale bars: 20 μm. (B) Myoblast cell viability after 48 h of culturing on 12 wt % 6, 17, and 31 kDa PVA hydrogels containing either empty lipogels or paclitaxel loaded liposomes and normalized to pristine PVA hydrogel controls (n = 3; *p < 0.05 and ***p < 0.001).
From a different perspective, it is highly important to endow vascular implants with tools to accelerate the restoration of the natural endothelial lining.35 In the case of spontaneously eroding PVA matrices, it can be envisioned that proliferating endothelial cells will ultimately replace the degrading hydrogel matrix—a prised scenario for tissue engineering. Toward this goal, we investigate PVA hydrogels toward bioconjugation of specific growth factors to present these strong proliferation promoters directly to the adhering cells. For bioconjugation, we use polymer terminal groups, Figure 1. Growth factor protein was modified using 2-iminothiolane to convert a fraction of amine functionalities into thiols and subsequently achieve conjugation to the PVA hydrogel under physiological conditions. Conjugation was carried out at 4 °C—conditions favoring stability of the hydrogels (Figure 4). The so assembled hydrogels with incorporated growth factors were used as substrates for proliferation of human umbilical vein endothelial cells (HUVECs). Results of the direct cell count and measured levels of metabolic activity were compared to the culture conditions in the absence of growth factors as well as growth factors added to the media, the latter being the standard cell culture approach (Figure 9). For the 31 kDa sample, data are taken from ref (17) and used herein for comparison. These data illustrate that, in the absence of growth factors, proliferation of HUVECs is 2–3-fold lower than in the presence of the protein. For polymer samples with amine terminal groups (that is, not suited for conjugation with growth factors via thiol disulfide exchange), immobilization of growth factors was inefficient. As a result, proliferation of cells was observed at a rate near identical to the case of growth factor-free cell culture. In contrast, growth factors were effective when administered into cell media and when incorporated into the hydrogel matrix through bioconjugation, the latter opportunity lending itself for site specific therapeutic effects and being the goal of this study. For the 6 kDa sample, this approach to delivery of growth factors proved inefficient—most likely due to fast erosion of the matrix and elimination of growth factors from the vicinity of cells. In turn, for the 17 kDa sample, hydrogel-based presentation of growth factors is highly efficient and proliferation of HUVECs is nearly doubled as compared to the administration of growth factors to cell media. Quantitative data are supported by the microscopy observations (Figure 10), illustrating that the 6 kDa sample supports adhesion and proliferation of a low number of cells and the cytoskeleton of cells is ill-organized. In turn, 17 and 31 kDa samples support adhesion and proliferation of HUVECs with a well-organized cytoskeleton.
Figure 9.

PVA physical hydrogels stabilized using PEG400, surface coated with PDA, and used as substrates for culturing of HUVECs for 72 h. The cell viability and average cell count per sample are normalized against cells cultured in growth factor supplemented media on nonfunctionalized PVA hydrogels (n = 3; *p < 0.05, **p < 0.01, and ***p < 0.001).
Figure 10.
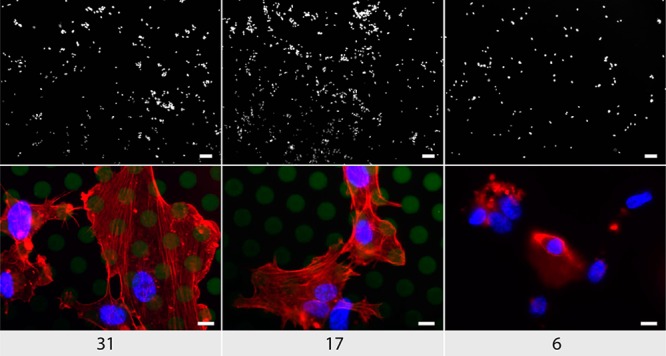
Visualization of HUVECs cultured for 48 h on PDA coated growth factor immobilized hydrogels with different molecular weights. Scale bars: top row, 100 μm; bottom row, 50 μm. Blue, DAPI; red, phalliodin; green, PVA-FITC.
A factor drastically limiting the utility of growth factors in biomedicine is their fragility. Cold chain of transportation and storage also tremendously increases the costs associated with handling of these potent therapeutics. Poly-ols are widely used as protein stabilizing agents36,37 acting through a “water replacement mechanism” whereby the polyol makes hydrogen bonds with the solute in place of bonds originally made by water. PVA too is known for its cryo-preservative properties38 although due to prevention of water crystallization (formation of ice). We aimed to investigate if PVA—which is a poly-ol—will stabilize growth factors during storage. To this end, 31 kDa PVA was used to assemble hydrogels and conjugate the growth factors, as described above. Following the conjugation, all liquid was removed and the samples were washed in nonbuffered pure water, dried, and stored in sealed containers for 1 month at room temperature (22 °C) or 4 °C. Following this, hydrogels were characterized per their instructive effect on the proliferating HUVECs, Figure 11. The most important observation is that the growth factor containing hydrogels supported adhesion and proliferation of cells significantly better than the pristine PVA hydrogels (without growth factors). This implies that a significant fraction of the protein molecules remained functional upon storage within PVA hydrogels over a month. The samples stored at room temperature supported cell growth to a lower degree, revealing that proteins possibly underwent a greater degree of deactivation. However, in this case too, protein-containing gels provided cell growth cues to the adhering cells. Further optimization of this system is underway to design hydrogels with an enhanced capacity to sustain structural activity of the immobilized growth factors.
Figure 11.
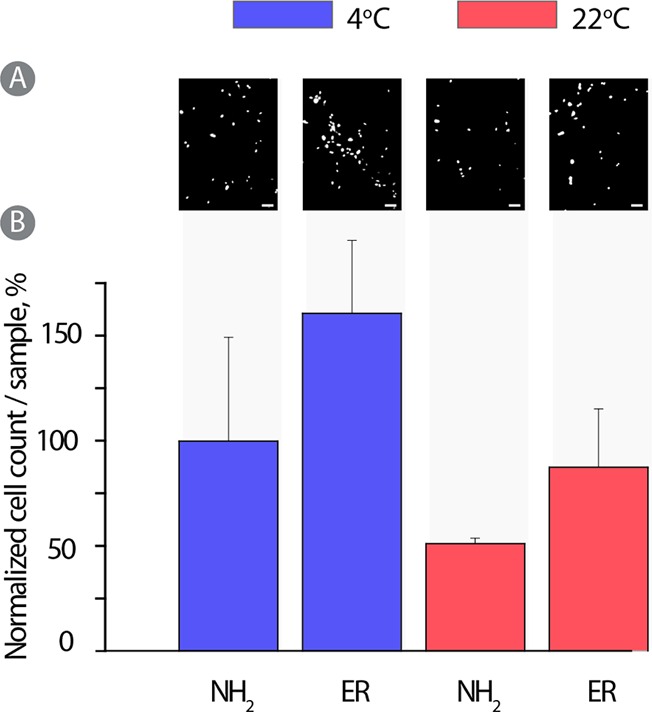
PVA physical hydrogels stabilized using PEG400, surface coated with PDA, and used as substrates for culturing of HUVECs for 72 h. (A) Fluorescent images of DAPI stained cells and (B) average cell count per sample were normalized against cells cultured in growth factor supplemented media.
Conclusions
In this work, we investigated opportunities in engineering hydrogel biomaterials based on PVA whereby the gels are stabilized through physical interactions between polymer chains. Hydrogels were characterized in terms of their spontaneous dissolution under physiological conditions and their capacity toward controlled incorporation of liposomes and growth factors. Hydrogels were investigated with regard to effects exerted onto adhering macrophages (inflammatory responses), myoblasts (antiproliferative activity), and endothelial cells (promotion of proliferation). Taken together, this work represents a systematic fundamental study illustrating effects of PVA molar mass on the properties of physical hydrogels made thereof. In light of prior biomedical success of PVA hydrogels and regulatory approval for several modes of utility in humans, our data are poised to open novel opportunities for applications of these hydrogels in biomedicine, specifically as therapeutically active, resorbable implantable biomaterials.
Acknowledgments
We wish to acknowledge financial support from the European Research Council Consolidator grant (ANZ, ERC-2013-CoG 617336 BTVI).
The authors declare no competing financial interest.
References
- Seliktar D. Designing Cell-Compatible Hydrogels for Biomedical Applications. Science 2012, 336, 1124–1128. 10.1126/science.1214804. [DOI] [PubMed] [Google Scholar]
- Hamidi M.; Azadi A.; Rafiei P. Hydrogel Nanoparticles in Drug Delivery. Adv. Drug Delivery Rev. 2008, 60, 1638–1649. 10.1016/j.addr.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Lin C.-C.; Metters A. T. Hydrogels in controlled Release Formulations: Network design And Mathematical Modeling. Adv. Drug Delivery Rev. 2006, 58, 1379–1408. 10.1016/j.addr.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Caló E.; Khutoryanskiy V. V. Biomedical Applications of Hydrogels: A Review of Patents and Commercial Products. Eur. Polym. J. 2015, 65, 252–267. 10.1016/j.eurpolymj.2014.11.024. [DOI] [Google Scholar]
- Kharkar P. M.; Kiick K. L.; Kloxin A. M. Designing Degradable Hydrogels For Orthogonal Control of Cell Microenvironments. Chem. Soc. Rev. 2013, 42, 7335–7372. 10.1039/C3CS60040H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaughter B. V.; Khurshid S. S.; Fisher O. Z.; Khademhosseini A.; Peppas N. A. Hydrogels in Regenerative Medicine. Adv. Mater. 2009, 21, 3307–3329. 10.1002/adma.200802106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vlierberghe S.; Dubruel P.; Schacht E. Biopolymer-Based Hydrogels as Scaffolds for Tissue Engineering Applications: A Review. Biomacromolecules 2011, 12, 1387–1408. 10.1021/bm200083n. [DOI] [PubMed] [Google Scholar]
- Alves M. H.; Jensen B. E. B.; Smith A. A. A.; Zelikin A. N. Poly(Vinyl Alcohol) Physical Hydrogels: New Vista on a Long Serving Biomaterial. Macromol. Biosci. 2011, 11, 1293–1313. 10.1002/mabi.201100145. [DOI] [PubMed] [Google Scholar]
- Baker M. I.; Walsh S. P.; Schwartz Z.; Boyan B. D. A Review of Polyvinyl Alcohol and its Uses in Cartilage and Orthopedic Applications. J. Biomed. Mater. Res., Part B 2012, 100B, 1451–1457. 10.1002/jbm.b.32694. [DOI] [PubMed] [Google Scholar]
- Lozinsky V. I.; Zubov A. L.; Titova E. F. Swelling Behavior of Poly(Vinyl Alcohol) Cryogels Employed as Matrices For Cell Immobilization. Enzyme Microb. Technol. 1996, 18, 561–569. 10.1016/0141-0229(95)00148-4. [DOI] [Google Scholar]
- Lozinsky V. I.; Zubov A. L.; Titova E. F. Poly(vinyl alcohol) Cryogels Employed as Matrices for Cell Immobilization. 2. Entrapped Cells Resemble Porous Fillers in Their Effects on the Properties of PVA-Cryogel Carrier. Enzyme Microb. Technol. 1997, 20, 182–190. 10.1016/S0141-0229(96)00110-X. [DOI] [Google Scholar]
- Lozinsky V. I.; Plieva F. M. Poly(vinyl alcohol) Cryogels Employed as Matrices for Cell Immobilization. 3. Overview of Recent Research and Developments. Enzyme Microb. Technol. 1998, 23, 227–242. 10.1016/S0141-0229(98)00036-2. [DOI] [Google Scholar]
- Lewis A. L.; Holden R. R. DC Bead Embolic Drug-Eluting Bead: Clinical Application in the Locoregional Treatment of Tumours. Expert Opin. Drug Delivery 2011, 8, 153–69. 10.1517/17425247.2011.545388. [DOI] [PubMed] [Google Scholar]
- Chong S.-F.; Smith A. A. A.; Zelikin A. N. Microstructured, Functional PVA Hydrogels through Bioconjugation with Oligopeptides under Physiological Conditions. Small 2013, 9, 942–950. 10.1002/smll.201201774. [DOI] [PubMed] [Google Scholar]
- Fejerskov B.; Smith A. A. A.; Jensen B. E. B.; Hussmann T.; Zelikin A. N. Bioresorbable Surface-Adhered Enzymatic Microreactors Based on Physical Hydrogels of Poly(vinyl alcohol). Langmuir 2013, 29, 344–354. 10.1021/la3040903. [DOI] [PubMed] [Google Scholar]
- Jensen B. E. B.; Hosta-Rigau L.; Spycher P. R.; Reimhult E.; Stadler B.; Zelikin A. N. Lipogels: Surface-Adherent Composite Hydrogels Assembled From Poly(Vinyl Alcohol) and Liposomes. Nanoscale 2013, 5, 6758–6766. 10.1039/c3nr01662e. [DOI] [PubMed] [Google Scholar]
- Jensen B. E. B.; Edlund K.; Zelikin A. N. Micro-structured, Spontaneously Eroding Hydrogels Accelerate Endothelialization Through Presentation of Conjugated Growth Factors. Biomaterials 2015, 49, 113–124. 10.1016/j.biomaterials.2015.01.036. [DOI] [PubMed] [Google Scholar]
- Lutolf M. P.; Lauer-Fields J. L.; Schmoekel H. G.; Metters A. T.; Weber F. E.; Fields G. B.; Hubbell J. A. Synthetic Matrix Metalloproteinase-Sensitive Hydrogels for the Conduction of Tissue Regeneration: Engineering Cell-Invasion Characteristics. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 5413–5418. 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. A. A.; Hussmann T.; Elich J.; Postma A.; Alves M. H.; Zelikin A. N. Macromolecular Design of Poly(vinyl alcohol) by RAFT Polymerization. Polym. Chem. 2012, 3, 85–88. 10.1039/C1PY00429H. [DOI] [Google Scholar]
- Moad G.; Rizzardo E.; Thang S. H. Living Radical Polymerization by the RAFT Process. Aust. J. Chem. 2005, 58, 379–410. 10.1071/CH05072. [DOI] [Google Scholar]
- Rasmussen K. F.; Smith A. A. A.; Ruiz-Sanchis P.; Edlund K.; Zelikin A. N. Cholesterol Modification of (Bio)Polymers Using UV-Vis Traceable Chemistry in Aqueous Solutions. Macromol. Biosci. 2014, 14, 33–44. 10.1002/mabi.201300286. [DOI] [PubMed] [Google Scholar]
- Jensen B. E. B.; Smith A. A. A.; Fejerskov B.; Postma A.; Senn P.; Reimhult E.; Pla-Roca M.; Isa L.; Sutherland D. S.; Stadler B.; Zelikin A. N. Poly(vinyl alcohol) Physical Hydrogels: Noncryogenic Stabilization Allows Nano- and Microscale Materials Design. Langmuir 2011, 27, 10216–10223. 10.1021/la201595e. [DOI] [PubMed] [Google Scholar]
- Jensen B. E. B.; Alves M.-H.; Fejerskov B.; Stadler B.; Zelikin A. N. Surface Adhered Poly(Vinyl Alcohol) Physical Hydrogels as Tools for Rational Design of Intelligent Biointerfaces. Soft Matter 2012, 8, 4625–4634. 10.1039/c2sm07075h. [DOI] [Google Scholar]
- Jiang Y. J.; Schadlich A.; Amado E.; Weis C.; Odermatt E.; Mader K.; Kressler J. In-vivo Studies on Intraperitoneally Administrated Poly(vinyl alcohol). J. Biomed. Mater. Res., Part B 2010, 93B, 275–284. 10.1002/jbm.b.31585. [DOI] [PubMed] [Google Scholar]
- Fejerskov B.; Jensen B. E. B.; Jensen N. B. S.; Chong S.-F.; Zelikin A. N. Engineering Surface Adhered Poly(vinyl alcohol) Physical Hydrogels as Enzymatic Microreactors. ACS Appl. Mater. Interfaces 2012, 4, 4981–4990. 10.1021/am3013467. [DOI] [PubMed] [Google Scholar]
- Anderson J. M.; Rodriguez A.; Chang D. T. Foreign Body Reaction to Biomaterials. Semin. Immunol. 2008, 20, 86–100. 10.1016/j.smim.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocgozlu L.; Lavalle P.; Koenig G.; Senger B.; Haikel Y.; Schaaf P.; Voegel J. C.; Tenenbaum H.; Vautier D. Selective and Uncoupled Role of Substrate Elasticity in the Regulation of Replication and Transcription in Epithelial Cells. J. Cell Sci. 2010, 123, 29–39. 10.1242/jcs.053520. [DOI] [PubMed] [Google Scholar]
- Hoare T. R.; Kohane D. S. Hydrogels in Drug Delivery: Progress and Challenges. Polymer 2008, 49, 1993–2007. 10.1016/j.polymer.2008.01.027. [DOI] [Google Scholar]
- Hosta-Rigau L.; Jensen B. E. B.; Fjeldsø K. S.; Postma A.; Li G.; Goldie K. N.; Albericio F.; Zelikin A. N.; Städler B. Surface-Adhered Composite Poly(Vinyl Alcohol) Physical Hydrogels: Polymersome-Aided Delivery of Therapeutic Small Molecules. Adv. Healthcare Mater. 2012, 1, 791–795. 10.1002/adhm.201200092. [DOI] [PubMed] [Google Scholar]
- Torchilin V. P. Recent Advances with Liposomes as Pharmaceutical Carriers. Nat. Rev. Drug Discovery 2005, 4, 145–160. 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- Cutiongco M. F. A.; Anderson D. E. J.; Hinds M. T.; Yim E. K. F. In vitro and ex vivo Hemocompatibility of off-the-shelf Modified Poly(vinyl alcohol) Vascular Grafts. Acta Biomater. 2015, 25, 97–108. 10.1016/j.actbio.2015.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaouat M.; Le Visage C.; Baille W. E.; Escoubet B.; Chaubet F.; Mateescu M. A.; Letourneur D. A Novel Cross-linked Poly(vinyl alcohol) (PVA) for Vascular Grafts. Adv. Funct. Mater. 2008, 18, 2855–2861. 10.1002/adfm.200701261. [DOI] [Google Scholar]
- Waugh J.; Wagstaff A. J. The Paclitaxel (TAXUS)-Eluting Stent: a Review of its Use in the Management of De Novo Coronary Artery Lesions. Am. J. Cardiovasc. Drugs 2004, 4, 257–68. 10.2165/00129784-200404040-00006. [DOI] [PubMed] [Google Scholar]
- Kaul U.; Bangalore S.; Seth A.; Arambam P.; Abhaychand R. K.; Patel T. M.; Banker D.; Abhyankar A.; Mullasari A. S.; Shah S.; Jain R.; Kumar P. R.; Bahuleyan C. G. Paclitaxel-Eluting versus Everolimus-Eluting Coronary Stents in Diabetes. N. Engl. J. Med. 2015, 373, 1709–1719. 10.1056/NEJMoa1510188. [DOI] [PubMed] [Google Scholar]
- Seifu D. G.; Purnama A.; Mequanint K.; Mantovani D. Small-Diameter Vascular Tissue Engineering. Nat. Rev. Cardiol. 2013, 10, 410–421. 10.1038/nrcardio.2013.77. [DOI] [PubMed] [Google Scholar]
- Carpenter J. F.; Crowe J. H. The mechanism of Cryoprotection of Proteins by Solutes. Cryobiology 1988, 25, 244–55. 10.1016/0011-2240(88)90032-6. [DOI] [PubMed] [Google Scholar]
- Arakawa T.; Prestrelski S. J.; Kenney W. C.; Carpenter J. F. Factors Affecting Short-Term and Long-Term Stabilities of Proteins. Adv. Drug Delivery Rev. 2001, 46, 307–326. 10.1016/S0169-409X(00)00144-7. [DOI] [PubMed] [Google Scholar]
- Deller R. C.; Vatish M.; Mitchell D. A.; Gibson M. I. Synthetic Polymers Enable Non-Vitreous Cellular Cryopreservation By Reducing Ice Crystal Growth During Thawing. Nat. Commun. 2014, 5, 3244. 10.1038/ncomms4244. [DOI] [PubMed] [Google Scholar]