

Summary
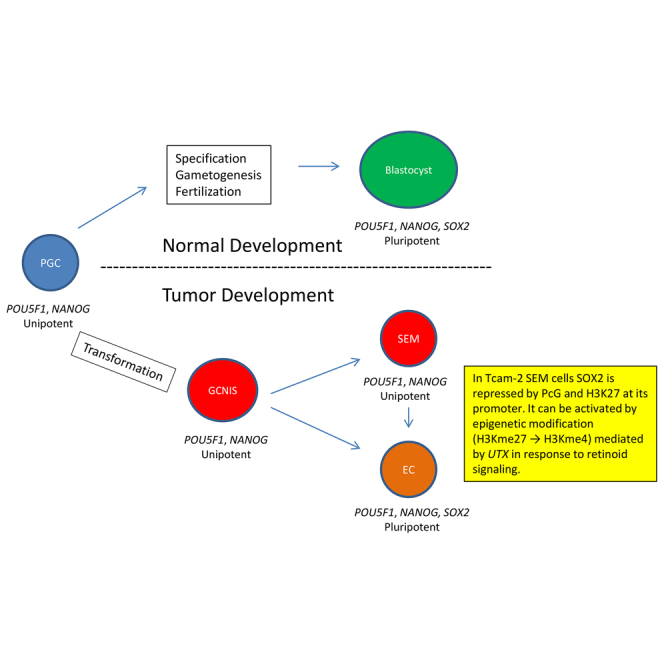
Human male germ cell tumors (GCTs) are derived from primordial germ cells (PGCs). The master pluripotency regulator and neuroectodermal lineage effector transcription factor SOX2 is repressed in PGCs and the seminoma (SEM) subset of GCTs. The mechanism of SOX2 repression and its significance to GC and GCT development currently are not understood. Here, we show that SOX2 repression in SEM-derived TCam-2 cells is mediated by the Polycomb repressive complex (PcG) and the repressive H3K27me3 chromatin mark that are enriched at its promoter. Furthermore, SOX2 repression in TCam-2 cells can be abrogated by recruitment of the constitutively expressed H3K27 demethylase UTX to the SOX2 promoter through retinoid signaling, leading to expression of neuronal and other lineage genes. SOX17 has been shown to initiate human PGC specification, with its target PRDM1 suppressing mesendodermal genes. Our results are consistent with a role for SOX2 repression in normal germline development by suppressing neuroectodermal genes.
Graphical Abstract
Highlights
-
•
SOX2 is repressed in hPGC, germ cell neoplasia in situ, and seminoma
-
•
SOX2 repression is mediated by PcG and H3K27me3 enrichment at its promoter
-
•
Retinoid signaling recruits UTX to SOX2 promoter leading to reactivation of SOX2
-
•
These studies shed light on the role of SOX2 in germline development
In this article, Chaganti and colleagues show that SOX2 repression in human germ cell tumors and possibly also their precursor hPGC is mediated by PcG and H3Kme27 at its promoter. The repression can be reversed by recruitment of UTX, a H3K27 demethylase, leading to upregulation of neuronal lineage genes.
Introduction
Human male germ cell tumors (GCTs) are thought to originate in primordial germ cells (PGCs) most likely by a mechanism similar to that recently described for the origin of teratocarcinomas in strain 128 family mice (Heaney et al., 2012). The key driver for this process is suggested to be upregulation of genes in the pathways controlling pluripotency and proliferation, such as NANOG, CCND2, and RASK2 that map to chromosome 12p (Chaganti and Houldsworth, 2000, Korkola et al., 2006). GCTs comprise two main subsets, seminoma (SEM) and nonseminoma (NS), with a common precursor, germ cell neoplasia in situ (GCNIS). SEM is unipotent whereas the NS subset embryonal carcinoma (EC) is pluripotent, analogous to the blastocyst (Andrews et al., 2005), and has a gene-expression profile (GEP) similar to that of embryonic stem cells (ESCs) (Sperger et al., 2003, Josephson et al., 2007). EC differentiates to extraembryonic (choriocarcinoma, yolk sac tumor) and embryonic (teratoma) lineages (Chaganti and Houldsworth, 2000). Comparison of GEPs of human PGC (hPCG)-like cells derived in vitro from ESCs, gonadal GCs, and the SEM cell line TCam-2 suggested that SEM arises in PGCs and hence is a good model system to investigate hPGC biology (Irie et al., 2015). SOX17 was shown to be the key specifier of hPGC fate, with the downstream PRDM1 repressing mesendodermal genes (Irie et al., 2015).
The core pluripotency regulatory master transcription factors (TFs) POU5F1 and NANOG are expressed in both EC and SEM, whereas SOX2 is repressed in hPGCs (Perrett et al., 2008, Irie et al., 2015), GCNIS, and SEM (Korkola et al., 2006). The molecular mechanism of SOX2 repression in the hPGC-GCNIS-SEM lineage has so far not been characterized. We show here that SOX2 repression in TCam-2 cells is due to the co-occupation by the Polycomb group (PcG) proteins and the repressive chromatin mark H3K27me3 near its transcription start site (TSS). We further show that the occupancy of H3K27me3 decreases when UTX, a H3K27-specific demethylase, is recruited to the SOX2 promoter in response to retinoid signaling, leading to SOX2 transcriptional derepression and induction of neuronal genes, consistent with its function as a neuroectodermal effector (Thomson et al., 2011, Zhang and Cui, 2014). Thus, SOX2 repression in TCam-2/SEM is imposed by PcG and its derepression is regulated by UTX. These data are consistent with a model of hPGC development initiated by SOX17, with PRDM1 repressing mesodermal genes and SOX2 repression inhibiting neuroectodermal genes.
Although murine and human PGCs re-express pluripotency genes following specification, pluripotency remains latent and becomes functional only when PGCs are cultured in vitro as embryonic germ cells or transform in vivo as GCTs (Leitch et al., 2013). By analysis of GEPs of SEM and EC, we show here that the functional pathways of SEM reflect their derivation from PGCs, while those of EC, also derived from PGCs, reflect re-establishment of pluripotency in the transformed PGCs. These data are of value in understanding the biology of hPGCs and regulation of the pluripotency state in the unique GCT system.
Results
Functional Programs in SEM and EC Reflect Their Development from PGCs following Malignant Transformation and Re-establishment of Pluripotency
Despite their common origin from transformed hPGCs, SEM retains the germline characteristic of latent pluripotency while EC attains embryonal-like pluripotency. As such, SEM and EC provide an opportunity to identify the functional pathways that underlie the latent and patent pluripotency of the two PGC-derived tumor states. Toward this end, we performed significance analysis for microarray (SAM) and gene ontology (GO biological process) analyses of the upregulated and downregulated genes in the GEPs of 41 EC and 16 SEM tumors in comparison with those of five normal testis controls. These GEPs were a subset of the GEP data of a larger cohort of GCTs representing all histologic and developmental categories and normal testis biopsies that we previously published (Korkola et al., 2005, Korkola et al., 2006, Korkola et al., 2009). SAM analysis showed that upregulated genes in SEM included the GC genes KIT, CD38, TNAP, SOX17, NANOS, TFAP2C, and UTF1 consistent with their PGC derivation as previously shown in the TCam-2 SEM cell line (Irie et al., 2015) (Tables S1 and S2). GO analysis identified significantly upregulated categories in SEM related to DNA integrity (p = 4.5 × 10−4) and damage response (4.5 × 10−5), regulation of cell morphogenesis (p = 4.86 × 10−4), and RNA processing (3.7 × 10−10), whereas those in EC related to stem cell maintenance (p = 0.003), cell morphogenesis (p = 4.2 × 10−7), multicellular organismal process (p = 0.0002), anatomic structure morphogenesis (p = 8.38 × 10−7), and response to wounding (p = 8.59 × 10−10) (Figure 1). In addition, upregulated categories representing cell proliferation (pSEM = 1.13 × 10−3; pEC = 0.0038) and negative regulation of apoptosis (pSEM = 3.34 × 10−4; pEC = 4.05 × 10−5) were common to both subsets, reflecting their transformed state (Figure 1). The downregulated categories were remarkably similar in both subsets and were represented by gamete generation (pSEM = 6.24 × 10−44; pEC = 2.28 × 10−45), spermatogenesis (pSEM = 7.24 × 10−52; pEC = 3.43 × 10−57), spermatid differentiation (pSEM = 2.68 × 10−12; pEC = 2.8 × 10−13), meiotic cycle (pSEM = 3.24 × 10−12; pEC = 3.16 × 10−12), and reproduction (pSEM = 7.58 × 10−31; pEC = 1.06 × 10−30) (Figure 1). These data represent elucidation of regulatory pathways in these two subsets.
Figure 1.

Top GO Categories Enriched in GEPs of SEM and EC in Comparison with Normal Testis
The graphs were prepared using the Cytoscape plugin (BiNGO).
(A and B) Upregulated categories in SEM (A) and EC (B) compared with normal testis.
(C and D) Downregulated categories in SEM (C) and EC (D) compared with normal testis.
Computationally Identified SOX2 Targets Involved in Pluripotency and Differentiation Pathways Are Significantly Enriched in EC Compared with SEM
We recently assembled a complete in vivo GCT TF interactome (GCTNet) based on the GEPs of tumor biopsies using the Algorithm for the Reconstruction of Accurate Cellular Networks (ARACNe) (Kushwaha et al., 2015). GCTNet, comprising 1,305 TFs and ∼250,000 interactions, which encompassed all the functional pathways operating in this tumor system and inferred all the expressed target genes of the entire complement of TFs. This analysis showed that POU5F1, NANOG, and SOX2 had 338, 376, and 307 ARACNe-inferred individual target genes, respectively. In addition, POU5F1 and NANOG shared 127 common targets, whereas POU5F1 and SOX2, and NANOG and SOX2, shared 40 common targets. We validated the ARACNe-inferred targets of these three genes by analysis of GEP following their short hairpin RNA-mediated knockdown (KD) and by genome sequencing following chromatin immunoprecipitation (ChIP) in EC-derived NT2/D1 cells, thus establishing their role as mechanistic transcriptional targets (Kushwaha et al., 2015). We now performed GEP analysis of NT2/D1 and TCam-2 cells and assessed the enrichment of the previously identified ARACNe-inferred targets of POU5F1, NANOG, and SOX2 in genes that are differentially expressed between the two cell lines by gene set enrichment analysis (GSEA) (Mootha et al., 2003, Subramanian et al., 2005). As shown in Figure 2A, this analysis clearly shows that whereas target enrichment was significant for all three TFs between NT2/D1 and TCam-2 cell lines, SOX2 targets showed enrichment in overexpressed genes in NT2/D1 compared with TCam-2, while NANOG and POU5F1 showed opposite enrichment, i.e., in underexpressed genes. Whereas a GO analysis of GSEA targets of SOX2 overexpressed in TCam-2 cells identified no significant categories, that of NT2/D1 cells showed stem cell development (p = 5.11 × 10−4), cellular developmental process (p = 4.61 × 10−4), nervous system development (p = 1.67 × 10−4), multicellular organismal development (p = 6.99 × 10−5), system development (p = 1.94 × 10−5), anatomical structure development (p = 1.7 × 10−5), developmental process (p = 9.93 × 10−6), and stem cell differentiation (p = 2.09 × 10−6) among the highly enriched categories. GSEA analysis of the three TFs from the 41 EC and 16 SEM showed consistent results (Figure 2B). Indeed, our previous analysis of POU5F1, NANOG, and SOX2 targets in GCTNet showed that whereas NANOG and POU5F1 had largely overlapping programs related to cellular organization and DNA and cellular metabolism categories, SOX2 controlled a relatively independent program enriched for categories representing regulation of histone methylation and modification, stem cell differentiation, and a variety of differentiation-associated programs including those associated with neuronal, axonal, and chondrocyte differentiation (Kushwaha et al., 2015). Taken together, these results establish that functional pathways related to stem cell and neuronal development regulated by SOX2 are curtailed in SEM but restored in EC following re-establishment of pluripotency.
Figure 2.

ARACNe-Predicted SOX2 Targets Are Enriched in NT2D/1 Cells and EC Compared with TCam-2 Cells and SEM
(A) GSEA enrichment analysis of targets of (a) SOX2 (p = 0.05), (b) POU5F1 (p = 0.04), and (c) NANOG (p = 0.0) in TCam-2 versus NT2/D1 cells.
(B) GSEA enrichment analysis of targets in EC versus SEM of (a) SOX2 (0.027), (b) POU5F1 (0.5), and (c) NANOG (0.4). All p values are by two-tailed t test.
PRDM1 Does Not Regulate SOX2 in TCam-2 and NT2/D1 Cells
A recent study showed that ectopic upregulation of PRDM1 in the H9 hES cells and the PA-1, NTERA-2, and NCCIT EC cells led to downregulation of SOX2 expression, suggesting direct PRDM1 regulation of SOX2 (Lin et al., 2014). We investigated the potential role of PRDM1 in regulating SOX2 expression in TCam-2 and NT2/D1 cells, the latter as SOX2-expressing control, by KD of the gene in these two cell lines using the relevant SMARTpool small interfering RNAs (siRNAs) and measuring SOX2 mRNA expression by qRT-PCR at 72 hr following KD. SOX2 expression was neither upregulated in TCam-2 cells nor downregulated in NT2/D1 cells. We further assayed for PRDM1 and SOX2 expression by immunofluorescence (IF) in NT2/D1 and PRDM1-KD TCam-2 cells, which confirmed the mRNA analysis (Figure 3). These results show that PRDM1 does not directly regulate SOX2 and that some other mechanism may be involved in keeping the gene repressed in hPGCs and SEM.
Figure 3.

SOX2 Expression Is Not Altered in PRDM1-KD TCam-2 Cells
(A) qRT-PCR analysis of (a) PRDM1 and (b) SOX2 expression in PRDM1-KD cells following normalization to PGK1 signal. Data represent mean ± SD of three independent experiments. ∗∗∗p > 0.001 by Student’s t test; ns, not significant.
(B) Representative IF images of (a) NT2D-1, (b) TCam-2, and (c) PRDM1-KD TCam-2 cells stained for DAPI (blue), PRDM1 (green), and SOX2 (red), and merged images. Scale bars represent 100 μm.
SOX2 Promoter in TCam-2 Cells Is a Target of PcG
We first investigated promoter methylation as a possible mechanism for SOX2 repression in SEM. We compared the methylation status of SOX2 promoter in bisulfite-converted DNA from five each of SEM and EC tumors along with eight EC cell lines (NT2/D1, 27X-1, NCCIT, 169A, 218A, 228A, 2101ep, TERA-1) and the TCam-2 SEM cell line, quantitating the degree of methylation by mass spectrometry of amplification products of eight primer sets that covered the entire CpG island of the SOX2 promoter (1,000 kbp upstream and 1,000 kbp downstream of the TSS), using the Sequenom EpiTYPER assay. No methylation was detected affecting the SOX2 promoter of SEM or EC tumors, or their derived cell lines (Figures S1A and S1B), ruling out promoter methylation as the basis for SOX2 repression in SEM.
We then investigated the possibility of epigenetic modification as the mechanism of SOX2 repression. In genome-wide mapping studies using human embryonic fibroblast cells, the SOX2 promoter has previously been recognized as a target of PcG, being enriched for SUZ12 (Polycomb repressive complex 2 [PRC2]), CBX8 (Polycomb repressive complex 1 [PRC1]), and H3K27me3 (Bracken et al., 2006). To investigate whether SOX2 promoter is a target for PcG in human germline cells, we assayed for the co-occupancy of SUZ12, BMI1 (PRC1), and H3K27me3 at the SOX2 promoter in TCam-2 cells by ChIP-qPCR. All three components of PcG were enriched at the SOX2 promoter, confirming epigenetic modification as the basis for its transcriptional repression in TCam-2 cells (Figure 4).
Figure 4.

PcG Proteins SUZ12 and BMI1 and the Repressive Chromatin Mark H3K27me3 Are Enriched at SOX2 Promoter in TCam-2 Cells
ChIP analyses were performed using antibodies for (A) SUZ12, (B) BMI1, (C) H2K27me3, or immunoglobulin G (IgG) (as nonspecific control) in Tcam-2 cells. Plotted values are relative enrichment to 10% input and measured for indicated site in the SOX2 promoter and GUSB (β-glucuronidase) gene (control). Data represent mean ± SD of three independent experiments. Student’s t test: ns, not significant; ∗∗p < 0.01; ∗p < 0.05.
SOX2 Transcription in TCam-2 Cells Is Regulated by UTX during Retinoid Signaling
UTX, a member of the JumonjiC family TFs, is a di- and trimethyl H3K27 demethylase that also associates with mixed-lineage leukemia 2/3 (MLL) complexes possessing H3K4 methyltransferase activity; it occupies the promoters of HOX gene clusters and modulates their transcriptional output by regulating PRC1 and monoubiquitination of H2A in HEK 293 cells (Lee et al., 2007). During retinoid-induced signaling in NT2/D1 cells, recruitment of UTX to HOX genes results in H3K27 demethylation and H3K4 methylation, leading to HOXA13 and HOXC4 gene transcription (Lee et al., 2007). UTX is endogenously expressed in TCam-2 cells and we reasoned that it may regulate SOX2 in these cells as well, when challenged to differentiate. Since retinoid signaling is known to be a major regulator of differentiation in stem cells (Gudas and Wagner, 2011), we induced differentiation in TCam-2 cells by treatment with all-trans retinoic acid (RA). Western blot and IF analysis of RA-treated cells showed SOX2 expression increasing and POU5F1 and NANOG expression decreasing from day 2 to day 6 following RA treatment (Figures 5A and 5C). UTX expression remained constant in treated as well as untreated cells (Figure 5B). However, ChIP-qPCR analysis showed significant enrichment of UTX at SOX2 promoter in day-6 RA-treated cells compared with control cells (Figure 5D).
Figure 5.

SOX2 Is Expressed in TCam-2 Cells following RA Treatment
(A) Immunoblot of RA-treated and untreated cells showing increased SOX2 (35 kDa) expression and decreased POU5F1 (39 kDa) and NANOG (34 kDa) expression over a 6-day time course. Tubulin (50 kDa) is loading control.
(B) Immunoblot showing unchanged UTX (150 kDa) expression over a 6-day time course. Tubulin (50 kDa) is loading control.
(C) IF showing SOX2, POU5F1, and NANOG expression following RA treatment. (a) Untreated TCam-2 cells showing POU5F1 (green), NANOG (magenta), and DAPI (blue), but not SOX2 (red) expression. (b) RA-treated TCam-2 cells showing SOX2 but not POU5F1 and NANOG expression. BF is brightfield image. Scale bars represent 100 μm.
(D) RA-treated and untreated TCam-2 cells were subjected to ChIP-qPCR using UTX antibody or an IgG antibody (nonspecific control). The immunoprecipitated DNA was subjected to PCR using primers specific to the SOX2 promoter region. Data represent mean ± SD of three independent experiments. Student’s t test: ns, not significant; ∗∗p < 0.01; ∗p < 0.05.
To investigate the role of UTX in SOX2 repression/derepression, we performed UTX KD in TCam-2 cells using SMARTpool siRNA and treated the cells with RA for 6 days, and assayed for expression of UTX and SOX2. SOX2 failed to be upregulated in response to RA in UTX-depleted cells, indicating UTX requirement for SOX2 derepression (Figures 6A and 6B). We then investigated the mechanism of UTX-regulated SOX2 derepression by assaying the changes in the enrichment of the histone marks H3K27me3 and H3K4me3 at the SOX2 promoter by ChIP-qPCR in control (scramble) and UTX-KD cells following 6 days of RA treatment. As expected, the H3K27me3 mark was enriched in control cells and was reduced upon RA treatment (Figure 6C). In the UTX-KD cells, however, the H3K27me3 mark enrichment remained unchanged between treated and untreated cells, confirming UTX dependence of the repressive mark change in response to RA at the SOX2 promoter (Figure 6C). RA treatment elicited a highly significant enrichment of the H3K4me3 mark at the SOX2 promoter in control cells (Figure 6D). In the UTX-KD cells the enrichment was significant, but less than that in the control cells (Figure 6D). Although UTX is known to associate with MLL complexes that possess H3K4 methyltransferase activity, it is not known to directly regulate H3K4me3. Thus, these results overall demonstrate that recruitment of UTX to SOX2 promoter in TCam-2 cells is associated with a decrease in occupancy of the repressive H3K27me3 and an increase in occupancy of the activating H3K4me3 mark, thereby enabling SOX2 derepression in response to RA signal.
Figure 6.

Epigenetic Modifications of SOX2 Promoter Leading to RA-Induced Derepression Are Modulated by UTX
(A) Expression of SOX2, POU5F1, and NANOG in the RA-treated UTX-KD cells compared with scramble (scr) cells in a 6-day time-course experiment.
(B) qRT-PCR showing expression level of SOX2, NANOG, POU5F1, and UTX in the untreated scr versus UTX-KD cells and treated scr versus UTX-KD cells. Data represent mean ± SD of the three independent experiments. ∗∗p < 0.01 between treated UTX-KD and scr by Student’s t test.
(C) ChIP-qPCR showing loss of H3K27me3 enrichment at SOX2 promoter of RA-treated scr cells compared with untreated cells, whereas the enrichment is unchanged in RA-treated UTX-KD cells. Data represent mean ± SD of three independent experiments. Student’s t test: ns, not significant; ∗∗p < 0.01; ∗p < 0.05.
(D) ChIP-qPCR with H3K4me3 antibody showing enrichment at SOX2 promoter of RA-treated scr cells compared with untreated scr cells and RA-treated UTX-KD cells compared with untreated KD cells. Data represent mean ± SD of three independent experiments. Student’s t test: ns, not significant; ∗∗p < 0.01; ∗p < 0.05.
Derepression of SOX2 Leads to Induction of Neuronal and Other Lineage Genes in TCam-2 Cells
We obtained the GEP of day-14 RA-treated TCam-2 cells, which by now were completely differentiated morphologically, using Affymetrix U133 A plus B arrays, and analyzed the expression of marker genes related to lineage development (Figure S2). The major lineage expressed was neuronal, with genes such as NEFM, PAX6, NEFL, NESTIN, TUJI, and TRKC showing significantly higher expression than in untreated cells. Epithelial and mesodermal genes such as CDH1, EPCAM, FOXN1, and T were downregulated in the RA-treated cells, although KRT was upregulated. Interestingly, several endodermal genes (DAB2, NODAL, SPARC) were upregulated while others (SOX17, EPAS1, GATA4) were downregulated. In addition, smooth muscle lineage genes such as MEF2C and MYH6 were also expressed in the differentiated cells. We confirmed the GEP results on neuronal gene expression by IF analysis using NEFM, NESTIN, TUJI, and TRKC antibodies (Figure 7A). We further evaluated by qRT-PCR the status of the neuronal markers NEUROD4, NEFM, NES, TRKC, and TUJI in control (scramble) and UTX-KD TCam-2 cells treated with RA to test the effect of UTX depletion that prevented SOX2 upregulation. The expression of each of these genes was significantly lower in the UTX-KD cells compared with control cells in response to RA treatment, confirming the role of SOX2 in regulating neuronal gene expression (Figure 7B).
Figure 7.

Neuronal Marker Genes Are Upregulated following SOX2 Derepression in TCam-2 Cells in Response to Retinoid Signaling
(A) Representative IF showing staining with (a) neurofilament (NEFM), (b) nestin (NES), (c) neuron-specific class III β-tubulin (TUJI), and (d) neurotrophic tyrosine kinase receptor type 3 (TRKC). Left image, DAPI; middle image, neuronal gene; right image, merged. For each antibody the upper panel represents untreated cells and the lower panel RA-treated cells. Scale bars represent 100 μm.
(B) Expression of neuronal genes in RA-treated UTX-KD cells compared with RA-treated scramble cells by qRT-PCR. Data represent mean ± SD of three independent experiments. ∗∗ p < 0.01, ∗p < 0.05 by Student’s t test.
(C) GO categories enriched in ARACNe-predicted SOX2 targets of TCam-2 cells following RA treatment. Blue bar represents actual enriched number of genes; red bar represents expected enrichment by chance.
We also performed GO analysis of the expressed SOX2 targets predicted by ARACNe in the GEP of the day-14 RA-treated TCam-2 cells. As shown in Figure 7C, the upregulated target genes represented a variety of developmental processes, with nervous system categories predominating: axonogenesis (p = 1.20 × 10−3), tube development (p = 1.07 × 10−3), gliogenesis (p = 8.03 × 10−4), nervous system development (p = 6.59 × 10−4), neuron projection regeneration (p = 6.13 × 10−4), axon cargo transport (p = 5.26 × 10−4), axon regeneration (p = 4.46 × 10−4), system development (p = 2.24 × 10−4), stem cell differentiation (p = 1.07 × 10−4), anterograde axon cargo transport (p = 6.84 × 10−5), multicellular organism development (p = 5.85 × 10−5), anatomical structural morphogenesis (p = 4.6 × 10−5), developmental process (p = 2.76 × 10−6), and anatomical structure development (p = 2.34 × 10−6). These results together confirm direct SOX2 regulation of neuroectodermal genes.
Taken together, the data presented above are consistent with the hypothesis that SOX2 transcription, which is repressed in TCam-2 as well as in its progenitor hPGC cells, is mediated by enrichment of PcG and H3K27me3 at its bivalent promoter and is regulated by UTX.
Discussion
In this study we show that SOX2 repression in TCam-2 cells is due to the presence of PcG and H3K27me3 at its promoter and that the repression can be reversed by recruitment of UTX, a H3K27 demethylase to the promoter. SOX2 repression is a feature of hPGCs, from which SEM and other GCTs are derived by transformation and upregulation of pluripotency- and proliferation-promoting genes.
PGC specification has been investigated in the greatest detail in the mouse, where it is regulated by a PGC-specific transcriptional network composed of the TFs Prdm1, Prdm14, and Tfap2C, the so-called tripartite network (Magnúsdóttir et al., 2013, Nakaki et al., 2013). Recently, human GC fate determination has been shown to be different to that of the murine GC fate (Irie et al., 2015, Surani, 2015). Thus, hPGC specification is suggested to be initiated by SOX17 and its downstream target PRDM1, which represses mesendodermal genes, along with extensive epigenome resetting (Tang et al., 2015, Gkountela et al., 2015). Although the pluripotency factors NANOG, POU5F1, PRDM14, LIN28A, KLF4, and TFP2CL1 are expressed in hPGCs as early as 5.5 weeks of gestation (Tang et al., 2015), pluripotency is kept latent and PGCs remain unipotent or lineage restricted during their entire development.
Human fetal GCs express many of the pluripotency markers in common with murine PGCs (De Miguel et al., 2010); however, of the three core pluripotency master regulators POU5F1, NANOG, and SOX2, only the former two are expressed whereas SOX2 is repressed (Perrett et al., 2008, Tang et al., 2015). Nevertheless, post fertilization, human embryonic blastomeres as well as inner cell mass cells express SOX2 (Galán et al., 2010), implying that its repression is restricted to germline development. A recent study showed that in H9 hES cells and EC cell lines, ectopic expression of PRDM1 led to significant reduction in SOX2 mRNA and protein expression (Lin et al., 2014). The same study also showed that in a ChIP assay, PRDM1 bound to a previously identified 12-bp consensus binding site of human SOX2 gene and that PRDM1 suppressed the luciferase activity regulated by this genomic region. Based on these results, it was suggested that PRDM1 regulates SOX2 during human GC development and acts as a molecular switch to modulate between neural and germline fates (Lin et al., 2014). However, neither the mechanism of SOX2 repression nor its proposed PRDM1 regulation has been demonstrated in hPGCs, GCNIS, or SEM cells, in all of which SOX2 expression is constitutively repressed. Since TCam-2 cells are considered to be transformed hPGCs undergoing specification, we investigated the possible control of SOX2 by PRDM1 in TCam-2 and NT2/D1 cells, in both of which PRDM1 is expressed. Contrary to expectation, our results of PRDM1 silencing did not confirm its regulation of SOX2 in either cell type. The discrepancy in results between the previous study and our study may be related to the differing contexts of cellular biology that they represented. Because the SOX2 promoter is known to be a target of PcG in human embryonal fibroblast cells (Bracken et al., 2006), we reasoned that its regulation may also be mediated by an epigenetic modification in TCam-2 cells. Thus, after ruling out DNA methylation as the mechanism of repression, we assayed for occupancy of PRC2, PRC1, and H3K27me3 at the promoter and found enrichment, confirming their role in SOX2 repression. We then sought to alter the enrichment of the repressive epigenetic mark at the promoter through recruitment of UTX, an H3K27-specific demethylase, by invoking retinoid signaling in TCam-2 cells, as was done previously in the case of the HOX genes in NT2/D1 cells (Lee et al., 2007). RA treatment resulted in a change in the status of UTX and the chromatin marks at the SOX2 promoter, leading to derepression of its transcription and induction of neuronal and other lineage genes. These results thus show that UTX targets SOX2 for transcriptional regulation as in the case of HOX genes in human and Drosophila cells (Lee et al., 2007, Copur and Müller, 2013). Our data also are in agreement with the idea that SOX2 repression is an essential component of hPGC specification by preventing neuronal gene expression. Overall, then, hPGC specification appears to be mediated by three key TFs: SOX17, which initiates the GC lineage; PRDM1, which represses the mesendodermal lineage; and SOX2, whose repression prevents neuroectodermal lineage.
SOX2 is an essential factor in the maintenance of both human and murine pluripotency; however, it is upregulated, along with other pluripotency factors, in murine PGCs but not hPGCs. In this study we have clarified the mechanism of its repression in the hPGC-derived TCam-2 SEM cells and suggest that the same mechanism may be responsible for SOX2 repression in hPGCs. Although retinoids activated SOX2 expression in TCam-2 cells, this did not result in restoration of pluripotency as indicated by the differentiated phenotype and downregulation of POU5F1 and NANOG in SOX2-expressing TCam-2 cells. Therefore, our results are relevant specifically to SOX2 biology in human germline, but not necessarily to re-acquisition of pluripotency in EC. The mechanism of the latter is currently unknown, perhaps requiring epigenetic modification at several loci, including SOX2. SOX2 regulation overall is complex and invokes multiple pathways including the action of genes such as POU5F1, NANOG, STAT3, and SMAD3 (Thomson et al., 2011, Zhang and Cui, 2014) and, as shown previously and in this study, PcG.
Pluripotency comprises a spectrum of states that present in vitro and in vivo (Hackett and Surani, 2014). SEM and EC represent latent and patent versions of hPGC pluripotency; in addition, in vivo GCT pluripotency is distinct from embryonal pluripotency in being nontransient and associated with self-renewal. The functional pathways operating in SEM and EC in their respective states of pluripotency have not been characterized so far. In this study, by SAM of GEPs and GO biological process analyses of the downregulated and upregulated genes in the two subsets in comparison with normal testes, we show that whereas SEM retained pathways and processes similar to those in PGCs, EC was enriched for pathways that regulate stem cell and development categories; both subsets downregulate meiosis and spermatogenesis categories and upregulate categories associated with proliferation and self-renewal. These data provide an insight into their biology and may represent the starting point for further studies of the unique sensitivity of GCTs to DNA-damaging agents and the regulation of pluripotency and self-renewal in this tumor system.
Experimental Procedures
GCT and Cell Line GEPs
We have previously reported the GEPs of 141 GCTs comprising 135 tumors of all histological types and six normal testes obtained by using Affymetrix U133 A plus B microarrays (Korkola et al., 2005, Korkola et al., 2006, Korkola et al., 2009). A subset of these GEPs comprising 16 SEMs and 41 ECs was used in the current analysis using previously described methods (Kushwaha et al., 2015). GEPs of NT2/D1 and TCam-2 cells were obtained in triplicate using the Affymetrix U133 A plus B microarrays as previously described (Kushwaha et al., 2015). GEP of TCam-2 cells treated with RA for 14 days were obtained from three biological replicates using Affymetrix A plus B microarrays.
Cell Lines, Cell Culture, and ATRA Treatment
TCam-2 and NT2/D1 cells were cultured and maintained as described previously (Eckert et al., 2008, Houldsworth et al., 2002). For RA treatment, TCam-2 cells were plated at a density of 2 × 106 per 10-cm plate and treated on the following day with ATRA (10 μM/ml in medium) (Sigma-Aldrich). Cells were collected on days 0, 2, 4, and 6 for downstream experiments. Every 2–3 days, cells were reseeded in the presence of ATRA. The entire time-course experiment was carried out in triplicate with untreated cultures as controls.
IF Analysis
TCam-2 cells treated with or without ATRA were fixed in 4% paraformaldehyde. Fixed cells were permeabilized in 0.3% Triton X-100 in PBS containing 5% normal goat serum and subjected to immunocytochemical analyses as previously described (Kushwaha et al., 2015). Primary antibodies used for staining were goat anti-SOX2 (1:100) (catalog #sc17319; Santa Cruz Biotechnology), rabbit anti-NANOG (1:100) (#ab21624; Abcam), mouse anti-POU5F1 (1:200) (#sc5279; Santa Cruz), rabbit anti-BLIMP1 (#9115; Cell Signaling Technology), mouse anti-Nestin (#sc23927; Santa Cruz), rabbit anti-neurofilament (#ab-9034; Abcam), goat anti-TRKC (#ab188592; Abcam), and mouse anti-TUJ1 (#801201; BioLegend). Appropriate fluorescence-labeled secondary antibodies were used for visualization.
Gene Silencing Using a Transient Assay
For silencing of PRDM1 or UTX genes, 0.04 × 106 TCam-2 cells were plated in 24-well plates 1 day prior to transfection. 80 nM PRDM1 (ON-TARGETplus Human PRDM1; Dharmacon) siRNA-SMARTpool or UTX (Dharmacon) siRNA-SMARTpool and control siRNA (ON-TARGETplus Non-targeting Pool; Dharmacon) were used to transfect each well using the DharmaFECT1 Transfection Reagent (Dharmacon). Media were changed after 24 hr of transfection and cells were collected for studies at different time intervals. RNA was isolated using Qiazol lysis reagent (Qiagen) and reverse transcribed using the SuperScript VILO cDNA Synthesis Kit (Life Technologies). mRNA expression was analyzed by qRT-PCR using Taqman probes and primer sets in a 7500 Real-Time PCR system (Applied Biosystems) according to the manufacturer's instructions. PGK1 mRNA level was used as internal normalization control.
SOX2 Promoter Methylation Assay by EpiTYPER Analysis
ChIP and ChIP-qPCR
ChIP assay was performed on TCam-2 cells according to methods previously described by us (Kushwaha et al., 2015). The following antibodies and reagents were used for immunoprecipitation: UTX (Abcam #36938), SUZ12 (Millipore #17661), BMI1 (Abcam #14389), H3K4me3 (pAb) (Active Motif #39159), anti-trimethyl-histone H3 (Lys 27) (Millipore #07-449), and anti-trimethyl-histone H3 (Lys4) (Millipore #07-437). A Dynabeads Protein G immunoprecipitation kit (Life Technologies #10007D) was used. Immunoprecipitated DNA was purified by phenol-chloroform extraction, resuspended in 50 ml of Tris-EDTA, and amplified using oligonucleotides (Table S3). ChIP-qPCR was performed using Power SYBR Green PCR Master Mix (Life Technologies #4367659) on an ABI 7500 cycler.
Author Contributions
R.S.K.C. and R.K. developed and designed the study. R.K., N.J., G.M., R.S., and J.E.K. performed experiments. V.T., M.K., and A.C. performed data analysis. M.S., A.C., and G.J.B. participated in manuscript development. R.S.K.C. and R.K. wrote the manuscript.
Acknowledgments
Supported by the Byrne Fund and the NCI grant CA008784. We thank the MSKCC Molecular Cytology Core Facility for help with the IF assays and Drs. Sohei Kitazawa and Janet Shipley for the TCam-2 cells. The help of Adriana Heguy in EpiTYPER analysis of promoter methylation is gratefully acknowledged.
Published: April 28, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, two figures, and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2016.04.002.
Supplemental Information
References
- Andrews P.W., Matin M.M., Bahrami A.R., Damjanov I., Gokhale P., Draper J.S. Embryonic stem (ES) cells and embryonal carcinoma (EC) cells: opposite sides of the same coin. Biochem. Soc. Trans. 2005;6:1526–1530. doi: 10.1042/BST0331526. [DOI] [PubMed] [Google Scholar]
- Bracken A.P., Dietrich N., Pasini D., Hansen K.H., Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006;20:1123–1136. doi: 10.1101/gad.381706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaganti R.S., Houldsworth J. Genetics and biology of adult human male germ cell tumors. Cancer Res. 2000;60:1475–1482. [PubMed] [Google Scholar]
- Copur Ö., Müller J. The histone H3-K27 demethylase Utx regulates HOX gene expression in Drosophila in a temporally restricted manner. Development. 2013;140:3478–3485. doi: 10.1242/dev.097204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Miguel M.P., Fuentes-Julián S., Alcaina Y. Pluripotent stem cells: origin, maintenance and induction. Stem Cell Rev. 2010;6:633–649. doi: 10.1007/s12015-010-9170-1. [DOI] [PubMed] [Google Scholar]
- Eckert D., Biermann K., Nettersheim D., Gillis A.J., Steger K., Jäck H.M., Müller A.M., Looijenga L.H., Schorle H. Expression of BLIMP1/PRMT5 and concurrent histone H2A/H4 arginine 3 dimethylation in fetal germ cells, CIS/IGCNU and germ cell tumors. BMC Dev. Biol. 2008;8:106. doi: 10.1186/1471-213X-8-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galán A., Montaner D., Póo M.E., Valbuena D., Ruiz V., Aguilar C., Dopazo J., Simón C. Functional genomics of 5- to 8-cell stage human embryos by blastomere single-cell cDNA analysis. PLoS One. 2010;5:e13615. doi: 10.1371/journal.pone.0013615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkountela S., Zhang K.X., Shafiq T., Liao W.-W., Hargan-Caluopina J., Chen P.-Y., Clark A.T. DNA methylation dynamics in the human prenatal germline. Cell. 2015;161:1425–1436. doi: 10.1016/j.cell.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudas L.J., Wagner J.A. Retinoids regulate stem cell differentiation. J. Cell Physiol. 2011;226:322–330. doi: 10.1002/jcp.22417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett J.A., Surani M.A. Regulatory principles of pluripotency: from the ground state up. Cell Stem Cell. 2014;15:416–430. doi: 10.1016/j.stem.2014.09.015. [DOI] [PubMed] [Google Scholar]
- Heaney J.D., Anderson E.L., Michelson M.V., Zechel J.L., Conrad P.A., Page D.C., Nadeau J.H. Germ cell pluripotency, premature differentiation and susceptibility to testicular teratomas in mice. Development. 2012;139:1577–1586. doi: 10.1242/dev.076851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houldsworth J., Heath S.C., Bosl G.J., Studer L., Chaganti R.S. Expression profiling of lineage differentiation in pluripotential human embryonal carcinoma cells. Cell Growth Differ. 2002;13:257–264. [PubMed] [Google Scholar]
- Irie N., Weinberger L., Tang W.W., Kobayashi T., Viukov S., Manor Y.S., Dietmann S., Hanna J.H., Surani M.A. SOX17 is a critical specifier of human primordial germ cell fate. Cell. 2015;160:253–268. doi: 10.1016/j.cell.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephson R., Ording C.J., Liu Y., Shin S., Lakshmipathy U., Toumadje A., Love B., Chesnut J.D., Andrews P.W., Rao M.S., Auerbach J.M. Qualification of embryonal carcinoma 2102ep as a reference for human embryonic stem cell research. Stem Cells. 2007;25:437–446. doi: 10.1634/stemcells.2006-0236. [DOI] [PubMed] [Google Scholar]
- Korkola J.E., Houldsworth J., Dobrzynski D., Olshen A.B., Reuter V.E., Bosl G.J., Chaganti R.S. Gene expression-based classification of nonseminomatous male germ cell tumors. Oncogene. 2005;24:5101–5107. doi: 10.1038/sj.onc.1208694. [DOI] [PubMed] [Google Scholar]
- Korkola J.E., Houldsworth J., Chadalavada R.S., Olshen A.B., Dobrzynski D., Reuter V.E., Bosl G.J., Chaganti R.S. Down-regulation of stem cell genes, including those in a 200-kb gene cluster at 12p13.31 is associated with in vivo differentiation of human male germ cell tumors. Cancer Res. 2006;66:820–827. doi: 10.1158/0008-5472.CAN-05-2445. [DOI] [PubMed] [Google Scholar]
- Korkola J.E., Houldsworth J., Feldman D.R., Olshen A.B., Qin L.X., Patil S., Reuter V.E., Bosl G.J., Chaganti R.S. Identification and validation of a gene expression signature that predicts outcome in adult men with germ cell tumors. J. Clin. Oncol. 2009;27:5240–5247. doi: 10.1200/JCO.2008.20.0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushwaha R., Jagadish N., Kustagi M., Tomishima M.J., Mendiratta G., Bansal M., Kim H.R., Sumazin P., Alvarez M.J., Lefebvre C. Interrogation of a context-specific transcription factor network identifies novel regulators of pluripotency. Stem Cells. 2015;33:367–377. doi: 10.1002/stem.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.G., Villa R., Trojer P., Norman J., Yan K.P., Reinberg D., Di Croce L., Shiekhattar R. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 2007;318:447–450. doi: 10.1126/science.1149042. [DOI] [PubMed] [Google Scholar]
- Leitch H.G., Nichols J., Humphreys P., Mulas C., Martello G., Lee C., Jones K., Surani M.A., Smith A. Rebuilding pluripotency from primordial germ cells. Stem Cell Rep. 2013;1:66–78. doi: 10.1016/j.stemcr.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin I.Y., Chiu F.L., Yeang C.H., Chen H.F., Chuang C.Y., Yang S.Y., Hou P.S., Sintupisut N., Ho H.N., Kuo H.C. Suppression of the SOX2 neural effector gene by PRDM1 promotes human germ cell fate in embryonic stem cells. Stem Cell Rep. 2014;2:189–204. doi: 10.1016/j.stemcr.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnúsdóttir E., Dietmann S., Murakami K., Günesdogan U., Tang F., Bao S., Diamanti E., Lao K., Gottgens B., Surani M.A. A tripartite transcription factor network regulates primordial germ cell specification in mice. Nat. Cell Biol. 2013;15:905–915. doi: 10.1038/ncb2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Nakaki F., Hayashi K., Ohta H., Kurimoto K., Yabuta Y., Saitou M. Induction of mouse germ-cell fate by transcription factors in vitro. Nature. 2013;501:222–226. doi: 10.1038/nature12417. [DOI] [PubMed] [Google Scholar]
- Perrett R.M., Turnpenny L., Eckert J.J., O'Shea M., Sonne S.B., Cameron I.T., Wilson D.I., Rajpert-De Meyts E., Hanley N.A. The early human germ cell lineage does not express SOX2 during in vivo development or upon in vitro culture. Biol. Reprod. 2008;78:852–858. doi: 10.1095/biolreprod.107.066175. [DOI] [PubMed] [Google Scholar]
- Sperger J.M., Chen X., Draper J.S., Antosiewicz J.E., Chon C.H., Jones S.B., Brooks J.D., Andrews P.W., Brown P.O., Thomson J.A. Gene expression patterns in human embryonic stem cells and human pluripotent germ cell tumors. Proc. Natl. Acad. Sci. USA. 2003;100:13350–13355. doi: 10.1073/pnas.2235735100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surani M.A. Human germline: a new frontier. Stem Cell Rep. 2015;4:955–960. doi: 10.1016/j.stemcr.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W.W., Dietmann S., Irie N., Leitch H.G., Floros V.I., Bradshaw C.R., Hackett J.A., Chinnery P.F., Surani M.A. A unique gene regulatory network resets the human germline epigenome for development. Cell. 2015;161:1453–1467. doi: 10.1016/j.cell.2015.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson M., Liu S.J., Zou L.N., Smith Z., Meissner A., Ramanathan S. Pluripotency factors in embryonic stem cells regulate differentiation into germ layers. Cell. 2011;145:875–889. doi: 10.1016/j.cell.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Cui W. Sox2, a key factor in the regulation of pluripotency and neural differentiation. World J. Stem Cells. 2014;6:305–311. doi: 10.4252/wjsc.v6.i3.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.