

SUMMARY
BRCA1/2 proteins function in homologous recombination (HR)-mediated DNA repair and cooperate with Fanconi anemia (FA) proteins to maintain genomic integrity through replication fork stabilization. Loss of BRCA1/2 proteins results in DNA repair deficiency and replicative stress, leading to genomic instability and enhanced sensitivity to DNA damaging agents. Recent studies have shown that BRCA1/2-deficient tumors upregulate Polθ-mediated alternative end-joining (alt-EJ) repair as a survival mechanism. Whether other mechanisms maintain genomic integrity upon loss of BRCA1/2 proteins is currently unknown. Here we show that BRCA1/2-deficient tumors also upregulate FANCD2 activity. FANCD2 is required for fork protection and fork restart in BRCA1/2-deficient tumors. Moreover, FANCD2 promotes Polθ recruitment at sites of damage and alt-EJ repair. Finally, loss of FANCD2 in BRCA1/2-deficient tumors enhances cell death. These results reveal a synthetic lethal relationship between FANCD2 and BRCA1/2, and identify FANCD2 as a central player orchestrating DNA repair pathway choice at the replication fork.
ETOC BLURB
Kais et al. show that BRCA1/2-deficient tumors have a compensatory increase in FANCD2 activity. FANCD2 stabilizes stalled replication forks and promotes alternative end-joining (alt-EJ) in BRCA1/2-deficient tumors. Loss of FANCD2 in these tumors results in severe DNA repair defects and enhanced cell death.
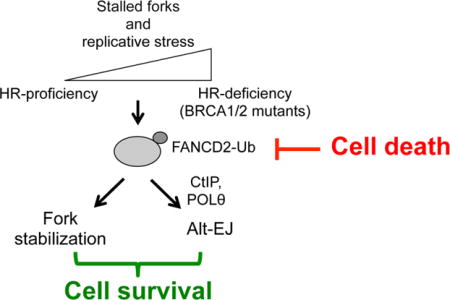
INTRODUCTION
Multiple mechanisms cooperate in cells to ensure the fidelity of DNA replication and to maintain genome integrity. Exogenous DNA damage and/or endogenous replication stress cause stalling of replication forks, leading to the recruitment of multiple proteins which stabilize stalled forks, repair DNA lesions, and restart replication (Branzei and Foiani, 2007, 2010; Michel et al., 2004). Failure to arrest replication forks at damaged sites or to restart replication once the repair is completed affects both genomic stability and cell survival (Cox et al., 2000). Indeed, damaged DNA, such as double strand breaks (DSBs) or interstrand crosslinks (ICLs), and replication fork collapse are the main forces that drive genome instability (Aparicio et al., 2014; Deans and West, 2011).
BRCA1 and BRCA2 (BRCA1/2) proteins have a dual role in protecting genomic integrity. On the one hand, BRCA1/2 proteins promote homologous recombination (HR)-mediated DNA repair (Moynahan et al., 1999; Moynahan et al., 2001). On the other hand, these proteins also limit replication stress by controlling the stability of stalled replication forks (Lomonosov et al., 2003; Pathania et al., 2014; Schlacher et al., 2011; Willis et al., 2014). Another DNA repair pathway carrying repair-independent functions during replication is the Fanconi anemia (FA) pathway (Gari et al., 2008; Kim and D’Andrea, 2012). Indeed, BRCA1/2 and some FA proteins such as FANCD2 localize to stalled replication forks, protect nascent strands from excessive nucleolytic degradation (Lossaint et al., 2013; Schlacher et al., 2011; Schlacher et al., 2012), and facilitate replication restart once DNA repair is complete (Lossaint et al., 2013; Schwab et al., 2015). For these reasons, the FA and BRCA1/2 proteins play a central role in limiting replication stress (Chan et al., 2009; Howlett et al., 2005; Naim and Rosselli, 2009). According to a conventional model, FANCD2 and BRCA1/2 proteins cooperate in an epistatic pathway, namely the FA/BRCA pathway, to both repair DNA lesions and stabilize replication forks (Kim and D’Andrea, 2012).
In accordance with the DNA repair and fork stabilization functions of BRCA1/2 proteins, BRCA1/2-deficient tumor cells exhibit both increased genomic instability and replicative stress (Cancer Genome Atlas Research, 2011; Schlacher et al., 2011; Zeman and Cimprich, 2014). As a result, BRCA1/2-deficient cells are hypersensitive to chemotherapeutic agents such as PARP inhibitors (PARPi) (Bryant et al., 2005; Farmer et al., 2005; Konstantinopoulos et al., 2015) and to replication stress inducing poisons (Howlett et al., 2005).
In BRCA1/2-deficient cells, unstable replication forks lead to chromosomal translocation and copy number variation (Hastings et al., 2009). Although genomic instability is critical to tumor progression, its excess can limit cell survival (Bartkova et al., 2005; Negrini et al., 2010). As a consequence, BRCA1/2-deficient cells have evolved mechanisms to tolerate replication stress and genomic instability, with the ultimate goal of ensuring DNA replication and cell survival (Ceccaldi et al., 2015b). As an example, BRCA1/2-deficient cells upregulate the error-prone Polθ/PARP1-mediated alternative end-joining (alt-EJ) DNA repair pathway, thereby compensating for defective HR (Ceccaldi et al., 2015a; Mateos-Gomez et al., 2015). Polθ is a translesion synthesis polymerase (Yousefzadeh and Wood, 2013) that prevents RAD51 assembly on single-stranded DNA (Ceccaldi et al., 2015a; Newman et al., 2015), and concurrently mediates PARP1-dependent alt-EJ to resume DNA replication (Kent et al., 2015). As a consequence, BRCA1/2-deficient cells are dependent on alt-EJ for survival. Inhibition of proteins functioning in alt-EJ, such as PARP1 or Polθ, is synthetically lethal in tumors with inactivated BRCA1/2 (Bryant et al., 2005; Ceccaldi et al., 2015a; Farmer et al., 2005; Mateos-Gomez et al., 2015). Besides intrinsically promoting tumor cell survival, the hyperactivation of mechanisms counteracting the onset of genomic instability can also lead to drug resistance (Bouwman and Jonkers, 2014; Lord and Ashworth, 2013). For example, secondary intragenic BRCA1/2 mutations can restore enzyme functionality and rescue HR, thus representing the most common acquired mechanism of resistance to cisplatin or PARPi (Edwards et al., 2008; Sakai et al., 2008).
Despite extensive research on mechanisms conferring resistance to PARPi (Bouwman and Jonkers, 2014; Lord and Ashworth, 2013), how BRCA1/2-deficient tumors limit their replication stress remain unknown. Many DNA repair proteins are indeed recruited to stalled forks upon replication stress (Alabert et al., 2014; Sirbu et al., 2013). For instance PARP1, which function is required for BRCA1/2-deficient cell survival, is present at the stalled replication fork and contributes to fork stabilization and recovery (Bryant et al., 2009; Ying et al., 2015; Ying et al., 2012). Whether other factors contribute to fork stabilization and survival of BRCA1/2-deficient cells is currently unknown.
Here, we demonstrate that BRCA1/2-deficient tumors exhibit a compensatory increase in FANCD2 expression and activity. Upon loss of BRCA1/2 proteins, FANCD2 localizes to stalled replication forks where it promotes replication fork protection and restart. FANCD2 recruits Polθ and CtIP to stalled forks and is required for alt-EJ. As a consequence, FANCD2 overexpression in BRCA1/2-mutant cells confers resistance to PARPi through replication fork stabilization. Finally, double knockdown experiments of FANCD2 and BRCA1/2 lead to tumor cell death in vitro and in vivo, revealing a synthetic lethal relationship between these proteins. Our findings identify FANCD2 as a critical factor required for the viability of BRCA1/2-deficient tumors, and for orchestrating DNA repair pathway choice at replication forks.
RESULTS
FANCD2 is upregulated in BRCA1/2-deficient cells
In order to identify gene candidates essential for the survival of BRCA1/2-deficient tumors, we examined gene expression profiles in cancers frequently associated with defects in HR (i.e. mutations in BRCA1/2 genes) (Cancer Genome Atlas, 2012; Cancer Genome Atlas Research, 2011). Publicly available microarray databases revealed specific overexpression of FANCD2 in subgroups of ovarian, breast, and uterine cancers associated with HR deficiency and high genomic instability (Figure 1A). FANCD2 expression was upregulated in a grade-dependent manner, correlated with KI67 expression, and increased in tumors harboring alterations (mutations or epigenetic silencing) in BRCA1 or BRCA2 (Figures S1A–D). Since FANCD2 and BRCA1/2 proteins are known to function epistatically in DNA repair and fork protection (Kim and D’Andrea, 2012; Lossaint et al., 2013; Schlacher et al., 2011; Schlacher et al., 2012), we analyzed the relationship between FANCD2 activation and BRCA1/2 alterations. Knockdown of BRCA1 or BRCA2 (BRCA1/2) with siRNA resulted in increased FANCD2 monoubiquitination (FANCD2-Ub), similar to the level observed by depletion of the deubiquitinase enzyme USP1 (Nijman et al., 2005) (Figures S1E–F). Subcellular fractionation of HeLa cells revealed that BRCA2 depletion increased FANCD2-Ub enrichment on the chromatin (Figure 1B). Knockdown of BRCA1/2 proteins increased basal and damage-inducible FANCD2 expression and foci formation (Figures 1C and S1G–I). Taken together, these data suggest FANCD2 expression and activation are increased in the setting of BRCA1/2 deficiency.
Figure 1. Upregulated FANCD2 protects replication forks in BRCA2-deficient tumor cells.
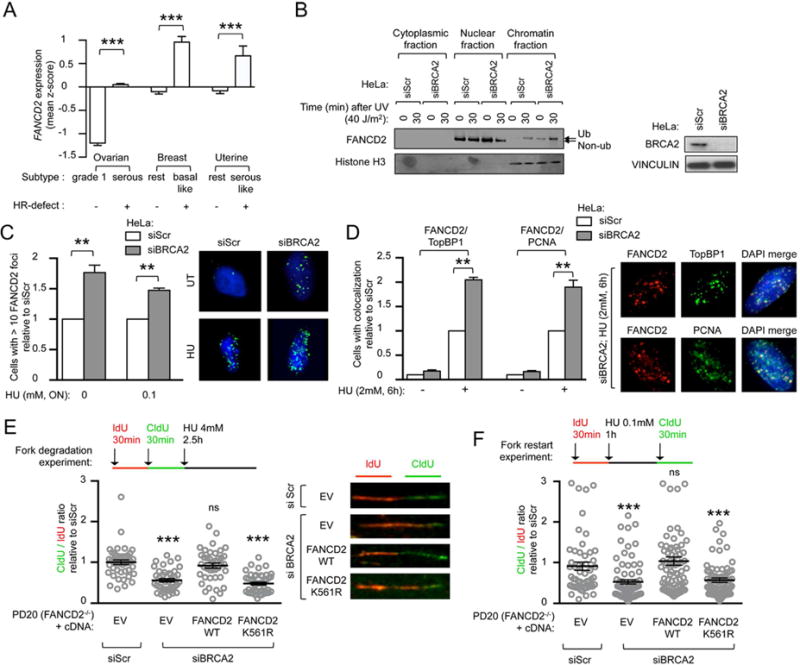
(A) FANCD2 gene expression in subtypes of ovarian, breast and uterine cancers. For each tumor group, expression values are represented as the mean of z-scores. (B) FANCD2 immunoblot in HeLa cells after siRNA depletion of BRCA2, at indicated time points after UV treatment and cellular fractionation. Immunoblot showing BRCA2 depletion efficiency. (C) Quantification of baseline and damage (HU)-induced FANCD2 foci in HeLa cells after siRNA depletion of BRCA2. Representative images are shown. Satistics were performed on n≥150 cells per condition and expressed as relative to siScr that was conventionally set to 1. (D) Baseline and damage (HU)-induced FANCD2, TopBP1 and PCNA immunofluorescence in HeLa cells after siRNA depletion of BRCA2. Representative images are shown. Representative images are shown. Satistics were performed on n≥150 cells per condition and expressed as relative to siScr that was conventionally set to 1. (E, F) Schematic for the labeling of FANCD2-deficient (PD20) cells with ldU and CIdU for fork degradation (E) and for fork restart experiments (F). Scatter dot plot for CIdU to ldU ratio of cells expressing indicated cDNA and transfected with indicated siRNA. Satistics were performed on n≥100 fibers per condition and expressed as relative to EV, that was conventionally set to 1. Representative images are shown. Data in C–F represent mean ± s.e.m. over n=3 independent experiments. Data in A, C–F were analyzed using Student’s t test. Abbreviations: ON, over-night; ns, non significant; EV, empty vector; WT, wild-type.
FANCD2 stabilizes replication forks in BRCA1/2-deficient cells
Loss of BRCA1/2 hinders fork stability and increases replication stress (Schlacher et al., 2011). Given that FANCD2 has recently emerged as a key factor in stabilizing replication forks (Lossaint et al., 2013; Schlacher et al., 2012), and since we observed a compensatory increase in FANCD2 activity in cells lacking BRCA1/2, we tested whether FANCD2 could have a primary role in stabilizing replication forks in this genetic setting. Two known replication stress proteins, TopBP1 and PCNA, form nuclear foci at sites of halted DNA replication (Howlett et al., 2009; Yan and Michael, 2009). We found that, in response to hydroxyurea (HU)-induced replicative stress, FANCD2 foci colocalize with TopBP1, similarly to what is observed with PCNA foci (Howlett et al., 2009). Interestingly, BRCA2 knockdown increased FANCD2 colocalization with TopBP1 and PCNA (Figure 1D). Moreover, siRNA-mediated BRCA1 and BRCA2 depletion in FANCD2-deficient cells (PD20) caused a significant increase in fork degradation and fork collapse after fork stalling by HU (Figures 1E–F and S1J–K). In order to assess the importance of FANCD2 monoubiquitination in the stabilization of the replication fork in BRCA1/2-deficient cells, we evaluated the ability of wild-type or the ubiquitination mutant of FANCD2 (K561R-FANCD2) to complement the siBRCA1/2-mediated fork instability. Expression of wild-type FANCD2, in FANCD2-deficient (PD20) cells, rescued fork degradation and fork restart, while K561R-FANCD2 did not (Figures 1E–F and S1J–K). Collectively, these data indicate that loss of BRCA1/2 triggers localization of FANCD2-Ub to stalled replicative sites where it maintains fork stability and promotes replication fork restart.
Synthetic lethal interaction between BRCA1/2 and FANCD2
Given that the stability of replication forks is essential for genomic integrity (Zeman and Cimprich, 2014), we hypothesized that FANCD2 is required for maintaining genomic stability in BRCA1/2-deficient cells. HR-deficient ovarian tumour cell lines, A2780-shBRCA1 and A2780-shBRCA2 cells, were generated and subjected to siRNA-mediated FANCD2 depletion, and clonogenic survival was measured (Figure S2A). FANCD2 depletion reduced the survival of BRCA1/2-deficient cells but had no effect on the isogenic HR-proficient cells (Figure 2A). Moreover, tumor cells expressing shRNAs against both BRCA2 and FANCD2 exhibited increased chromosomal aberrations and reduced survival after MMC treatment as compared to shScr control cells (Figures 2B–C and S2B). Consistent with these findings, a BRCA2-depleted (VU423) cell line showed increased basal and MMC-induced chromosomal aberrations together with reduced clonogenic survival after siRNA-mediated FANCD2 depletion (Figures 2D and S2C). Since FANCD2 forms a heterodimer with FANCI (Smogorzewska et al., 2007), we evaluated the potential synthetic lethality between FANCI and BRCA1/2 proteins. Similarly to FANCD2, FANCI depletion specifically reduced BRCA1/2-deficient cell survival without affecting the survival of BRCA1/2-proficient cells (Figures S2D). Collectively, our data show that FANCD2 limits the extent of genomic instability in BRCA1/2-deficient cells and is required for their survival.
Figure 2. FANCD2 maintains genomic stability in BRCA1 and BRCA2-deficient tumors.
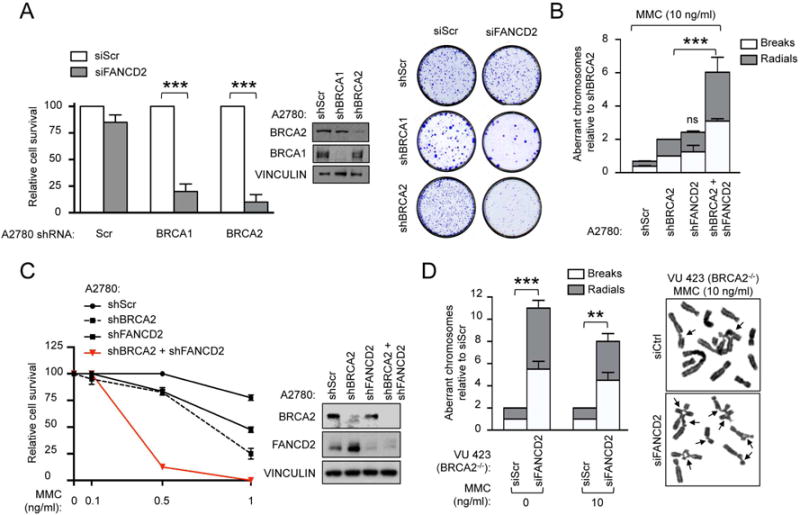
(A) Clonogenic formation of A2780 cells expressing the indicated shRNA together with the indicated siRNA. Immunoblot showing knockdown efficiencies of siRNAs and representative images for clonogenic formation are shown. (B) Chromosome breakage analysis of A2780 cells expressing the indicated shRNA and treated with MMC. (C) Clonogenic formation of A2780 cells expressing the indicated shRNA under increasing concentration of MMC. Survival is shown as relative to the untreated sample (MMC 0 ng/mL). Immunoblot showing silencing efficiency of shRNAs. (D) Chromosome breakage analysis of BRCA2-deficient (VU 423) cells transfected with the indicated siRNA. Representative images are shown. Arrows indicate chromosomal aberrations. Data in A–D represent mean ± s.e.m. over n=3 independent experiments and were analyzed by using the Chi-squared test for trend in proportions (A) or Student’s t test (B, D). Data in A, D are displayed as relative to siScr samples, while in B they are displayed as relative to shBRCA2 sample.
FANCD2 depletion reduces survival of BRCA1/2-deficient tumors in vitro and in vivo
To further analyze the synthetic lethal relationship between FANCD2 and BRCA1/2, we measured the clonogenic survival of a panel of 18 breast and 36 ovarian cell lines (Table S1) subjected to FANCD2 depletion. Depletion of FANCD2 primarily affected survival of cell lines with BRCA1/2 alterations (Figure 3A and S3A). FANCD2 depletion also affected BRCA1-deficient tumor growth in vivo. The BRCA1-mutated (MDA-MB-436) breast cell line, expressing a doxycycline-inducible FANCD2 shRNA, was xenotransplanted into athymic nude mice. Once tumors reached palpable sizes (100–200 mm3), mice were treated with doxycycline and tumor volumes were measured for 6 weeks. FANCD2 depletion significantly impaired tumor growth in vivo (Figures 3B–C). Taken together, these data confirm that BRCA1/2-deficient tumors are hypersensitive to the loss of FANCD2.
Figure 3. BRCA1 and BRCA2-deficient tumor cells are hyperdependent on monoubiquitinated FANCD2 for survival.
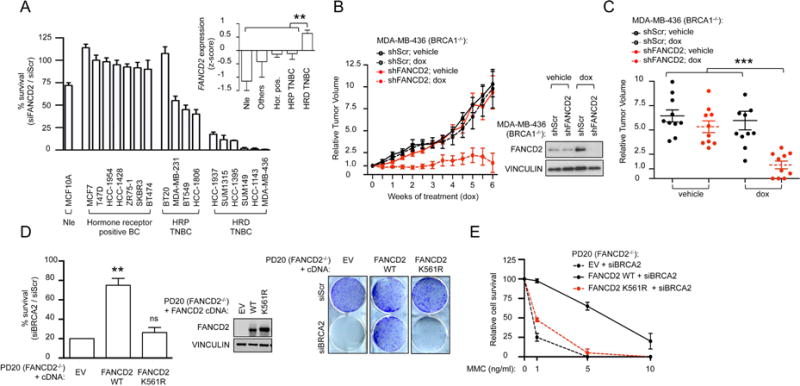
(A) Clonogenic formation of a panel of 18 breast cell lines transfected with Scr or FANCD2 siRNA. Percent survival of cells transfected with siFANCD2 versus siScr is plotted. The genetic status of the cell line analyzed is indicated (Nle: normal, non-transformed mammary epithelial cell line; TNBC: triple negative breast cancer; HRP: HR-proficient; HRD: HR-deficient). FANCD2 gene expression in the 58 breast cell lines from the CCLE collection that includes 16 of the 18 breast cell lines tested in Figure 3A. Cell lines are grouped based on their genetic status. For each group, expression values are represented as the mean of z-scores. (Hor. pos.: hormone positive breast cancer). A nonparametric test (Mann-Withney-Wilcoxon) was used to compare the value of the HRD TNBC group with the rest of the groups. (B) Growth of MDA-MB-436 cells expressing doxycycline (dox)-inducible FANCD2 or scrambled (Scr) shRNA in athymic nude mice. Immunoblot showing silencing efficiency. (C) Relative tumour volumes (RTV) for individual mice treated in (B) after five weeks of doxycycline (dox) treatment. Each group represents n≥10 tumors from n≥6 mice while each dot represents data from one tumor. Data are shown as mean ± s.e.m. Student’s t test was used to compare each sample to “shFANCD2; dox”. (D) Clonogenic formation of FANCD2-deficient (PD20) cells expressing empty vector (EV) or FANCD2 cDNA constructs and transfected with BRCA2 siRNA. Percent survival of cells transfected with BRCA2 versus Scr siRNA is plotted. An Immunoblot shows expression of FANCD2 cDNA constructs. Representative images are shown. (E) Clonogenic formation of FANCD2-deficient (PD20) cells expressing EV or FANCD2 cDNA constructs and transfected with BRCA2 siRNA in increasing concentration of MMC. Percent survival relative to non-treated cells (MMC 0 ng/mL) is plotted. Data in A, D–E represent mean ± s.e.m. over n=3 independent experiments. Data in D was analyzed using the Chi-squared test for trend in proportions. Abbreviations: ns, non significant; EV, empty vector; WT, wild-type.
To determine whether FANCD2 monoubiquitination is required for the survival of BRCA1/2-depleted cells, we performed complementation studies in BRCA1/2-deficient cells. Expression of wild-type, but not K561R-FANCD2, in FANCD2-deficient (PD20) cells, in which BRCA2 had been depleted, rescued clonogenic survival and MMC toxicity (Figures 3D–E). Similarly, in BRCA1-mutated (MDA-MB-436) cells, in which the endogenous FANCD2 had been depleted, expression of wild-type, but not K561R-FANCD2, rescued clonogenic survival (Figures S3B). Taken together, our findings indicate that FANCD2 monoubiquitination is required to maintain genomic stability and cell survival in BRCA1/2-deficient tumors.
FANCD2 is required for Polθ and CtIP foci assembly and promotes alt-EJ in BRCA1/2-deficient tumors
FANCD2 expression highly correlates with Polθ expression (Figure S4A), an alt-EJ factor strongly upregulated in BRCA1/2-deficient cells (Ceccaldi et al., 2015a). Moreover, tumors with high FANCD2 expression exhibit an increase in point mutation frequency and copy number alteration, suggesting an increase in error-prone alt-EJ repair (Figures S4B–C). Knockdown of BRCA2 increased DNA damage-inducible Polθ foci formation (Figure 4A), confirming that alt-EJ activity is upregulated in BRCA1/2-deficient tumors (Ceccaldi et al., 2015a). Recent evidence has suggested a role of FA proteins in alt-EJ (Howard et al., 2015). Moreover, the FA protein FANCA is required for class switch recombination (Nguyen et al., 2014), a process that relies, at least in part, on alt-EJ for rejoining DSBs (Deriano and Roth, 2013). Nonetheless, the mechanism by which the FA pathway interacts with alt-EJ remains unknown. In accordance with previous findings in a cell-based assay which measures the efficiency of recombination of two GFP alleles (alt-EJ assay) (Bennardo et al., 2008) (Howard et al., 2015). FANCD2 depletion reduced alt-EJ efficiency similarly to the reduction observed following PARP1 or Polθ depletion (Figure 4B). Conversely, FANCD2 depletion had no effect on single strand annealing (SSA), another double strand break repair mechanism, thus arguing for a specific role of FANCD2 in alt-EJ (Figure S4D).
Figure 4. Monoubiquitinated FANCD2 is required for Polθ and CtIP foci formation and for alt-EJ.
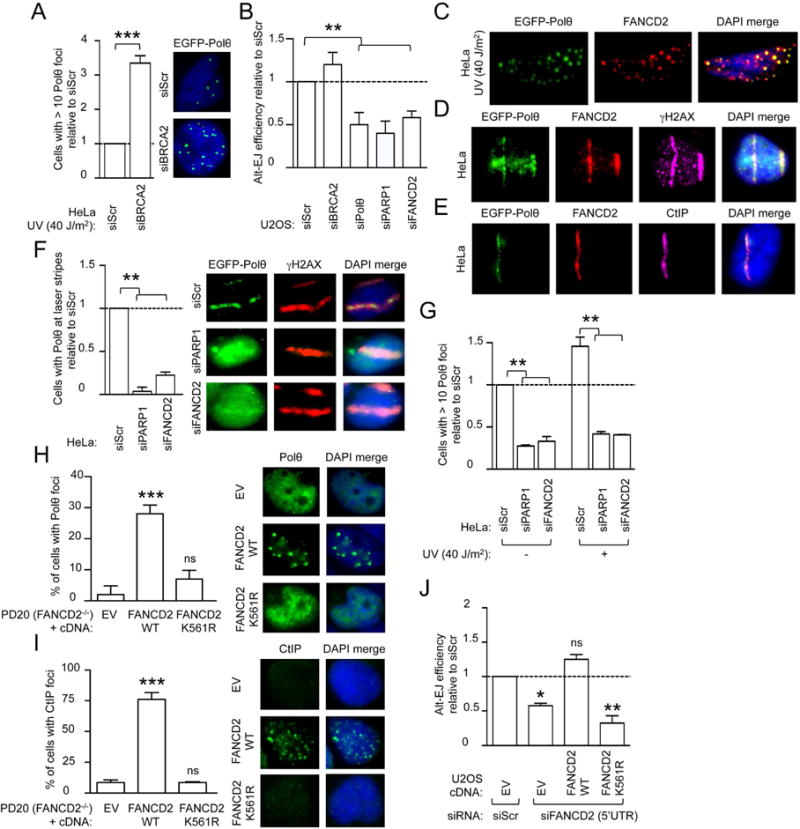
(A) Quantification of damage (UV)-induced Polθ foci in HeLa cells after siRNA depletion of BRCA2. (B) End-joining reporter assay in EJ2-U2OS cells transfected with the indicated siRNA. (C) FANCD2 immunofluorescence of HeLa cells transfected with GFP-tagged full length Polθ and subjected to UV damage. Representative images are shown. (D) FANCD2 and γH2AX immunofluorescence in HeLa cells transfected with GFP-tagged full length Polθ after laser micro-irradiation. Representative images are shown. (E) FANCD2 and CtIP immunofluorescence in HeLa cells transfected with GFP-tagged full length Polθ after laser micro-irradiation. Representative images are shown. (F) Quantification of Polθ localization at laser micro-irradiation sites in HeLa cells transfected with GFP-tagged full length Polθ and with indicated siRNA. γH2AX immunofluorescence serves as a marker of laser-induced DNA breaks. Representative images are shown. (G) Quantification of baseline and damage (UV)-induced Polθ foci in HeLa cells transfected with indicated siRNA. (H, I) Quantification of Polθ foci (H) and CtIP foci (I) formation in FANCD2-deficient (PD20) cells expressing EV, wild-type or K561R FANCD2 cDNA constructs. Representative images are shown. (J) End-joining reporter assay in EJ2-U2OS cells expressing indicated cDNAs and transfected with Scr or 5′UTR-FANCD2 siRNA. Data in A–B, F–J represent mean ± s.e.m. over n=3 independent experiments. Data in A–B, F–G, J are displayed as relative to siScr transfected sample, which value is set to 1 and were analyzed using Student’s t test. Data in H and I were analyzed using the Chi-squared test for trend in proportions. Abbreviations: ns, non significant; EV, empty vector; WT, wild-type.
Since FANCD2 and the Polθ-mediated alt-EJ pathway are upregulated and essential for BRCA1/2-deficient cell survival (Ceccaldi et al., 2015a; Mateos-Gomez et al., 2015), we next evaluated whether FANCD2 could cooperate with Polθ in promoting alt-EJ. Interestingly, FANCD2 and Polθ colocalized at damage foci and at laser-induced γH2AX-positive DNA breaks (Figure 4C–D). The endonuclease CtIP cooperates with BRCA1 to promote DNA-end resection and HR repair at DSBs (Escribano-Diaz et al., 2013; Sartori et al., 2007), but is also required for alt-EJ (Badie et al., 2015; Zhang and Jasin, 2011). In addition, CtIP and FANCD2 proteins physically interact (Murina et al., 2014; Unno et al., 2014; Yeo et al., 2014). Based on these findings, we determined whether CtIP might cooperate with FANCD2 and Polθ in alt-EJ repair. CtIP, like FANCD2, colocalized with Polθ at sites of DNA breaks (Figure 4E). We next aimed at better elucidating the functional hierarchy among these alt-EJ factors. Of note, FANCD2 inhibition reduced Polθ accumulation at laser-induced DNA breaks and Polθ foci (Figures 4F–G and S4E), similar to the reduction observed upon depletion of the alt-EJ factor PARP1 (Audebert et al., 2004; Ceccaldi et al., 2015a; Mateos-Gomez et al., 2015). Analogously to MMC treatment (Murina et al., 2014), knockdown of FANCD2 impaired ultraviolet (UV)-light and HU-induced CtIP foci formation, together with its localization to laser-induced DNA breaks (Figure S4F–G). Knockdown of Polθ did not affect FANCD2 foci formation (Figure S4H) and depletion of CtIP by siRNA neither affected FANCD2 or Polθ foci formation, nor their localization to DNA breaks (Figure S4I–J). Taken together, these data establish a functional hierarchy among alt-EJ factors and suggest that FANCD2 functions upstream of Polθ and CtIP in the alt-EJ pathway by orchestrating their recruitment to sites of DSB repair.
To further dissect how FANCD2 mediates alt-EJ, we asked whether its monoubiquitination is necessary. To this goal, we first assessed the ability of wild-type or K561R-FANCD2 to complement the siFANCD2-mediated decrease in Polθ and CtIP foci and their localization to laser-induced DNA stripes. Wild-type FANCD2, but not K561R-FANCD2, complemented the cells (Figures 4H–I and S4K). In parallel, to evaluate whether FANCD2 monoubiquitination is necessary for alt-EJ activity, we assessed the ability of wild-type or K561R-FANCD2 to complement the siFANCD2-mediated decrease alt-EJ activity. Wild-type FANCD2, but not K561R-FANCD2, rescued the alt-EJ activity, suggesting that FANCD2-Ub is required for alternative end-joining (Figure 4J). Collectively, these data show that FANCD2 monoubiquitination promotes alt-EJ at damaged sites in a mechanism dependent on Polθ and CtIP recruitment.
Since a synthetic lethal relationship exists between BRCA1/2 and FANCD2 (Figure 2A) as well as between BRCA1/2 and Polθ (Ceccaldi et al., 2015a; Mateos-Gomez et al., 2015), we evaluated whether also CtIP knockdown affected BRCA1/2-deficient cells survival. Unlike FANCD2 depletion, CtIP depletion did not reduce BRCA2-deficient (VU 423) cell survival (Figure S5A). These data were recapitulated in isogenic pairs of BRCA1/2-proficient and deficient HeLa cells, subjected to siRNA-mediated FANCD2 or CtIP depletion and assessed for clonogenic survival (Figure S5B). All together, our data support a role for FANCD2 as an upstream alt-EJ regulatory factor required for both CtIP and Polθ recruitment to damage sites and for BRCA1/2-deficient cell survival.
FANCD2 overexpression confers resistance to PARP inhibitors through replication fork stabilization
In BRCA1/2-deficient tumors, reduction of genomic instability results in resistance to PARP inhibitors (PARPi) (Bouwman and Jonkers, 2014; Lord and Ashworth, 2013). Since FANCD2 enables BRCA1/2-deficient cell survival, we determined whether FANCD2 overexpression could confer resistance to PARPi. We assessed the clonogenic survival of BRCA1- and BRCA2-mutated cells (MDA-MB-436 and VU 423, respectively) overexpressing wild-type or K561R-FANCD2 cDNAs and grown in the presence of PARPi. Overexpression of wild-type, but not K561R-FANCD2, increased the PARPi resistance of BRCA1/2-mutated cells (Figures 5A and S5C). Restoration of HR functionality represents the most common acquired mechanism of PARPi resistance in BRCA1/2-deficient tumors (Edwards et al., 2008; Sakai et al., 2008). We thus assessed whether FANCD2 overexpression restored HR in BRCA1-mutated (MDA-MB-436) cells. Neither wild-type nor K561R-FANCD2 improved HR function as measured by ionizing radiation (IR)-induced RAD51 foci formation (Figure 5B), suggesting that FANCD2-mediated PARPi resistance occurs independently of HR restoration.
Figure 5. FANCD2 overexpression in BRCA1-mutated breast cell line confers resistance to PARP inhibitor.
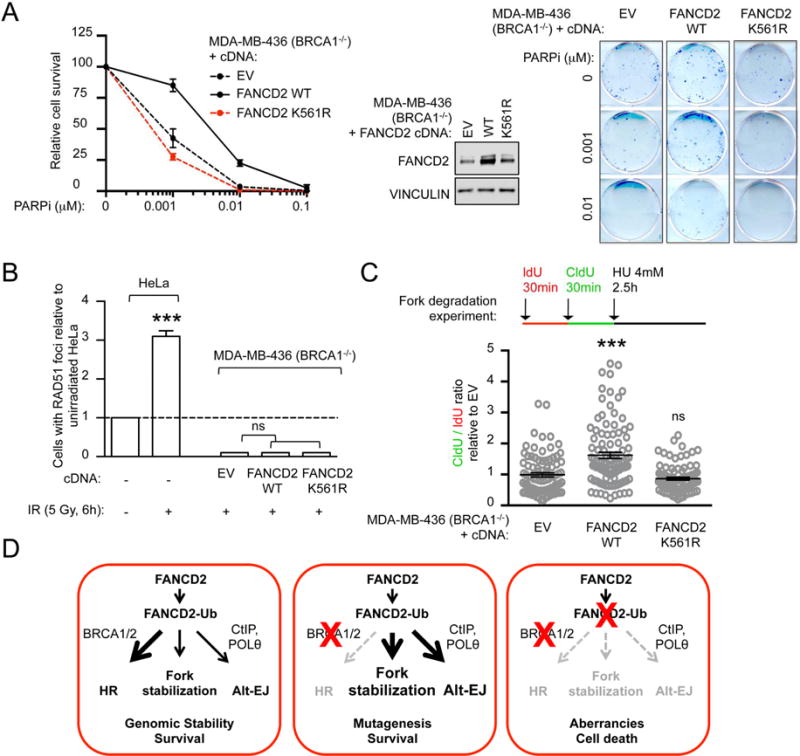
(A) Clonogenic formation assay of BRCA1-mutated (MDA-MB-436) breast cell line expressing EV or FANCD2 cDNA constructs treated with increasing concentration of PARPi. FANCD2 immunoblot showing cDNA constructs expression. Representative images for clonogenic formation assays are shown. Survival of each sample is expressed as percent relative to non-treated (PARPi: 0 μM). (B) Quantification of baseline and damage (IR)-induced RAD51 foci in HeLa and BRCA1-mutated (MDA-MB436) cells expressing indicated cDNA. Data are displayed as relative to unirradiated HeLa cells. (C) Schematic for the labeling of MDA-MB-436 cells with ldU and CIdU for fork degradation experiments. Scatter dot plot for CIdU to ldU ratio upon HU treatment for BRCA1-mutated (MDA-MB-436) breast cell line expressing indicated cDNA. Satistics were performed on n≥100 fibers per condition and expressed as relative to EV, that was conventionally set to 1. (D) Model for FANCD2 functions in BRCA1- and BRCA2-deficient tumors. FANCD2 functions in HR and alt-EJ repair pathways and also participates in genomic maintenance by protecting fork stability. Although all pathways function in cycling cells, HR is the predominant pathway under normal conditions and the alt-EJ pathway is though to play only a minor role (left panel). Inactivation of BRCA1 or BRCA2 has no consequence on cell survival but induces both the localization of FANCD2 to stalled forks and the hyperactivation of the alt-EJ pathway (middle panel). As a consequence, loss of both BRCA1/2 and FANCD2 pathways induces cell death, defining a synthetic lethal interaction between the BRCA and the FA pathways (right panel). Data in A–C represent mean ± s.e.m. over n=3 independent experiments and were analyzed using Student’s t test. Abbreviations: ns, non significant; EV, empty vector; WT, wild-type.
Given that FANCD2-Ub stabilizes replication forks in BRCA1/2-deficient cells (Figure 1E), we tested whether the FANCD2-mediated PARPi resistance correlated with increased fork protection. Of note, wild-type, but not K561R-FANCD2, improved fork stabilization in BRCA1-mutated (MDA-MB-436) cells, as measured by fork degradation after HU exposure (Figure 5C). Accordingly, in BRCA1/2-deficient cells, increased FANCD2-Ub represents a mechanism of PARPi resistance mediated by replication fork protection.
DISCUSSION
The ability to stabilize and restart stalled replication forks is essential to maintain genomic stability and cell survival. The importance of this survival mechanism is even more pronounced in cancer cells, characterized by increased replication stress levels (Bartkova et al., 2005; Gorgoulis et al., 2005). BRCA1/2 and FA proteins are known to cooperate in the repair of DNA lesions causing replication stalling such as ICLs, by promoting end resection and HR (Garcia-Higuera et al., 2001; Wang et al., 2004). In the context of replication stress, BRCA1/2 and FA proteins also cooperate in the protection of stalled replication forks by limiting the endonuclease-dependent degradation of nascent DNA strands (Schlacher et al., 2011; Schlacher et al., 2012). Besides cooperating with the BRCA1/2, FANCD2 also cooperates with BLM and FAN1 to promote fork recovery (Chaudhury et al., 2013; Chaudhury et al., 2014; Lachaud et al., 2016).
BRCA1/2 mutations, and other HR gene mutations, are found in a large proportion of ovarian and breast cancers (Cancer Genome Atlas, 2012; Cancer Genome Atlas Research, 2011). Importantly, these tumors are characterized by high levels of replication stress (Cancer Genome Atlas Research, 2011; Pathania et al., 2014; Schlacher et al., 2011; Zeman and Cimprich, 2014). Elevated replication stress renders these tumors hyperdependent on additional protective mechanisms that might reduce the stress thus promoting tumor cell survival.
In order to identify new survival mechanisms of BRCA1/2-deficient cancers, we analyzed gene expression profiles in subsets of tumors frequently associated with defects in HR. FANCD2 mRNA and protein were specifically overexpressed in BRCA1- and BRCA2-mutated ovarian and breast tumors, compared to HR-proficient cancers. While FANCD2 normally functions epistatically with BRCA1/2 to promote repair of ICL lesions (Figure 5D, left panel), these results reveal a nonepistatic function of FANCD2 and BRCA1/2 proteins in fork stabilization. Indeed, in the case of tumors defective in HR (BRCA1/2-mutated), FANCD2 localizes at sites of stalled replication and is monoubiquitinated (FANCD2-Ub) to a higher extent. Moreover, the upregulation of FANCD2-Ub at stalled forks prevents fork degradation and facilitates fork restart (Figure 5D, middle panel). These results are consistent with previous studies that show that FANCD2 has a role in fork stabilization (Lossaint et al., 2013; Schlacher et al., 2012), and fork restart by cooperating with the BLM helicase (Chaudhury et al., 2013; Lossaint et al., 2013). Concomitant with replication fork stabilization, FANCD2 mediates the survival of BRCA1/2-mutated tumors by counteracting the onset of genome instability events. These events arise from the collapse or the instability of stalled forks. Indeed, loss of FANCD2 in BRCA1/2-deficient cells hinders the stability of stalled replication forks and causes severe and disabling damage that compromises genomic integrity and cell survival. These findings reveal a mechanism of synthetic lethality between BRCA1/2 and FANCD2 (Figure 5D, right panel). Of note, the survival dependency of BRCA1/2-mutated cancers on FANCD2 is displayed in a large panel of breast and ovarian cancer cell lines and in in vivo models of breast tumor xenografts.
While our results reveal a requirement for FANCD2-mediated stabilization and restart of replication forks in BRCA1/2-deficient cells, the mechanism by which these cells repair stalled forks was also examined. FANCD2 was required for Polθ and CtIP recruitment to damaged sites in order to promote alternative end-joining (alt-EJ) (Figure 5D, middle panel). Alt-EJ is a highly error-prone DNA repair pathway, requiring CtIP-mediated end resection and PARP1/Polθ-mediated microhomology based repair (Kent et al., 2015; Lee-Theilen et al., 2011; Simsek and Jasin, 2010; Wang et al., 2006; Zhang and Jasin, 2011). While alt-EJ is a backup pathway for the repair of DSBs in cells deficient for classical non-homologous end-joining (C-NHEJ), it has recently emerged as an integral DSB repair pathway contributing to the survival of HR-deficient cancer cells (Aparicio et al., 2014; Bunting and Nussenzweig, 2013; Ceccaldi et al., 2015a; Deriano and Roth, 2013; Mateos-Gomez et al., 2015).
While usually relying on HR for error-free repair (Couch et al., 2013), stalled forks are also repaired through mutagenic pathways, such as C-NHEJ or alt-EJ itself in the absence of HR (Ceccaldi et al., 2015b). This hypothesis is supported by evidence for a direct role of PARP1 in fork stabilization and recovery after replicative stress (Bryant et al., 2009; Ying et al., 2015; Ying et al., 2012). In this study, we show that FANCD2 also acts as an alt-EJ factor. Indeed, FANCD2 colocalizes with CtIP and Polθ at bona fide DNA damage sites and promotes microhomology-mediated DNA repair in a GFP-based cell reporter system for alt-EJ, similar to PARP1 and Polθ. We next investigated the role of FANCD2 in alt-EJ by establishing a functional hierarchy among alt-EJ proteins. FANCD2 precedes and mediates the binding of CtIP and Polθ to DNA damages sites. Interestingly, siRNA-mediated CtIP depletion does not limit the number of FANCD2 and Polθ foci after damage, nor does it cause synthetic lethality in BRCA1/2-deficient cells. These data are in accordance with previous studies on the mutual contributions of CtIP and FANCD2 in the control of fork recovery (Yeo et al., 2014). They also support the hypothesis of a specific role for FANCD2 in fork protection and ultimately survival of HR-deficient cells.
While BRCA1 and BRCA2 proteins harbor distinct functions in HR-mediated repair (Prakash et al., 2015), their deficiency similarly leads to fork instability and high replicative stress (Pathania et al., 2014; Schlacher et al., 2011). Moreover, FANCD2 is analogously upregulated in BRCA1- and BRCA2-deficient tumors. For these reasons, we examined the role of FANCD2 upon loss of BRCA1 or BRCA2 as a whole. Previous data showed that BRCA1 was required for FANCD2 activation. For instance, loss of BRCA1 leads to a decrease in ionizing radiation (IR)-dependent induction of FANCD2 foci (Bunting et al., 2012; Duquette et al., 2012; Garcia-Higuera et al., 2001), and in Xenopus egg extracts, BRCA1 is required for FANCD2 recruitment to a site-specific ICL (Long et al., 2014). Nevertheless, BRCA1-deficient cells retain the ability to form distinct FANCD2 foci (Garcia-Higuera et al., 2001), and loss of BRCA1 increases the level of FANCD2-Ub. Importantly, these data suggest that even in the absence of functional BRCA1, FANCD2-Ub is still loaded onto chromatin. While FANCD2 is required for the survival of BRCA1-deficient cells independently of the established BRCA1 role in activating FANCD2, further studies should address whether BRCA1 is upstream or downstream of FANCD2. A possibility could be that FANCD2 and BRCA1 present different functional interendependencies based on the genome stability pathway taken into consideration. For instance, while BRCA1 seems to function upstream of FANCD2 in ICL repair (Long et al., 2014), our data suggest that, in fork protection, FANCD2 functions upstream of BRCA1.
Overall, our study demonstrates a dependency of HR-deficient tumors on FANCD2 for cellular survival. In accordance with previous studies (Ceccaldi et al., 2015a; Mateos-Gomez et al., 2015), survival of HR-deficient tumors is promoted by mutagenic alt-EJ DNA repair pathways, of which FANCD2 is identified as an upstream component. Moreover, we propose that the dependency on FANCD2 of these tumors is mediated by its fork stability and recovery functions. In contrast with the common view where BRCA1/2 and FA proteins cooperate in the repair of DNA lesions by HR, we here present evidence for a compensatory fork stabilization activity of FANCD2 in the absence of BRCA1/2 proteins. Aware of the limitations of the genetic, repair deficient context that we have investigated, we believe that characterizing the role of FANCD2 and its partners in fork protection might shed light on the unexplored mechanisms of fork stabilization as a widespread mechanism of tumor cell survival. More broadly, it will be of interest to investigate whether FANCD2 represents a fork stabilization and/or a survival factor only in BRCA1/2-mutated cancers or also in other cancers that are characterized by high replication stress levels, for example as a consequence of oncogene activation.
Other questions remain to be answered; for example whether an inverse compensatory mechanism exists between the BRCA1/2 and FA proteins where BRCA1/2 proteins could compensate for FA pathway deficiency, as in the case of FA patients. Further studies should also aim to understand in greater depth what aspect of FANCD2 (fork stabilization or alt-EJ repair) is responsible for the survival of BRCA1/2-deficient tumors. In this regard, our data indicate that FANCI depletion induces synthetic lethality in BRCA1/2-deficient cells, similarly to FANCD2 depletion. Since FANCI was recently shown to promote fork restart upon replication stress (Chen et al., 2015), our results enforce the hypothesis that the fork protection function of FA pathway members is a major regulator of BRCA1/2-deficient cells survival. In addition, while previous studies on Polθ suggest that the alt-EJ pathway is required for BRCA1/2 survival (Ceccaldi et al., 2015a; Mateos-Gomez et al., 2015), this is not the case for other alt-EJ factors such as CtIP, whose depletion, while decreasing alt-EJ efficiency, does not confer synthetic lethality with BRCA1/2 deficiency. Clinical applications relying on synthetic lethality relationships are just beginning to emerge and understanding these relationships can shed light on unexpected connections between biological pathways. Therefore, it is of critical importance to dissect their molecular basis from both a basic and a translational perspective.
EXPERIMENTAL PROCEDURES
Bioinformatic analysis
TCGA data sets were accessed through the public TCGA data portal (https://tcga-data.nci.nih.gov/tcga/). For mutation count, we accessed data from tumors included in the TCGA data sets for which gene expression and whole-exome DNA sequencing was available. Data were accessed through the public TCGA data portal and the cBioPortal for Cancer Genomics (http://www.cbioportal.org). For each TCGA data set, non-synonymous mutation count was assessed in tumors with the high FANCD2 expression (top 50%) and compared to tumors with low FANCD2 expression (the remaining 50%). In the uterine TCGA, we curated all tumors except the ultra and hyper-mutated group (that is, POLE and MSI tumors). In the ovarian and breast TCGA datasets, all tumors were analysed. FANCD2 expression analysis shown in Figure S1C was extracted from the ovarian serous carcinoma datasets (GSE14001, GSE14007, GSE18520, GSE16708, GSE10971) and compared to the ovarian clear cell carcinoma dataset (GSE29450).
Immunoblot analysis, cellular fractionation and immunofluorescence
Cells were lysed with 1% NP40 lysis buffer (1% NP40, 300 mM NaCl, 0.1 mM EDTA, 50 mM Tris (pH 7.5)) supplemented with protease inhibitor cocktail (Roche), resolved by NuPAGE (Invitrogen) gels and transferred onto nitrocellulose membrane, followed by detection using the LAS-4000 Imaging system (GE Healthcare Life Sciences). For cellular fractionation, cells were incubated with low-salt permeabilization buffer (10 mM Tris (pH 7.3), 10 mM KCl, 1.5 mM MgCl2) with protease inhibitor on ice for 20 min. Following centrifugation, nuclei were resuspended in 0.2 M HCl and the soluble fraction was neutralized with 1 M Tris-HCl (pH 8.0). Nuclei were lysed in 150 mM NaCl and, following centrifugation, the chromatin pellet was digested by micrococcal nuclease (Roche) for 5 min at room temperature. See the Supplemental Information for details on immunofluorescence procedures.
Antibodies, chemicals, plasmids and transfection
Antibodies used in this study included: anti-FANCD2 (FI-17, for immunoblotting), anti-histone H3 (FL-136), anti-PCNA (PC-10), anti-TopBP1 (B-7), anti-vinculin (H-10), anti-RAD51 (H-92) (all from Santa Cruz); anti-BRCA1 (Ab-1, Calbiochem); anti-BRCA2 (Ab-1, Calbiochem), anti-FANCD2 (NB100-182, for immunofluorescence) (Novus), anti-phospho-Histone-H2AX (p-Ser139) (05-636, EMD Millipore and 2572, Cell Signaling), anti-CtIP (14-1, Active Motif). Mitomycin C (MMC), and Hydroxyurea (HU) were purchased from Sigma. See the Supplemental Information for details on siRNAs, shRNAs and plasmids.
Cell Culture
HeLa, U2OS, VU 423 (BRCA2−/−), PD20 (FANCD2−/−), and PD20-corrected cells were maintained in Dulbecco’s modified Eagle’s medium plus 10% fetal calf serum (FCS), L-glutamine (2 mM), and penicillin-streptomycin (1%). MDA-MB-436 (BRCA1-mutated) cells were maintained in RPMI medium plus 10% FCS, L-glutamine (2 mM), and penicillin-streptomycin (1%). Breast and ovarian cell lines used in the Figures 3A and S3A were culture according to recommendations present in the literature (Table S1). HR, alt-EJ and SSA efficiency was measured using U2OS cell line expressing the DR-GFP (HR efficiency), EJ2-GFP (alt-EJ efficiency) and SA-GFP (SSA efficiency) reporter assay and, performed as previously described (Bennardo et al., 2008; Nakanishi et al., 2005).
Cell survival assays
In order to assess clonogenic survival, cells were transfected twice with the indicated siRNAs for 48 h, then harvested and seeded at low density into 6-well plates in triplicates. For clonogenic survival under MMC or PARPi (rucaparib), cells were treated continuously with the indicated drug concentrations. Survival at each drug concentration was plotted as a percentage relative to the survival in drug-free media. Colony formation was scored 14 days after drug treatment or siRNA transfection using 0.5% (w/v) crystal violet in methanol. Survival curves were expressed as a percentage ± s.e.m. over three independent experiments and relative to control cells (siScr). Mitomycin C (MMC) was purchased from Sigma while the PARPi rucaparib (AG-014699) was purchased from Selleckchem.
Chromosomal breakage analysis
A2780 expressing the indicated shRNAs and VU 423 cells twice-transfected with the indicated siRNAs were incubated for 48 h with or without the indicated concentrations of MMC. Cells were exposed for 2 h to 100 ng/ml of colcemid and treated with a hypotonic solution (0.075 M KCl) for 20 min and fixed with 3:1 methanol/acetic acid. Slides were stained with Wright’s stain and 50 metaphase spreads were scored for aberrations. The relative number of chromosomal breaks and radials was calculated relative to control cells (si Scr) or shBRCA2 as indicated in the figure legends.
FANCD2 gene expression
RNA samples extracted using the TRIzol Reagent (Invitrogen) were reverse transcribed using the Transcriptor Reverse Transcriptaze kit (Roche) and oligo dT primers. The resulting cDNA was used to analyze FANCD2 expression by RT-qPCR using the QuantiTect SYBRGreen kit (Qiagen) in an iCycler machine (Bio-Rad). FANCD2 gene expression values were normalized to expression of the housekeeping gene GAPDH, using the ΔCt method and are shown on a log2 scale. See the Supplemental Information for details on primers and for the FANCD2 expression data shown in Figure 3A.
DNA fiber analysis
For the fork degradation analysis, PD20 cells expressing empty vector (EV), wild-type (WT) FANCD2, or K561R-FANCD2 were incubated with 250 μM iododeoxyuridine (IdU) (Sigma, I7125) for 30 min followed by 25 μM chlorodeoxyuridine (CldU) (Sigma, C6891) for 30 min. Cells were then incubated with 4 mM HU for 2.5 h. For the fork restart analysis, cells were incubated with 250 μM ldU for 30 min. Cells were then treated with 2 mM hydroxyurea (HU) for 1 h and incubated in 25 μM CldU for 30 min after washout of the drug. Spreading of DNA fibers on glass slides was done as previously reported (Jackson and Pombo, 1998). Glass slides were then washed in distilled water and in 2.5 M HCl for 120 min followed by three washes in PBS. The slides were incubated for 1 h in blocking buffer (PBS with 1% BSA) and then for 1 h at 37°C in rat anti-BrdU antibody (Abcam, ab6326) and mouse anti-BrdU antibody (BD Biosciences, 347580) diluted in blocking buffer. After washes in PBS, the slides were incubated for 1 h at 37°C in goat anti-rat Alexa 488 antibody (Life Tech nologies, A-11006) and chicken anti-mouse Alexa 594 (Life Technologies, A-21201) diluted in blocking buffer. The slides were then washed with PBS and then mounted with DAPI. At least 100 fibers were counted per condition. Pictures were taken with an Olympus confocal microscope and fiber length was measured by ImageJ software.
Studies of xenograft-bearing CrTac: NCr-Foxn1nu mice
In vivo studies were performed as previously described (Ceccaldi et al., 2015a), under approval of The Animal Resource Facility at The Dana-Farber Cancer Institute. See the Supplemental Information for more details.
Statistical analysis
Data are represented as mean ± s.e.m. over n=3 independent experiments. Unless otherwise stated, significance was calculated using the unpaired Student’s t-test or the Chi-squared test for trend in proportions. Analyses were performed by the use of GraphPad Prism 6 software (GraphPad Software). A P value less that 0.05 was considered as statistically significant (asterisks indicate ns= not significant; *P<0.05; **P<0.01; ***P<0.001). All the in vivo experiments were run with at least 10 tumors from 6 mice for each condition.
Supplementary Material
HIGHLIGHTS.
Loss of FANCD2 and BRCA1/2 is synthetic lethal
FANCD2 maintains fork stability in BRCA1/2-deficient cells
FANCD2 promotes alternative end-joining (alt-EJ)
FANCD2 overexpression confers resistance to PARP inhibitors
Acknowledgments
We would like to thank R. Drapkin, A. de Fazio, D. Chowdhury, P.A. Konstantinopoulos, K. Strickland and T. Taniguchi for providing ovarian cell lines, L. Moreau for chromosomal breakage analysis and K.W. O’Connor, K.W. Mouw, P. Sarangi for critical reading of the manuscript, S.V. Cardenas and K.W. O’Connor for early contributions to the project, J.B. Lazaro and C. Clairemont for assistance on DNA fiber analysis. We thank A. Constantinou and G. Lossaint for providing the dox-inducible pTripZ shFANCD2 construct; M. Cohn for providing the Poz-N-FANCD2 construct. Z.K. is a recipient of the Ovarian Cancer Research Fellowship (OCRF), B.R. is a recipient of the Italian Association for Cancer Research (AIRC) Fellowships for Abroad. This research was supported by a Claudia Adams Barr Program award (to R.C.) and a Stand Up To Cancer – Ovarian Cancer Research Fund-Ovarian Cancer National Alliance-National Ovarian Cancer Coalition Dream Team Translational Research Grant (Grant Number: SU2C-AACR-DT16-15). Stand Up To Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C. This work was also supported by grants from the U.S. National Institutes of Health (R01DK43889, R37HL052725), the Breast Cancer Research Foundation, and the Fanconi Anemia Research Fund (to A.D.D.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Z.K., B.R., A.D.D. and R.C. designed the study, performed experiments, and wrote the manuscript. A.H. performed chromosomal breakage analysis. Z.K., C.O. and D.K. performed mice work. A.D.D. and R.C. supervised the study. All authors approved the final version of the manuscript.
References
- Alabert C, Bukowski-Wills JC, Lee SB, Kustatscher G, Nakamura K, de Lima Alves F, Menard P, Mejlvang J, Rappsilber J, Groth A. Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nature cell biology. 2014;16:281–293. doi: 10.1038/ncb2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio T, Baer R, Gautier J. DNA double-strand break repair pathway choice and cancer. DNA repair. 2014;19:169–175. doi: 10.1016/j.dnarep.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audebert M, Salles B, Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. The Journal of biological chemistry. 2004;279:55117–55126. doi: 10.1074/jbc.M404524200. [DOI] [PubMed] [Google Scholar]
- Badie S, Carlos AR, Folio C, Okamoto K, Bouwman P, Jonkers J, Tarsounas M. BRCA1 and CtIP promote alternative non-homologous end-joining at uncapped telomeres. The EMBO journal. 2015;34:828. doi: 10.15252/embj.201570610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS genetics. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman P, Jonkers J. Molecular pathways: how can BRCA-mutated tumors become resistant to PARP inhibitors? Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:540–547. doi: 10.1158/1078-0432.CCR-13-0225. [DOI] [PubMed] [Google Scholar]
- Branzei D, Foiani M. Interplay of replication checkpoints and repair proteins at stalled replication forks. DNA repair. 2007;6:994–1003. doi: 10.1016/j.dnarep.2007.02.018. [DOI] [PubMed] [Google Scholar]
- Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nature reviews. Molecular cell biology. 2010;11:208–219. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- Bryant HE, Petermann E, Schultz N, Jemth AS, Loseva O, Issaeva N, Johansson F, Fernandez S, McGlynn P, Helleday T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. The EMBO journal. 2009;28:2601–2615. doi: 10.1038/emboj.2009.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Kozak ML, Kim JM, Wong N, Lopez-Contreras AJ, Ludwig T, Baer R, Faryabi RB, Malhowski A, et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Molecular cell. 2012;46:125–135. doi: 10.1016/j.molcel.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Nussenzweig A. End-joining, translocations and cancer. Nature reviews. Cancer. 2013;13:443–454. doi: 10.1038/nrc3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI, O’Connor KW, Konstantinopoulos PA, Elledge SJ, Boulton SJ, et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature. 2015a;518:258–262. doi: 10.1038/nature14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Rondinelli B, D’Andrea AD. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2015b doi: 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nature cell biology. 2009;11:753–760. doi: 10.1038/ncb1882. [DOI] [PubMed] [Google Scholar]
- Chaudhury I, Sareen A, Raghunandan M, Sobeck A. FANCD2 regulates BLM complex functions independently of FANCI to promote replication fork recovery. Nucleic acids research. 2013;41:6444–6459. doi: 10.1093/nar/gkt348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury I, Stroik DR, Sobeck A. FANCD2-controlled chromatin access of the Fanconi-associated nuclease FAN1 is crucial for the recovery of stalled replication forks. Molecular and cellular biology. 2014;34:3939–3954. doi: 10.1128/MCB.00457-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Jones MJ, Yin Y, Crist SB, Colnaghi L, Sims RJ, 3rd, Rothenberg E, Jallepalli PV, Huang TT. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Molecular cell. 2015;58:323–338. doi: 10.1016/j.molcel.2015.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA, et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes & development. 2013;27:1610–1623. doi: 10.1101/gad.214080.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. The importance of repairing stalled replication forks. Nature. 2000;404:37–41. doi: 10.1038/35003501. [DOI] [PubMed] [Google Scholar]
- Deriano L, Roth DB. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annual review of genetics. 2013;47:433–455. doi: 10.1146/annurev-genet-110711-155540. [DOI] [PubMed] [Google Scholar]
- Duquette ML, Zhu Q, Taylor ER, Tsay AJ, Shi LZ, Berns MW, McGowan CH. CtIP is required to initiate replication-dependent interstrand crosslink repair. PLoS genetics. 2012;8:e1003050. doi: 10.1371/journal.pgen.1003050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–1115. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Molecular cell. 2013;49:872–883. doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D’Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Molecular cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- Gari K, Decaillet C, Stasiak AZ, Stasiak A, Constantinou A. The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Molecular cell. 2008;29:141–148. doi: 10.1016/j.molcel.2007.11.032. [DOI] [PubMed] [Google Scholar]
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr, Kastrinakis NG, Levy B, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS genetics. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard SM, Yanez DA, Stark JM. DNA damage response factors from diverse pathways, including DNA crosslink repair, mediate alternative end joining. PLoS genetics. 2015;11:e1004943. doi: 10.1371/journal.pgen.1004943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett NG, Harney JA, Rego MA, Kolling FWt, Glover TW. Functional interaction between the Fanconi Anemia D2 protein and proliferating cell nuclear antigen (PCNA) via a conserved putative PCNA interaction motif. The Journal of biological chemistry. 2009;284:28935–28942. doi: 10.1074/jbc.M109.016352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett NG, Taniguchi T, Durkin SG, D’Andrea AD, Glover TW. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Human molecular genetics. 2005;14:693–701. doi: 10.1093/hmg/ddi065. [DOI] [PubMed] [Google Scholar]
- Jackson DA, Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. The Journal of cell biology. 1998;140:1285–1295. doi: 10.1083/jcb.140.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent T, Chandramouly G, McDevitt SM, Ozdemir AY, Pomerantz RT. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase theta. Nature structural & molecular biology. 2015;22:230–237. doi: 10.1038/nsmb.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, D’Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes & development. 2012;26:1393–1408. doi: 10.1101/gad.195248.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer discovery. 2015;5:1137–1154. doi: 10.1158/2159-8290.CD-15-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachaud C, Moreno A, Marchesi F, Toth R, Blow JJ, Rouse J. Ubiquitinated Fancd2 recruits Fan1 to stalled replication forks to prevent genome instability. Science. 2016 doi: 10.1126/science.aad5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Theilen M, Matthews AJ, Kelly D, Zheng S, Chaudhuri J. CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nature structural & molecular biology. 2011;18:75–79. doi: 10.1038/nsmb.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomonosov M, Anand S, Sangrithi M, Davies R, Venkitaraman AR. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes & development. 2003;17:3017–3022. doi: 10.1101/gad.279003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long DT, Joukov V, Budzowska M, Walter JC. BRCA1 promotes unloading of the CMG helicase from a stalled DNA replication fork. Molecular cell. 2014;56:174–185. doi: 10.1016/j.molcel.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nature medicine. 2013;19:1381–1388. doi: 10.1038/nm.3369. [DOI] [PubMed] [Google Scholar]
- Lossaint G, Larroque M, Ribeyre C, Bec N, Larroque C, Decaillet C, Gari K, Constantinou A. FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Molecular cell. 2013;51:678–690. doi: 10.1016/j.molcel.2013.07.023. [DOI] [PubMed] [Google Scholar]
- Mateos-Gomez PA, Gong F, Nair N, Miller KM, Lazzerini-Denchi E, Sfeir A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature. 2015;518:254–257. doi: 10.1038/nature14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel B, Grompone G, Flores MJ, Bidnenko V. Multiple pathways process stalled replication forks. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:12783–12788. doi: 10.1073/pnas.0401586101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Molecular cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Molecular cell. 2001;7:263–272. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- Murina O, von Aesch C, Karakus U, Ferretti LP, Bolck HA, Hanggi K, Sartori AA. FANCD2 and CtIP cooperate to repair DNA interstrand crosslinks. Cell reports. 2014;7:1030–1038. doi: 10.1016/j.celrep.2014.03.069. [DOI] [PubMed] [Google Scholar]
- Naim V, Rosselli F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nature cell biology. 2009;11:761–768. doi: 10.1038/ncb1883. [DOI] [PubMed] [Google Scholar]
- Nakanishi K, Yang YG, Pierce AJ, Taniguchi T, Digweed M, D’Andrea AD, Wang ZQ, Jasin M. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:1110–1115. doi: 10.1073/pnas.0407796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability–an evolving hallmark of cancer. Nature reviews. Molecular cell biology. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- Newman JA, Cooper CD, Aitkenhead H, Gileadi O. Structure of the Helicase Domain of DNA Polymerase Theta Reveals a Possible Role in the Microhomology-Mediated End-Joining Pathway. Structure. 2015;23:2319–2330. doi: 10.1016/j.str.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TV, Riou L, Aoufouchi S, Rosselli F. Fanca deficiency reduces A/T transitions in somatic hypermutation and alters class switch recombination junctions in mouse B cells. The Journal of experimental medicine. 2014;211:1011–1018. doi: 10.1084/jem.20131637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D’Andrea AD, Bernards R. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Molecular cell. 2005;17:331–339. doi: 10.1016/j.molcel.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Pathania S, Bade S, Le Guillou M, Burke K, Reed R, Bowman-Colin C, Su Y, Ting DT, Polyak K, Richardson AL, et al. BRCA1 haploinsufficiency for replication stress suppression in primary cells. Nature communications. 2014;5:5496. doi: 10.1038/ncomms6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash R, Zhang Y, Feng W, Jasin M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harbor perspectives in biology. 2015;7:a016600. doi: 10.1101/cshperspect.a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–1120. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer cell. 2012;22:106–116. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab RA, Nieminuszczy J, Shah F, Langton J, Lopez Martinez D, Liang CC, Cohn MA, Gibbons RJ, Deans AJ, Niedzwiedz W. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Molecular cell. 2015;60:351–361. doi: 10.1016/j.molcel.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek D, Jasin M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nature structural & molecular biology. 2010;17:410–416. doi: 10.1038/nsmb.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, McDonald WH, Dungrawala H, Badu-Nkansah A, Kavanaugh GM, Chen Y, Tabb DL, Cortez D. Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. The Journal of biological chemistry. 2013;288:31458–31467. doi: 10.1074/jbc.M113.511337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D’Andrea AD, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unno J, Itaya A, Taoka M, Sato K, Tomida J, Sakai W, Sugasawa K, Ishiai M, Ikura T, Isobe T, et al. FANCD2 binds CtIP and regulates DNA-end resection during DNA interstrand crosslink repair. Cell reports. 2014;7:1039–1047. doi: 10.1016/j.celrep.2014.04.005. [DOI] [PubMed] [Google Scholar]
- Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic acids research. 2006;34:6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Andreassen PR, D’Andrea AD. Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Molecular and cellular biology. 2004;24:5850–5862. doi: 10.1128/MCB.24.13.5850-5862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis NA, Chandramouly G, Huang B, Kwok A, Follonier C, Deng C, Scully R. BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature. 2014;510:556–559. doi: 10.1038/nature13295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S, Michael WM. TopBP1 and DNA polymerase-alpha directly recruit the 9-1-1 complex to stalled DNA replication forks. The Journal of cell biology. 2009;184:793–804. doi: 10.1083/jcb.200810185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo JE, Lee EH, Hendrickson EA, Sobeck A. CtIP mediates replication fork recovery in a FANCD2-regulated manner. Human molecular genetics. 2014;23:3695–3705. doi: 10.1093/hmg/ddu078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Chen Z, Medhurst AL, Neal JA, Bao Z, Mortusewicz O, McGouran J, Song X, Shen H, Hamdy FC, et al. DNA-PKcs and PARP1 bind to unresected stalled DNA replication forks where they recruit XRCC1 to mediate repair. Cancer research. 2015 doi: 10.1158/0008-5472.CAN-15-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Hamdy FC, Helleday T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer research. 2012;72:2814–2821. doi: 10.1158/0008-5472.CAN-11-3417. [DOI] [PubMed] [Google Scholar]
- Yousefzadeh MJ, Wood RD. DNA polymerase POLQ and cellular defense against DNA damage. DNA repair. 2013;12:1–9. doi: 10.1016/j.dnarep.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nature cell biology. 2014;16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Jasin M. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nature structural & molecular biology. 2011;18:80–84. doi: 10.1038/nsmb.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.