

Abstract
Uterine carcinosarcoma is a clinically aggressive malignancy composed of a mix of carcinomatous and sarcomatous elements. We performed targeted next‐generation sequencing of 27 uterine cancer and sarcoma genes together with immunohistochemical analyses of selected proteins in 30 uterine carcinosarcomas. This included 13 cases in which the distinct carcinoma and sarcoma components were sequenced separately and 10 cases where the metastatic tumours were analysed in addition to the primary tumours. We identified non‐synonymous somatic mutations in 90% of the cases, with 27 of 30 cases (90%) harbouring TP53 alterations. The PI3K pathway was the most commonly mutated signalling pathway with mutations identified in PIK3CA, PTEN, PIK3R1, and/or PIK3R2 in two‐thirds of the cases. Mutations in FBXW7, PPP2R1A, ARID1A and KRAS were demonstrated in a minority of cases. In cases where the carcinomatous and sarcomatous components were separately analysed, most of the mutations identified were present in both components, indicating a common origin for the two components. Furthermore, the same TP53 alterations and/or PI3K pathway mutations seen in the primary tumours were also identified in the metastatic sites. Overall, carcinosarcomas exhibited heterogeneous molecular features that resemble the heterogeneity seen in endometrial carcinomas, with some showing endometrioid carcinoma‐like and others showing serous carcinoma‐like mutation profiles. While patients with serous‐like tumours presented more frequently with advanced‐stage disease compared to patients with endometrioid‐like tumours, there was no statistical difference in outcome between the two groups. Our results provide insights into the oncogenesis of uterine carcinosarcoma and identify targetable mutations that represent early oncogenic events. The findings of the different molecular types of uterine carcinosarcoma that parallel the different molecular types in endometrial carcinoma may have future treatment implications with targeted therapies.
Keywords: uterine carcinosarcoma, molecular profiles, sequencing, mutations, TP53, PI3K pathway, PIK3CA, PTEN
Introduction
Uterine carcinosarcoma 1 is a malignant mixed epithelial–mesenchymal tumour that accounts for about 3–4% of all uterine malignancies 2, 3. Although uncommon, this tumour follows an aggressive clinical course and accounts for 16% of deaths caused by uterine malignancy 4, 5. Patients with International Federation of Gynecology and Obstetrics (FIGO) stage 1–2 disease have a 5‐year disease‐specific survival of 59%, while those with stage 3 and 4 disease have a 5‐year disease‐specific survival of 22% and 9%, respectively 4. Histologically, uterine carcinosarcoma is composed of carcinoma and sarcoma elements that impart a biphasic appearance. Both the carcinoma and sarcoma components are typically high grade. The carcinoma component may resemble known histologic subtypes of endometrial cancer such as endometrioid or serous carcinoma in some cases 6, while the sarcoma component can display homologous or heterologous differentiation 7.
Although initially considered as a subtype of uterine sarcomas 8, it is now thought that most uterine carcinosarcomas represent sarcomatous transformation/transdifferentiation of endometrial carcinoma (metaplastic carcinoma) rather than a collision between two biologically distinct tumour types 9. The clinical behaviour of carcinosarcomas supports the carcinoma origin hypothesis 10, as carcinosarcomas like most carcinomas preferentially follow lymphatic dissemination 4, 11. A number of prior studies compared patterns of X‐chromosome inactivation, microsatellite instability (MSI), types of TP53 and KRAS mutations and loss of heterozygosity (LOH) status between the carcinoma and sarcoma elements and demonstrated shared molecular features in the majority of cases 12, 13, 14, 15. However, these results also suggest a bi‐clonal nature (collision tumour) in a minority (10–20%) of uterine carcinosarcomas 13, 14.
Our group has previously performed mutational analysis on a series of uterine carcinosarcomas using in a nine gene panel, though we did not analyse the carcinoma and sarcoma elements separately 16. While prior studies have shown evidence for the monoclonal nature in most uterine carcinosarcomas, there has not been a comprehensive molecular study of the carcinoma and sarcoma components of carcinosarcomas. In this study, we employed next‐generation targeted gene sequencing to decipher and compare the mutational profiles between the different components of uterine carcinosarcomas, as well as between primary and metastatic tumours.
Methods
Study samples
This study examined tissues from 30 uterine carcinosarcomas with available formalin‐fixed paraffin‐embedded (FFPE) tumour and normal tissue, fresh frozen tumour (FFT) samples obtained from resection specimens, as well as matched normal tissue (buffy coat). The tissue samples were obtained from the Vancouver General Hospital pathology archives (FFPE tumour and normal tissue) and the BC Cancer Agency OvCaRe Tumour Biobank (FFT tissue and buffy coat). This study has received institutional research board approval. All patients were approached for written informed consent, before undergoing surgery, to donate tissue surplus to diagnostic requirements plus a blood sample. The histology slides from the 30 hysterectomy specimens were reviewed by the study pathologists (LH and CHL). FFPE block(s) that contained the areas of interest were identified for DNA extraction.
DNA extractions
For FFPE tissue blocks, the tumour containing areas in the primary and metastatic tumours were marked and cored (3–4 cores at 0.6 mm diameter). DNA was extracted from the tissue cores using the Qiagen FFPE DNA kit (Valencia, CA, USA) as per manufacturer's protocol. In 14 of the 30 cases, the carcinoma and sarcoma elements formed spatially distinct areas in the primary tumour, such that DNA from the separate components was extracted for comparison. The carcinoma and the sarcoma elements in the remaining 16 cases were closely admixed in the primary tumour and the extracted DNA represented a mixture of the two components. In 10 cases, DNA from the metastatic tumour was also extracted.
For FFT samples, all tumours were verified by frozen section to ensure adequate viability and cellularity of tumour tissue. The FFT tissue was then cryosectioned for DNA extraction using the Gentra Puregene kit (Qiagen) (Valencia, CA, USA) as per manufacturer's protocol. Germline DNA was extracted from buffy coat. In the cases where matched buffy coat was not available, DNA was extracted from normal FFPE tissue. All DNA was quantified using the Qubit fluorometer using Molecular Probes broad range Qubit quantification kit (Life Technologies) (Carlsbad, CA, USA).
Discovery targeted sequencing and analysis
An Illumina custom Truseq amplicon panel (version 1) (Illumina, San Diego, CA, USA) was designed using the Illumina Design studio software to amplify 175bp libraries. This included 1519 amplicons in 27 genes that were previously found to be recurrently mutated in endometrial cancer and/or uterine sarcoma 16, 17, 18, 19, 20, 21: ABCC9, AKT1, AKT2, AKT3, ARHGAP35 (GRLF1), CCND1, CHD4, CTCF, CSMD3, EP300, FGFR2, KRAS, MAP3K4, MED12, ARID1A, CTNNB1, PTEN, PIK3CA, PIK3R1, PIK3R2, POLE, PPP2R1A, FBXW7, SPOP, TP53, TSPYL2, ZFHX3. The Illumina TruSeq custom library preparation was utilized using 250 ng of DNA for FFPE DNA. The Illumina protocol was followed for library preparation (includes sample PCR and barcoding); however, the protocol was modified for pooling and normalization. Before library pooling, libraries were quantified using the Qubit fluorometer, then pooled at equal concentrations. Each library pool was then quantified for amplifiable products using the KAPA Illumina SYBR qPCR quantification kit using the ABI7900 fast real‐time instrument. Each pool was run on the Illumina MiSeq using 300 cycle version 2 kits, and all bam and VCF files were generated using Illumina MiSeq reporter. Analysis was performed using the VCF files generated by the somatic variant caller 3.2.3.0, then filtered based on reads passing filter, non‐synonymous, somatic mutations with >5% variant allele frequency. All potential mutations were then manually interrogated using the Integrated Genome Viewer (IGV).
Mutation validations
All non‐synonymous somatic mutations (except for CSMD3) underwent secondary validation using either Fluidigm 48X48 Access Arrays (Fluidigm, Markhma, ON, Canada), then barcoded and sequenced on a MiSeq, or by Sanger sequencing. In brief, primer sets were designed using Primer 3 to amplify the specific mutations, tagged with CS1 (5′‐ACACTGACGACATGGTTCTACA‐3′) and CS2 (5′‐TACGGTAGCAGAGACTTGGTCT‐3′) sequencing tags, and synthesized by IDT Technologies (Coralville, IA, USA). PCR products (150–200bp) were generated using the Fluidigm 48X48 Access Arrays, as per manufacturer's protocol, with input of 100 ng for FFPE derived DNA, and 50 ng for high‐quality DNA from buffy coat or frozen tumour DNA. DNA barcodes (10bp) with Illumina cluster‐generating adapters were added to the amplified libraries as previously described 22, then purified using Agencourt AMpure XP beads (Beckman Coulter, Mississauga, ON, Canada), quantified and pooled as described by our methods for the custom discovery targeted panel sequencing. In total, 96 libraries were sequenced using a MiSeq 300 cycle V2 kit on the Illumina MiSeq for ultra‐deep validations. Analysis was performed as described for discovery targeted panel sequencing. Sanger sequencing was utilized if the amplicon of interest failed MiSeq validation sequencing, and was performed as previously described 23; however, CS1 and CS2 primers were used as a universal sequencing primers on the ABI 3130xl Genetic Analyzer (Applied Biosystems). In the case of TP53, the Truseq panel sequencing did not cover the entire region of exons 4 and 5. Therefore, Sanger sequencing of these exons was performed when the immunohistochemistry and sequencing results did not concur. These included re‐sequencing for cases 1, 13, 16, 17, 18, 21 and 29.
Immunohistochemistry and analysis
All immunohistochemistry was performed on tissue microarrays and/or whole sections using the Ventana Discovery XT and the Ventana Benchmark XT automated systems (Ventana Medical Systems, Tucson, AZ, USA). Immunohistochemical stains for mismatch repair (MMR) proteins (MLH1, PMS2, MSH2, MSH6) were performed as previously described 24. Primary incubations were performed as follows: p53 (Dako, mouse monoclonal antibody, clone DO‐7) at 1:400 dilution, 32 minutes, 37 °C, with heat induced antigen retrieval; β‐catenin (Cell Marque, mouse monoclonal antibody, clone 14) at 1:100 dilution, 32 min, 37 °C, with heat induced antigen retrieval; BAF250 (ARID1A) (Sigma, rabbit polyclonal antibody) at 1:100 dilution, 64 min, 37 °C, WT1 (Dako, mouse monoclonal antibody, clone 6F‐H2) at 1:100 dilution, 32 min, 37 °C. The Ventana Universal Secondary Antibody was used for 32 min at 37 °C. The detection systems were the Ventana OptiView DAB kit (p53, β‐catenin) and Ventana ChromoMap DAB kit (BAF250/ARID1A).
Results
Clinical and pathologic features of study samples
We performed targeted sequencing of 30 uterine carcinosarcomas with matched FFT and FFPE tumour DNA samples. The mutation findings in this study are summarized in Figure 1, and all mutations (except CSMD3), were orthogonally validated and confirmed to be somatic (supplementary material Tables 1 and 2). We performed sequencing in both matched frozen and FFPE primary tumour samples in 28 of 30 cases, as two cases did not have sufficient frozen tumour tissue available for analyses. In 14 of 30 tumours, the carcinoma and sarcoma components formed spatially distinct tumour areas such that the separate components were sequenced separately. In addition to the analysis of the primary tumour, extrauterine metastases were analysed separately in 10 cases.
Figure 1.
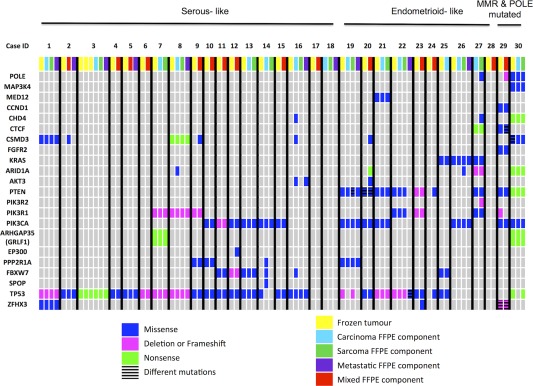
Mutation profiles of all uterine carcinosarcomas. Uterine carcinosarcoma patients are grouped into columns and are separated by black lines. The genes that were sequenced are found in rows. Coloured boxes indicate the presence of a DNA alteration (point mutations, frameshifts, deletions). The grey boxes indicate that no alteration was identified in the specific gene and specific patient. The cases are grouped into serous‐like molecular subtype and endometrioid‐like molecular subtype based on the pattern of mutations. The last two cases (ID 29, 30) are tumours with MMR and POLE mutated, respectively.
Table 1.
Summary of the clinicopathologic features of the study series (n = 30)
| Age (range) | 65 years (50–83 years) |
| Menopausal status | |
| Pre‐menopausal | 1 |
| Post‐menopausal | 29 |
| Tumour size (range) | 6.0 cm (2.3 – 15.0 cm) |
| Sarcoma component | |
| Homologous | 14 |
| Heterologous | 16 |
| Rhabdomyosarcoma | 5 |
| Chondrosarcoma | 3 |
| Osteosarcoma | 3 |
| >1 sarcoma type | 5 |
| Metastases | |
| Absent | 19 |
| Present | 11 |
| Pelvic metastases | 6 |
| Carcinoma only | 3 |
| Sarcoma only | 1 |
| Carcinoma and sarcoma | 2 |
| Distant metastases (beyond pelvis) | 5 |
| Carcinoma only | 5 |
| Sarcoma only | 0 |
| Carcinoma and sarcoma | 0 |
| Lymph nodes | |
| Benign | 12 |
| Positive for metastases | 3 |
| Not taken | 15 |
| Pelvic washings | |
| Negative | 17 |
| Positive | 9 |
| Not taken | 4 |
| Clinical Stage | |
| I | 15 |
| II | 0 |
| III | 10 |
| IV | 5 |
Table 2.
Immunohistochemical features of 30 uterine carcinosarcomas
| Case No. | p53 | MMR | ARID1A | B‐catenin |
|---|---|---|---|---|
| 1 | Abnormal (loss) | Normal | Normal | Membranous |
| 2 | Abnormal (diffuse) | Normal | Normal | Membranous |
| 3 | Abnormal (loss) | Normal | Normal | Membranous |
| 4 | Abnormal (diffuse) | Normal | Normal | Membranous |
| 5 | Abnormal (diffuse) | Normal | Normal | Membranous |
| 6 | Abnormal (loss) | Normal | Normal | Membranous & cytoplasmic |
| 7 | Abnormal (diffuse) | Normal | Normal | Membranous |
| 8 | Abnormal (loss) | Normal | Normal | Membranous |
| 9 | Abnormal (diffuse) | Normal | Normal | Membranous |
| 10 | Abnormal (diffuse) | Normal | Normal | Membranous & cytoplasmic |
| 11 | Abnormal (diffuse) | Normal | Normal | Membranous |
| 12 | Abnormal (diffuse) | Normal | Normal | Membranous |
| 13 | Abnormal (diffuse) | Normal | Normal | Membranous |
| 14 | Abnormal (diffuse) | Normal | Normal | Membranous |
| 15 | Abnormal (diffuse) | Normal | Normal | Membranous |
| 16 | Abnormal (diffuse) | Normal | Normal | Membranous |
| 17 | Abnormal (diffuse) | Normal | Normal | Membranous & cytoplasmic |
| 18 | Abnormal (loss) | Normal | Normal | Membranous |
| 19 | Normal | Normal | Normal | Membranous |
| 20 | Abnormal (diffuse) | Normal | Normal | Membranous & cytoplasmic |
| 21 | Abnormal (diffuse) | Normal | Normal | Membranous |
| 22 | Abnormal (loss) | Normal | Normal | Membranous |
| 23 | Abnormal (diffuse) | Normal | Normal | Membranous |
| 24 | Abnormal (diffuse) | Normal | Normal | Membranous |
| 25 | Abnormal (diffuse) | Normal | Normal | Membranous |
| 26 | Normal | Normal | Normal | Membranous & cytoplasmic |
| 27 | Normal | Normal | Loss | Membranous |
| 28 | Normal | Normal | Normal | Membranous |
| 29 | Abnormal (diffuse) | Abnormal (Loss of MLH1 & PMS2) | Normal | Membranous |
| 30 | Normal | Normal | Loss | Membranous |
MMR: mismatch repair protein (MLH1, MSH2, MSH6 and PMS2).
The clinical features of the study cohort are summarized in Table 1 (detailed information in supplementary material Table 3). Pre‐operative biopsy was performed in 28 of 30 patients and the diagnoses were carcinosarcoma in 24 patients, high‐grade serous carcinoma in three patients and grade 3 endometrioid carcinoma in one patient. Half of the patients presented with uterine confined disease, while the other half presented with extrauterine tumour spread. The sarcoma component was high‐grade in all except one case (case 30; Figure 2A–D). Heterologous differentiation was present in 16 of 30 primary tumours with rhabdomyoblastic differentiation being the most common. In the primary tumour, the proportion of the carcinoma component varied from <5% to > 95% (average of 50%). The extrauterine (metastatic foci) tumours (10 patients) consisted only of carcinoma in six cases, sarcoma in one case, and a mix in three cases. The carcinoma elements of the metastatic tumour resembled the carcinoma component of the primary in all except one case (case 22; Figure 2E–F).
Table 3.
Clinical feature stratified by molecular subtypes
| Molecular subtype | Number of cases | Age (years) | Tumour size (cm) | Sarcoma type | Stage |
|---|---|---|---|---|---|
| Serous‐like | 18 | 63.1 (50–80) | 6.5 (2.3–15) | Homologous: 7 Heterologous: 11 | Low (stages 1 and 2): 6 High (stages 3 and 4): 12 |
| Endometrioid‐like | 11 | 70.2 (54–83) | 5.4 (2.5–11) | Homologous: 6 Heterologous: 5 | Low (stages 1 and 2): 8 High (stages 3 and 4): 3 |
| p‐value | 0.0521a | 0.452a | 0.129b | 0.0376b |
p‐value determined by student's t‐test.
p‐value determined by chi‐squared test.
Figure 2.
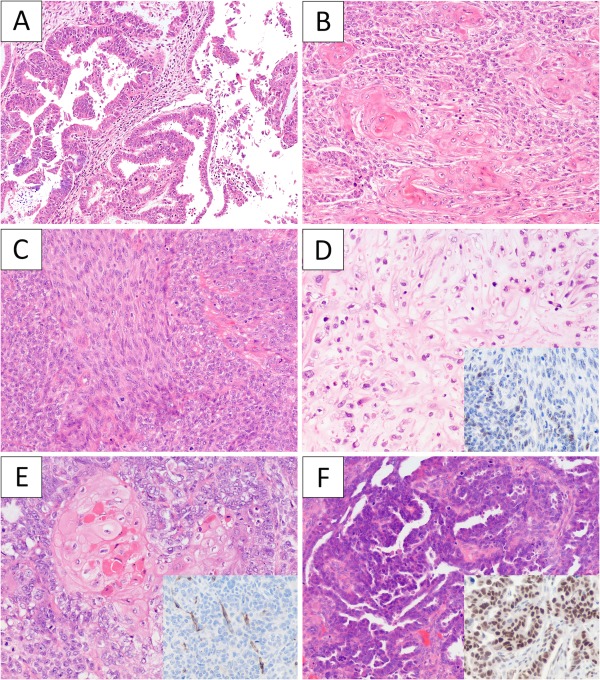
The histological features of selected carcinosarcoma cases. (A) Case 30 showed a low‐grade endometrioid component (A), solid areas with abundant squamous differentiation (B), low‐grade spindle cell component (C) and focal chondroid differentiation (D). p53 immunohistochemistry (D, inset) was focal in both the endometrioid and spindle cell components. Case 22 had uterine, bilateral ovarian and peritoneal tumour. The uterine tumour showed a carcinosarcoma with prototypical endometrioid histology with squamous differentiation (E) and negative staining for WT1 (E, inset). The ovarian tumour as well as the peritoneal tumour deposits were all pure carcinoma with high‐grade serous carcinoma histology (F) and strong nuclear staining for WT1 (F, inset).
Targeted sequencing shows frequent TP53 and PI3K pathway mutations in primary uterine carcinosarcoma samples
We identified non‐synonymous somatic mutations in 27 of 30 (90%) cases (Figure 1). There were 28 cases where both frozen and FFPE tumour samples were available for analysis. The mutation profiles were completely concordant in 25 samples after excluding low‐frequency findings (mutations found in < 5% of the reads) as likely minor clonal changes (intratumoural heterogeneity). Three cases (14, 20 and 24) showed some differences between the mutations identified in the frozen or FFPE tumour sample. Overall, there appeared to be good concordance in the detection of mutations in FFPE tumour tissue.
TP53 mutations were present in the majority of carcinosarcoma (24 of 30) (80%). Most of the tumours harbouring missense TP53 mutations showed diffuse nuclear p53 immunostaining, with the exception of two cases (7 and 21) that harboured TP53 deletions and showed diffuse nuclear p53 immunostaining (Table 2). Cases 19 and 30 both demonstrated TP53 mutations (frameshift and nonsense) in the sarcoma component but not the carcinoma component of the tumour, and both displayed a wild‐type pattern of p53 immunostaining in both components of the tumour. Loss of nuclear p53 immunohistochemistry was detected in 5 cases with indel or nonsense mutations, and 3 additional tumours (cases 17, 18 and 29) displayed abnormal patterns of staining with no detectable mutations (Table 2 and supplementary material Table 2). In total (27 of 30) 90% of all tumours harboured TP53 aberrations as assessed by either sequencing or immunohistochemistry.
In the primary tumours, mutations involving genes that encode the kinase or regulatory proteins of the PI3K pathway were identified in 20 of 30 tumours (67%), where multiple PI3K pathway proteins were mutated in 6 tumours. PIK3CA mutations were found in 12 of 30 (40%) tumours, which were scattered throughout the different functional domains of PIK3CA (Figure 3). PTEN mutations, most commonly missense in type, were observed in eight tumours (27%). PIK3R1 mutations were found in five tumours (17%), while only one tumour demonstrated a PIK3R2 mutation. Of note in this cohort, PIK3CA mutations were rarely found with concurrent PIK3R1 or PIK3R2 mutations. One tumour (case 20) with a PIK3CA mutation also harboured an AKT3 (E167D) mutation, while case 18 harboured an AKT3 (M336I) mutation at low frequency (5%) without concurrent PI3K pathway mutations. No mutations involving AKT1 or AKT2 were identified in this series.
Figure 3.
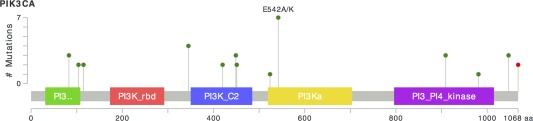
Schematic depiction of the PIK3CA mutations identified in uterine carcinosarcoma. Each identified PIK3CA mutation is indicated as a lollipop plot along the protein domains of PIK3CA. The number of mutations for all cases is indicated on the x‐axis. Green dots indicate point mutations, and red dots indicate frameshift mutations. Mutations can be identified along all regions of the PIK3CA gene and are not restricted to only the classical hotspot regions. The mutation plot was constructed using the cBioPortal 25, 26 visualization tool MutationMapper v1.0.
Other recurrently mutated genes included FBXW7 (six tumours, 20%), PPP2R1A (four tumours, 13%), KRAS (three tumours, 10%), ARID1A (three tumours, 10%), ZFHX3 (two tumours, 7%) and CSMD3 (seven tumours, 23%). By immunohistochemistry, only two cases showed loss of BAF250 (ARID1A), which corresponded to the presence of frameshift and nonsense mutations (Table 2). There was no evidence of CTNNB1 mutations by sequencing or by immunohistochemistry. A somatic MED12 mutation (D23Y) was identified in one tumour, which has been previously documented in a case of endometrial carcinoma 27; however, it was not the typical hotspot MED12 mutation found in uterine smooth muscle tumours 20, 21.
The expression of mismatch repair (MMR) proteins (MLH1, PMS2, MSH2 and MSH6) was evaluated by immunohistochemistry in all 30 primary tumours (Table 2). Only 1 of 30 tumours (3%) (case 29), showed MMR protein deficiency (MLH1 and PMS2 loss) while the remaining 29 tumours demonstrated intact expression of MMR proteins.
A somatic POLE exonuclease domain mutation (M444K) was identified in one tumour (case 30), which was reported in an ultra‐mutated endometrial carcinoma in a prior study 17. In keeping with the ultra‐mutator phenotype, this tumour also possessed 11 point mutations involving eight other genes. In retrospective histologic review, this tumour was re‐classified as a FIGO grade 2 endometrioid‐type carcinoma with spindle cell elements (and focal chondroid differentiation) (Figure 2A–D). This was the only histologic outlier in our present series as all other cases were confirmed to be carcinosarcoma. Therefore, in histologically confirmed uterine carcinosarcomas (n = 29 tumours), none of the tumours harboured POLE exonuclease domain mutations.
Carcinoma and sarcoma components of uterine carcinoma show concordant mutation profiles
In this series, 13 of 29 uterine carcinosarcomas displayed spatially distinct carcinoma and sarcoma components such that these paired components were amenable to separate analyses. Twelve of the 13 pairs demonstrated at least one mutation in common with eight cases being 100% identical, while one case (case 18) showed no detectable mutations in either component (Figure 1). Two cases exhibited additional mutation(s) in the sarcoma component compared to the carcinoma component: case 19 with a TP53 missense mutation, and case 27 with PIK3R2, CHD4 mutations, and a POLE non‐exonuclease domain mutation. In contrast, two cases displayed additional mutation(s) in the carcinoma component compared to the sarcoma component: case 8 with an ARID1A mutation, and case 14 with PPP2R1A, FBXW7 and SPOP mutations. Overall, there was complete concordance or partial overlap in mutations identified in the carcinoma and the sarcoma components in 11 of 12 cases, hence indicating clonal relatedness between the carcinoma and the sarcoma components.
Primary and metastatic tumours show concordant mutation profiles
Matched uterine primary and metastatic tumour samples were analysed in 10 cases, in one of which no mutations were identified. Of the nine cases with validated somatic mutations, five cases demonstrated identical mutations in the metastatic tumour compared to the primary. Three showed partially overlapping mutations but with fewer mutations identified in the metastatic tumour compared to the respective uterine primary. For case 22, the carcinoma and the sarcoma components of the uterine primary all harboured the same PTEN, PIK3R1 and TP53 (frameshift) mutations with loss of p53 immunostaining. In contrast, the metastatic tumour (bilateral ovaries and peritoneal deposits) displayed no evidence of PTEN, PIK3R1 mutations, and harboured a different TP53 missense mutation with diffuse nuclear p53 immunostaining (Figure 2E–F). STR testing confirmed that the DNA of the carcinosarcoma and the apparent metastatic tumour were derived from the same patient (supplementary material Table 4). Histologically, the carcinoma component of the primary carcinosarcoma displayed typical endometrioid features with squamous differentiation, while the ovarian/peritoneal tumour consisted purely of carcinoma with features consistent with ovarian high‐grade serous carcinoma. Additional immunohistochemistry analysis demonstrated nuclear WT1 positivity in the ovarian tumour but not the uterine tumour (Figure 2E–F) 28. These findings strongly indicate a synchronous uterine carcinosarcoma and ovarian high‐grade serous carcinoma in this patient. Further review of annotated clinical data revealed a personal history of bilateral high‐grade breast carcinomas and a strong breast cancer family history, thus raising the possibility of BRCA1/2 germline background.
Clinical significance of molecular profiles in uterine carcinosarcoma
We have previously identified different molecular types of uterine carcinosarcomas with some tumours showing endometrial serous carcinoma‐like mutation profiles (characterized by the presence of TP53 mutation with PPP2R1A and/or FBXW7 mutations and in the absence of PTEN, CTNNB1, KRAS or ARID1A mutations) and other tumours showing endometrioid carcinoma‐like mutation profiles (characterized by the presence of PTEN, CTNNB1, KRAS and/or ARID1A mutations) 16. Based on the combined genetic and immunohistochemical profiles in the present cohort, 18 tumours showed serous‐like while 11 tumours showed endometrioid‐like molecular profiles (Figure 1 and Table 2). There was good agreement between histologic subtyping (based on the morphology of the epithelial component) and molecular subtyping in 27 of 29 uterine carcinosarcomas (93% agreement; Figure 4).
Figure 4.
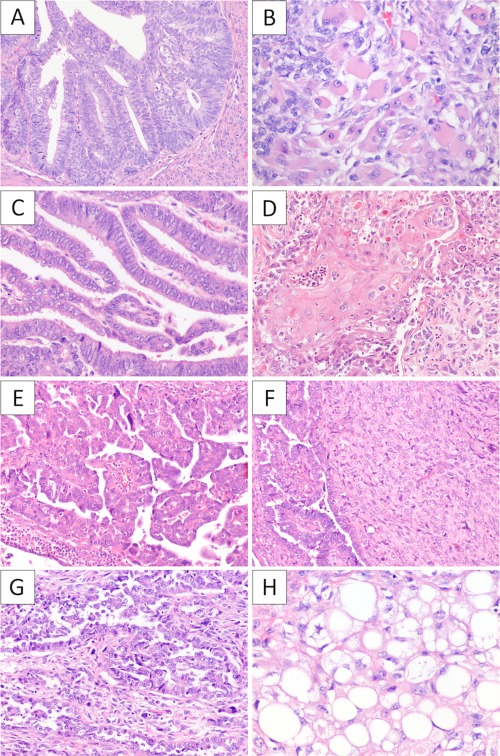
Representative histology of uterine carcinosarcoma with endometrioid‐like and serous‐like molecular features. (A) Tumours with endometrioid‐like molecular features typically displayed endometrioid‐type histology in the carcinoma component, as illustrated by case 27, which showed low‐grade endometrioid carcinoma component (A) with rhabdomyoblastic (B), oesteogenic and chondroid differentiation (not shown), and by case 21 that showed low‐grade endometrioid carcinoma component (C) with squamous differentiation (D). Tumours with serous‐like molecular features typically displayed serous‐type histology in the carcinoma component, as illustrated by case 3, which showed high‐grade serous carcinoma component (E) and homologous sarcoma (F), and case 9 that showed high‐grade serous areas (G) with lipogenic (H) and rhabdomyoblastic differentiation (not shown).
Clinically, the majority of patients with serous‐like tumour presented with advanced stage disease as 12 of 18 (67%) patients had stage III‐IV disease, while 8 of 11 (73%) patients with endometrioid‐like tumour presented with stage I disease (Table 3). Despite the differences in presenting disease stage, there was no significant difference in disease‐specific survival between patients with endometrioid‐type tumour and patients with serous‐type tumours (p = 0.98) (Figure 5).
Figure 5.
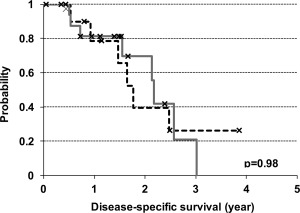
Disease‐specific survival analysis stratified by carcinosarcoma molecular subtypes. The Kaplan–Meier survival analysis for disease specific survival where the dotted black line represents the endometrioid‐like molecular subtype and the solid grey line represents the serous‐like molecular subtype.
Discussion
Uterine carcinosarcoma is a histologically intriguing biphasic tumour that contains both carcinoma and sarcoma within the same tumour mass. While the carcinoma and the sarcoma elements can be intimately admixed, they can also form spatially distinct components. This prompted different theories regarding the histogenesis of uterine carcinosarcomas (collision tumour versus metaplastic carcinoma). The prevailing view at the present is that the carcinoma and sarcoma components share a common origin in the majority of uterine carcinosarcoma 12, 13, 14, 15. Our findings here provide further objective and unambiguous support for the common origin hypothesis. This is evident in the cases where the two histologic components were separately analysed; all cases with detectable mutations shared at least one somatic mutation in common between the carcinoma and the sarcoma components.
While our results clearly demonstrated genetic similarities between the carcinoma and the sarcoma components, it did not offer definitive insights into the directionality of tumour progression for the different histologic components. Most of the cases analysed showed identical mutations profiles between the carcinoma and the sarcoma components. There were four tumours that showed differences in mutations in additional to shared mutations between the two components. These included two cases with additional mutations found in the sarcoma component and two cases with additional mutations found in the carcinoma component. These differences, however, may represent intratumoural genetic heterogeneity and it would not be prudent to infer directionality solely based on the presence of additional mutations in this small number of cases. Even though the directionality of progression cannot be concluded from these results, the overall mutation profiles seen in the present series and our previously published series of uterine carcinosarcoma resembles the mutation profiles of endometrial serous and endometrioid carcinomas 16. This provides indirect evidence that uterine carcinosarcomas most likely develop through sarcomatous transdifferentiation of the underlying endometrial carcinoma, as suggested by earlier studies 12, 13, 14, 15. Moreover, it is equally likely sarcomatous transdifferentiation represents a loose state of control in cell differentiation, which reflects a highly disrupted genetic/epigenetic cellular milieu of the tumour. As such, sarcomatous transdifferentiation can be regarded as a morphologic surrogate marker for more aggressive clinical behaviour of the endometrial carcinoma, rather than a sequential state of tumour progression with the sarcoma component representing the more biologically aggressive component of the tumour. This notion is in keeping with the clinical observation that the metastatic tumour in the great majority of cases is comprised either exclusively or predominantly of the carcinoma elements 4, 5.
We have identified mutations involving PI3K pathway in two‐thirds of uterine carcinosarcoma examined, most commonly involving PIK3CA and PTEN. In contrast to prior studies on uterine carcinosarcomas 29, 30, our present analyses identified both traditional PIK3CA hotspots (exons 9 and 20) mutations as well as mutations in the adaptor binding domain, helical domain, and C2 domain which can also enhance kinase enzymatic activity 31, 32. When present, these PI3K pathway mutations were found in the carcinoma and sarcoma components of the primary tumour, as well as in the metastatic tumour. This indicates that these PI3K pathway mutations occur relatively early in the tumourigenesis of carcinosarcoma and likely represent important oncogenic driver events that could be targeted with PIK3CA/mTOR inhibitors 33, 34, 35. We also observed a very high frequency of TP53 alterations (90%) in this series of uterine carcinosarcomas, which is supported by previous literature 5, 12, 16. The same TP53 aberrations were present in both carcinoma and sarcoma components of uterine carcinosarcoma, which also implicates an early event in tumourigenesis. In keeping with prior studies, MMR protein deficiency that suggests underlying microsatellite instability was rarely observed in our present series of uterine carcinosarcoma 12, 24. A recent study demonstrated frequent mutations in chromatin remodelling genes (ARID1A, ARID1B, MLL3, SPOP) with the identification of 4 mismatch repair deficient tumour in a cohort of 17 uterine and five ovarian carcinosarcomas 36. While these results are overall different from our present cohort, a closer examination reveals that nearly all of the identified ARID1A, ARID1B and MLL3 mutations were found in the four MMR‐deficient uterine carcinosarcomas and in four of the five ovarian carcinosarcomas. We did, however, identify a few cases in our cohort with ARID1A mutations and one with an SPOP mutation. Lastly, POLE exonuclease domain mutations do not appear to occur in bona fide uterine carcinosarcoma.
About two‐thirds of the present series of uterine carcinosarcoma demonstrates a serous carcinoma‐like mutation profile, while the remaining third demonstrated an endometrioid carcinoma‐like mutation profile. This apparent molecular heterogeneity is similar to what we have observed in a previous cohort of uterine carcinosarcomas 16. In addition to the serous carcinoma‐like uterine carcinosarcoma (all of which harboured TP53 aberrations), half of the uterine carcinosarcoma that displayed an endometrioid carcinoma‐like mutation profile also harboured TP53 aberrations. Based on the proposed molecular types of endometrial carcinoma by the recent TCGA (The Cancer Genome Atlas) study 17, our findings here indicate that a significant majority of uterine carcinosarcoma likely belong to the copy number high serous‐like molecular type, with a minor subset representing copy number low endometrioid molecular type and more rarely the microsatellite‐unstable hypermutated molecular type. With regards to clinical features, we did not observe a significant difference in patient survival between the two apparent molecular types of uterine carcinosarcoma, however our study series is relatively small. In contrast, De Jong et al found that uterine carcinosarcomas with non‐endometrioid carcinoma components showed a trend for worse disease‐specific survival compared to carcinosarcomas with endometrioid carcinoma components in 40 tumours, although the difference was not significant 5. Irrespective of the prognostic significance, there are clearly differences in the underlying tumourigenic mechanisms between the two apparent molecular types and this may have treatment implications with emerging therapies.
With regards to the POLE‐mutated cases, endometrioid carcinoma with spindle cell elements is a recognized histologic mimic of carcinosarcoma, as both tumour types display biphasic appearance and can include foci of heterologous differentiation 37, 38. Clinically, endometrioid carcinoma with spindle cell elements is associated with a much more favourable prognosis than uterine carcinosarcoma 37. Interestingly, endometrial carcinoma of POLE ultra‐mutated molecular type is also associated with excellent prognosis 17, 39. This patient with POLE mutated endometrioid carcinoma presented with stage 1B disease and underwent chemotherapy treatment, and is alive with no evidence of disease recurrence nearly seven years later. Further studies are needed to investigate whether there is increased propensity for POLE‐mutated endometrioid carcinoma to display spindle cell elements.
In summary, we performed targeted gene sequencing on a series of uterine carcinosarcomas, and identified frequent aberrations involving TP53 and PI3K pathway that may be therapeutically targeted. Our analyses demonstrate that the carcinoma and the sarcoma components of uterine carcinosarcoma are similar if not identical genetically; hence they are both parts of the same tumour. We also confirmed previous observations that most uterine carcinosarcomas display either endometrial serous carcinoma‐like or endometrioid carcinoma‐like molecular profiles. These findings altogether support the notion that uterine carcinosarcoma represents sarcomatous transdifferentiation of endometrial carcinoma, and the identification of the different molecular types of uterine carcinosarcoma (reflecting the underlying endometrial carcinoma molecular types) may have future clinical management implications.
Author contributions
MKM, LNH, BAC, CHL conceived and designed the study. MHC, LNH, JNM, BAC, CBG, CHL assisted in accruing samples for the study. MKM, LNH, NR, JS, WY, RM carried out the technical experimentation. LNH, CBG, CHL performed pathological reviews. MKM, LNH, CHL, BAC, JNM, CBH, DGH assisted in the interpretation of the data. MKM, LHN and CHL wrote the manuscript and created the figures. All authors have reviewed and approved the manuscript.
Supporting information
Additional Supporting Information may be found in the online version of this article.
Detailed materials and methods for STR analysis and statistical analysis.
Table S1. Details for all mutations identified in all samples including: genomic position, nucleotide changes, amino acid changes, depth, and frequency.
Table S2. A simpler version of Supplemental Table 2 with condensed amino acid changes, IHC data, histology and cellularity for each case.
Table S3. Includes all clinical data for each uterine carcinosarcoma case.
Table S4. Detailed results for the carcinosarcoma cases tested by STR analysis.
Acknowledgements
This study is partially supported by a sarcoma research funds from Liddy Shriver Sarcoma Initiative. CH Lee is supported by funding from Royal Alexandra Hospital foundation. MK McConechy is supported by the CIHR Frederick Banting and Charles Best Canada Graduate Scholarship Doctoral Research award. We would like to thank all the women who have donated their samples to enable our research. We would also like to thank Amy Lum, Christine Chow and Margaret Luk for their technical assistance.
References
- 1. Kurman RJ, International Agency for Research on Cancer, World Health Organization WHO Classification of Tumours of Female Reproductive Organs (4th edn). International Agency for Research on Cancer: Lyon, 2014. [Google Scholar]
- 2. Silverberg SG, Major FJ, Blessing JA, et al. Carcinosarcoma (malignant mixed mesodermal tumor) of the uterus. A Gynecologic Oncology Group pathologic study of 203 cases. Int J Gynecol Pathol 1990; 9: 1‐19. [DOI] [PubMed] [Google Scholar]
- 3. Kernochan LE, Garcia RL. Carcinosarcomas (malignant mixed Mullerian tumor) of the uterus: advances in elucidation of biologic and clinical characteristics. J Natl Compr Canc Netw 2009; 7: 550‐556; quiz 557. [DOI] [PubMed] [Google Scholar]
- 4. Gonzalez Bosquet J, Terstriep SA, Cliby WA, et al. The impact of multi‐modal therapy on survival for uterine carcinosarcomas. Gynecol Oncol 2010; 116: 419‐423. [DOI] [PubMed] [Google Scholar]
- 5. de Jong RA, Nijman HW, Wijbrandi TF, et al. Molecular markers and clinical behavior of uterine carcinosarcomas: focus on the epithelial tumor component. Mod Pathol 2011; 24: 1368‐1379. [DOI] [PubMed] [Google Scholar]
- 6. Ferguson SE, Tornos C, Hummer A, et al. Prognostic features of surgical stage I uterine carcinosarcoma. Am J Surg Pathol 2007; 31: 1653‐1661. [DOI] [PubMed] [Google Scholar]
- 7. Norris HJ, Taylor HB. Mesenchymal tumors of the uterus. 3. A clinical and pathologic study of 31 carcinosarcomas. Cancer 1966; 19: 1459‐1465. [DOI] [PubMed] [Google Scholar]
- 8. Ober WB. Uterine sarcomas: histogenesis and taxonomy. Ann N Y Acad Sci 1959; 75: 568‐585. [DOI] [PubMed] [Google Scholar]
- 9. McCluggage WG. Uterine carcinosarcomas (malignant mixed Mullerian tumors) are metaplastic carcinomas. Int J Gynecol Cancer 2002; 12: 687‐690. [DOI] [PubMed] [Google Scholar]
- 10. Zelmanowicz A, Hildesheim A, Sherman ME, et al. Evidence for a common etiology for endometrial carcinomas and malignant mixed mullerian tumors. Gynecol Oncol 1998; 69: 253‐257. [DOI] [PubMed] [Google Scholar]
- 11. Sreenan JJ, Hart WR. Carcinosarcomas of the female genital tract. A pathologic study of 29 metastatic tumors: further evidence for the dominant role of the epithelial component and the conversion theory of histogenesis. Am J Surg Pathol 1995; 19: 666‐674. [PubMed] [Google Scholar]
- 12. Taylor NP, Zighelboim I, Huettner PC, et al. DNA mismatch repair and TP53 defects are early events in uterine carcinosarcoma tumorigenesis. Mod Pathol 2006; 19: 1333‐1338. [DOI] [PubMed] [Google Scholar]
- 13. Wada H, Enomoto T, Fujita M, et al. Molecular evidence that most but not all carcinosarcomas of the uterus are combination tumors. Cancer Res 1997; 57: 5379‐5385. [PubMed] [Google Scholar]
- 14. Jin Z, Ogata S, Tamura G, et al. Carcinosarcomas (malignant Mullerian mixed tumors) of the uterus and ovary: a genetic study with special reference to histogenesis. Int J Gynecol Pathol 2003; 22: 368‐373. [DOI] [PubMed] [Google Scholar]
- 15. Watanabe M, Shimizu K, Kato H, et al. Carcinosarcoma of the uterus: immunohistochemical and genetic analysis of clonality of one case. Gynecol Oncol 2001; 82: 563‐567. [DOI] [PubMed] [Google Scholar]
- 16. McConechy MK, Ding J, Cheang MC, et al. Use of mutation profiles to refine the classification of endometrial carcinomas. J Pathol 2012; 228: 20‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kandoth C, Schultz N, Cherniack AD, et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013; 497: 67‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kuhn E, Wu RC, Guan B, et al. Identification of molecular pathway aberrations in uterine serous carcinoma by genome‐wide analyses. J Natl Cancer Inst 2012; 104: 1503‐1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gallo ML, O'Hara AJ, Rudd ML, et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin‐remodeling and ubiquitin ligase complex genes. Nat Genet 2012; 44: 1310‐1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perot G, Croce S, Ribeiro A, et al. MED12 alterations in both human benign and malignant uterine soft tissue tumors. PLoS One 2012; 7: e40015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ravegnini G, Marino‐Enriquez A, Slater J, et al. MED12 mutations in leiomyosarcoma and extrauterine leiomyoma. Mod Pathol 2013; 26: 743‐749. [DOI] [PubMed] [Google Scholar]
- 22. Forshew T, Murtaza M, Parkinson C, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med 2012; 4: 136ra168. [DOI] [PubMed] [Google Scholar]
- 23. McConechy MK, Anglesio MS, Kalloger SE, et al. Subtype‐specific mutation of PPP2R1A in endometrial and ovarian carcinomas. J Pathol 2011; 223: 567‐573. [DOI] [PubMed] [Google Scholar]
- 24. Hoang LN, Ali RH, Lau S, et al. Immunohistochemical survey of mismatch repair protein expression in uterine sarcomas and carcinosarcomas. Int J Gynecol Pathol 2014; 33: 483‐491. [DOI] [PubMed] [Google Scholar]
- 25. Cerami E, Gao J, Dogrusoz U, et al. The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2: 401‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Forbes SA, Tang G, Bindal N, et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res 2010; 38: D652‐657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Madore J, Ren F, Filali‐Mouhim A, et al. Characterization of the molecular differences between ovarian endometrioid carcinoma and ovarian serous carcinoma. J Pathol 2010; 220: 392‐400. [DOI] [PubMed] [Google Scholar]
- 29. Bashir S, Jiang G, Joshi A, et al. Molecular alterations of PIK3CA in uterine carcinosarcoma, clear cell, and serous tumors. Int J Gynecol Cancer 2014; 24: 1262‐1267. [DOI] [PubMed] [Google Scholar]
- 30. Growdon WB, Roussel BN, Scialabba VL, et al. Tissue‐specific signatures of activating PIK3CA and RAS mutations in carcinosarcomas of gynecologic origin. Gynecol Oncol 2011; 121: 212‐217. [DOI] [PubMed] [Google Scholar]
- 31. Gabelli SB, Echeverria I, Alexander M, et al. Activation of PI3Kalpha by physiological effectors and by oncogenic mutations: structural and dynamic effects. Biophys Rev 2014; 6: 89‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gabelli SB, Huang CH, Mandelker D, et al. Structural effects of oncogenic PI3Kalpha mutations. Curr Top Microbiol Immunol 2010; 347: 43‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shoji K, Oda K, Kashiyama T, et al. Genotype‐dependent efficacy of a dual PI3K/mTOR inhibitor, NVP‐BEZ235, and an mTOR inhibitor, RAD001, in endometrial carcinomas. PLoS One 2012; 7: e37431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. English DP, Bellone S, Cocco E, et al. Oncogenic PIK3CA gene mutations and HER2/neu gene amplifications determine the sensitivity of uterine serous carcinoma cell lines to GDC‐0980, a selective inhibitor of Class I PI3 kinase and mTOR kinase (TORC1/2). Am J Obstet Gynecol 2013; 209: 465 e461‐469. [DOI] [PubMed] [Google Scholar]
- 35. Weigelt B, Warne PH, Lambros MB, et al. PI3K pathway dependencies in endometrioid endometrial cancer cell lines. Clin Cancer Res 2013; 19: 3533‐3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jones S, Stransky N, McCord CL, et al. Genomic analyses of gynaecologic carcinosarcomas reveal frequent mutations in chromatin remodelling genes. Nat Commun 2014; 5: 5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Murray SK, Clement PB, Young RH. Endometrioid carcinomas of the uterine corpus with sex cord‐like formations, hyalinization, and other unusual morphologic features: a report of 31 cases of a neoplasm that may be confused with carcinosarcoma and other uterine neoplasms. Am J Surg Pathol 2005; 29: 157‐166. [DOI] [PubMed] [Google Scholar]
- 38. Tornos C, Silva EG, Ordonez NG, et al. Endometrioid carcinoma of the ovary with a prominent spindle‐cell component, a source of diagnostic confusion. A report of 14 cases. Am J Surg Pathol 1995; 19: 1343‐1353. [DOI] [PubMed] [Google Scholar]
- 39. Meng B, Hoang LN, McIntyre JB, et al. POLE exonuclease domain mutation predicts long progression‐free survival in grade 3 endometrioid carcinoma of the endometrium. Gynecol Oncol 2014; 134: 15‐19. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article.
Detailed materials and methods for STR analysis and statistical analysis.
Table S1. Details for all mutations identified in all samples including: genomic position, nucleotide changes, amino acid changes, depth, and frequency.
Table S2. A simpler version of Supplemental Table 2 with condensed amino acid changes, IHC data, histology and cellularity for each case.
Table S3. Includes all clinical data for each uterine carcinosarcoma case.
Table S4. Detailed results for the carcinosarcoma cases tested by STR analysis.