

Abstract
Amiodarone (AD) is an iodinated benzofuran derivative, especially known for its antiarrhythmic properties. It exerts serious side‐effects even in patients receiving low doses. AD is well‐known to induce apoptosis of type II alveolar epithelial cells (AECII), a mechanism that has been suggested to play an important role in AD‐induced lung fibrosis. The precise molecular mechanisms underlying this disease are, however, still unclear. Because of its amphiphilic nature, AD becomes enriched in the lysosomal compartments, affecting the general functions of these organelles. Hence, in this study, we aimed to assess the role of autophagy, a lysosome‐dependent homeostasis mechanism, in driving AECII apoptosis in response to AD. In vitro, AD‐treated MLE12 and primary AECII cells showed increased proSP‐C and LC3B positive vacuolar structures and underwent LC3B‐dependent apoptosis. In addition, AD‐induced autophagosome‐lysosome fusion and increased autophagy flux were observed. In vivo, in C57BL/6 mice, LC3B was localised at the limiting membrane of lamellar bodies, which were closely connected to the autophagosomal structures in AECIIs. Our data suggest that AD causes activation of macroautophagy in AECIIs and extensive autophagy‐dependent apoptosis of alveolar epithelial cells. Targeting the autophagy pathway may therefore represent an attractive treatment modality in AD‐induced lung fibrosis.
Keywords: autophagy, amiodarone, alveolar epithelial cells, lamellar bodies, LC3B, apoptosis
Introduction
Amiodarone (AD) is pharmacologically classified as a cationic amphiphilic drug. It is an efficient antiarrhythmic drug with typical class III Vaughan‐Williams properties. In spite of its beneficial effects against almost all kinds of arrhythmias, its clinical use is tempered because of its contraindications 1, 2, which include corneal micro deposits, dermatitis, symptomatic bradycardia, hypo‐ and hyper‐thyroidism and, most importantly, severe pulmonary toxicity 1. It is estimated that 10–17% of patients receiving 400 mg AD per day develop pulmonary toxicity, with fatalities occurring in about 10% of cases 3. Previous studies suggested that elderly patients with a pre‐existing lung disease are at a high risk of developing AD‐induced pulmonary toxicity. It was initially believed that a low dose of AD was relatively safe, but more recent case reports have revealed a high risk of pulmonary complications, especially confluent lung fibrosis, even with low dosage treatment of patients (200 mg per day). Acute respiratory distress syndrome and pneumonitis are also well‐documented side effects of AD 1, 4. Hence, according to the recent guideline for the management of patients with atrial fibrillation, AD is prescribed to treat atrial fibrillation in the absence of pre‐excitation only when other agents are unsuccessful 5.
At a cellular level, foamy macrophages and hyperplasia of type II alveolar epithelial cells (AECIIs) are the most common features of AD‐induced pulmonary fibrosis. In addition, it has been demonstrated that AD induces apoptosis of alveolar epithelial cells in vitro and that AD induces toxicity in several types of lung cells 6, 7. In full accordance, we have recently shown that intratracheal administration of AD induces lung fibrosis in mice and that AD induces surfactant accumulation, AECII apoptosis, lysosomal stress and ER stress in this model 8. Although the precise molecular events underlying AD‐induced lung fibrosis still remain to be clearly settled, mechanisms such as excessive intracellular phospholipidosis, direct cytotoxicity, oxidative stress and the angiotensin signalling pathway 9, 10, 11, 12, were previously suggested to contribute to the pathogenic chain of events.
It has been shown that AD becomes enriched in lysosomes (∼500‐fold as compared to serum) and causes accumulation of multilamellar bodies in the cytoplasm of various cell types 13, 14. Similarly, AD as well as its derivative dronedarone was shown to induce autophagy, a lysosome‐mediated degradation pathway that is extremely important in maintaining cellular homeostasis 15, 16. More recently, it was claimed that activating the autophagy pathway rescues AD‐induced lung fibrosis in rats 17.
Autophagy is a fundamental catabolic cellular process that degrades unnecessary proteins and damaged organelles, thereby helping in cell survival. At a molecular level, different types of autophagy pathways have been described. Macroautophagy is well characterised and is one of the important types of autophagy that involves sequestration of the cargo into a double‐membrane vesicle known as an autophagosome. This process involves complex interactions between several autophagy‐related (Atg) proteins. The autophagosome ultimately fuses with the lysosome to degrade its components. More detailed analyses of this process are given in previous reviews 18, 19. Several diseases such as cancer, neurodegenerative diseases, lysosomal storage disorders, cystic fibrosis, chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF) have been linked to altered autophagy 20.
We recently established a mouse model of AD‐induced lung fibrosis and reported surfactant alterations, severe AECII apoptosis and cellular stress in this model. We reported that AD‐induced AECII apoptosis is not mediated via Cathepsin D, and speculated on the involvement of other lysosomal pathways in driving AECII apoptosis in this model 8. In the current study, we aimed to carefully analyse the role of macroautophagy (referred as autophagy hereafter) in AD‐induced alveolar epithelial cell apoptosis and to identify if the autophagy marker protein microtubule‐associated protein 1 light chain‐3B (LC3B) is localised to the lamellar bodies of alveolar epithelial cells.
Methods
Cell culture
Mouse lung epithelial cell lines (MLE12) and isolation of primary mouse AECII and culture were performed as described before 21. Preparations of AD solution and the protocol for siRNA transfection are described elsewhere 8. For immunofluorescence analysis, MLE12 cells were plated in 8‐well chamber slides. After overnight adherence and following treatments, cells were washed and fixed with 4% paraformaldehyde, followed by washing and permeabilisation with 0.5% Triton X‐100. Permeabilised cells were then washed, blocked with 10% donkey serum and 3%BSA in PBS. Incubation with primary antibody (LC3B or LAMP2; abcam, Cambridge, UK) was performed overnight at 4 °C. Following washing, secondary antibody (Alexa Fluor 555, donkey anti rabbit or Alexa Fluor 488, donkey anti rat; Life Technologies, Darmstadt, Germany) was added for 1 h at RT in dark. Samples were then washed and mounted with mounting medium with DAPI for nuclear staining (VECTASHIELD mounting medium with DAPI, Vector Labs/Enzo Life Sciences GmbH, Lörrach, Germany) followed by microscopy (Leica Mirosystems, Germany). Protocols for RNA extraction, RT‐PCR and primers used are given in Supporting Information.
Autophagy flux
10 mg/ ml stock solution of chloroquine (CQ; Chloroquine diphosphate salt, Sigma‐Aldrich) was prepared by dissolving it in cell culture grade aqua dest. MLE12 cells were plated in 6‐well cell culture dishes. Next day, medium was replaced with fresh culture medium and CQ or vehicle (cell culture grade aqua dest) was added. 2 or 6 µl from CQ stock solution per 2ml culture medium was added to get a final concentration of 10 or 30 µg/ml, respectively. 4 h later, AD or vehicle was added and cells were harvested after 8 h, followed by Western blotting for LC3B. Similarly, MLE12 cells were pre‐incubated with DMSO or 10 µg/ml pepstatin and E64D (Enzo life sciences) dissolved in DMSO for 1 h followed by incubation with AD for 8 h, after which cells were harvested and the lysates were processed for Western blotting of LC3B.
Mice
Intratracheal administration of vehicle or AD (0.8 mg/kg body weight, Sigma‐Aldrich, Germany) was performed in C57BL/6 mice every fifth day as described elsewhere 8. Ten mice were included per group. Both the University Animal Care Committee and the Federal Authorities for Animal Research of the Regierungspraesidium Giessen (Hessen, Germany) approved the study protocol.
Electron microscopy
Mice were sacrificed 7 and 14 days after first AD or vehicle challenge for perfusion fixation. In order to represent the whole organ, systematic uniform sampling was performed. Detailed protocols are described in Supporting Information.
Biochemical and statistical analysis
Details of Western blot, immunohistochemistry and sources of antibodies are given in Supporting Information. All data are expressed as means ± SEM of at least five mice for in vivo studies. For in vitro experiments, three or more independent experiments were conducted for AD treatment and triplicate transfections were performed for siRNA studies. Statistical significance was assessed using the Mann–Whitney U test. Significance is indicated as: *p < 0.5, **p < 0.01, ***p < 0.001.
Results
AD induces macroautophagy and increases autophagy flux in murine alveolar epithelial cells
Mouse lung epithelial (MLE) 12 cells were treated with vehicle or with 10 µg/ml AD (according to 7, 8) for 8, 16 and 24 h. We observed a heavily vacuolated cytoplasm in MLE 12 cells treated with AD (Figure 1A). Immunofluorescence analysis for the macroautophagy marker, LC3B as shown in Figure 1B, revealed giant autophagosomes in AD‐treated cells decorated with LC3B; vehicle‐treated cells on the other hand showed occasional punctate staining, but more often a diffuse staining for LC3B. Western blot analysis showed an impressive increase in the conversion of LC3BI to LC3BII, p62 and LAMP2 in AD‐treated cells as compared to vehicle treated or untreated cells (Figure 1C, Supporting Information Figure S1a). It has been indicated that p62 cannot be used as a maker for autophagy in all contexts, especially if it is transcriptionally upregulated 22. Hence we aimed to analyse if transcriptional up‐regulation of p62 might explain the marked increase at the protein level under AD treatment and, not surprisingly, semi‐quantitative and q‐PCR analysis showed significant time‐dependent up‐regulation of p62 mRNA after treating cells with AD, as compared to vehicle‐treated or untreated cells (Supporting Information Figure S1b). Primary AECII treated with AD or vehicle for 24 h also showed significantly increased LC3B and proSP‐C after AD treatment as compared to vehicle‐treated cells (Figure 1D). As an attempt to understand if the fusion event between autophagosomes and lysosomes occurs in response to AD treatment, we performed immunofluorescence for LC3B and LAMP2. Indeed, AD‐treated MLE12 cells showed giant autophagosomes, decorated with LC3B and LAMP2, which co‐localised with each other (Figure 2A), demonstrating fusion between autophagosomes and lysosomes. Further, in order to evaluate the autophagy flux after AD treatment (according to current guidelines 23), MLE12 cells were pre‐treated with the vacuolar ATPase inhibitor bafilomycin A1, followed by AD versus vehicle treatment. Surprisingly, AD‐induced vacuolar morphology completely disappeared upon pre‐treatment of MLE12 cells with bafilomycin A1 (Supporting Information Figure S2a). This observation is in full accordance with a previous study, which reported that bafilomycin A1 significantly suppresses the uptake of AD in different cell types 15. Hence, we excluded bafilomycin A1 and used another protease inhibitor, chloroquine, and a combination of pepstatin and E64D, which have also been suggested for assessing autophagic flux 23. MLE12 cells were pre‐incubated with saturating concentrations of chloroquine (30 µg/ml) 23, for 4 h followed by AD or vehicle treatment. We observed a significant increase in LC3BII and a marked decrease in p62 after AD treatment in the presence of chloroquine (Figure 2B,C), thus clearly indicating a slight but significant increase in autophagy flux after AD treatment. Increase in autophagic flux was also observed when cells were pre‐incubated with pepstatin and E64D, followed by AD treatment (Supporting Information Figure S2b).
Figure 1.
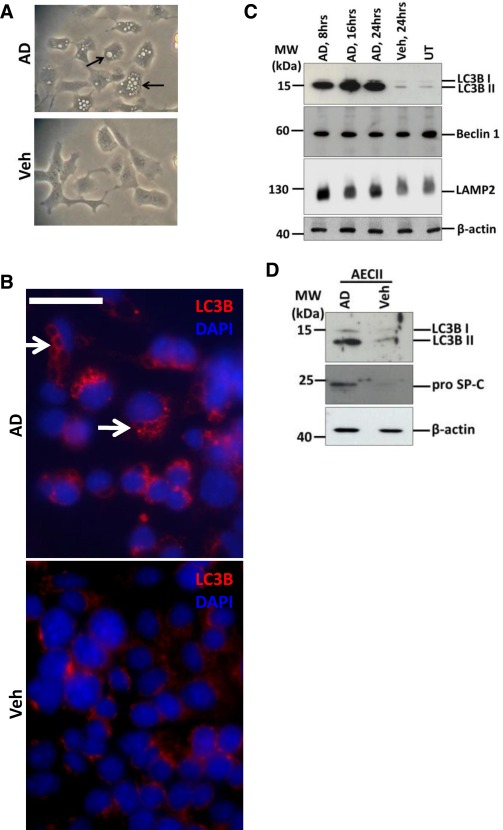
Increase in markers of autophagy in alveolar epithelial cells following AD treatment. (A) Phase contrast images of MLE12 cells 8 h post AD or vehicle treatment. Arrows indicate huge vacuolar structures in AD‐treated MLE 12 cells. (B) Immunofluorescence images for LC3B (red) from MLE 12 cells treated with AD or vehicle for 8 h. Arrows indicate LC3B‐labelled vacuolar structures in AD‐treated cells. (C) Representative Western blot images from cell lysates from AD (8, 16, and 24 h) or vehicle treated (24 h) or untreated MLE12 cells for LC3B, Beclin1, LAMP2 and ß‐actin. (D) Immunoblot images for LC3B and pro SP‐C from AECIIs isolated from C57Bl/6 mice and treated for 24 h with AD or vehicle.
Figure 2.
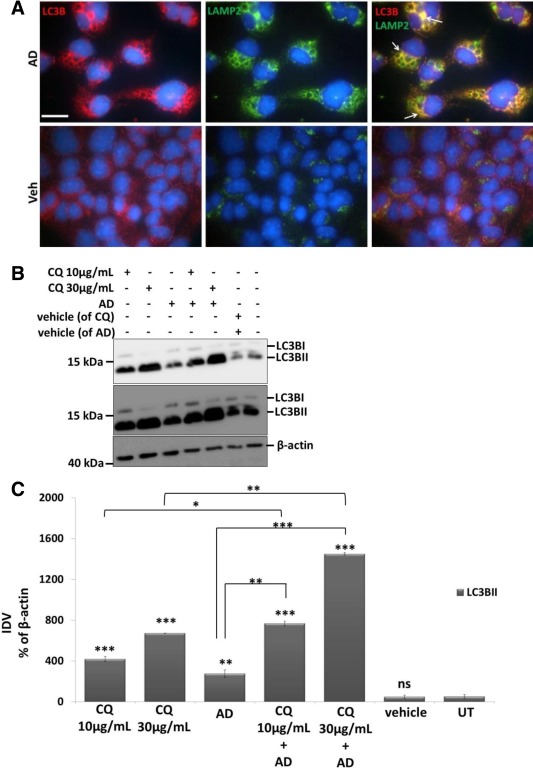
AD‐induced increased autophagy flux in MLE 12 cells. (A) Immunofluorescence images for LC3B (red), LAMP2 (green) and overlay images of both LC3B and LAMP2 from MLE 12 cells treated with AD or vehicle for 8 h. Arrows indicate huge vacuolar structures labelled with both LC3B and LAMP2 (yellow) in AD‐treated cells. Nuclei are stained with DAPI (blue). Scale bar = 50 µm. (B and C) Representative Western blot images for LC3B and β‐actin after pre‐incubation of MLE12 cells with chloroquine (CQ; 10 and 30 µg/ml) for 4 h followed by AD or vehicle treatment for 8 h. Upper and middle panels represent lower and higher exposures (to detect LC3BI), respectively, of the LC3B Western blot membrane and densitometry analysis performed from n = 3 independent experiments. Significance was calculated by comparing all treated groups with untreated cells and then between different treatment groups as indicated. *p < 0.05, **p < 0.01, ***p < 0.001.
AD‐induced AECII apoptosis is mediated by LC3B in vitro
In order to clarify the mechanistic role of macroautophagy in AD‐induced AECII injury and apoptosis, we transfected MLE12 cells for 48 h with a LC3B‐specific and a scrambled siRNA and then treated with AD or vehicle for 8 h. Quite strikingly, cleaved caspase 3 levels, being massively increased in scrambled siRNA‐transfected and AD‐treated cells, returned to almost normal levels in response to LC3B knockdown (Figure 3A,B). Additionally, specific knockdown of LC3B also reversed the AD‐induced proSP‐C elevation almost to control levels (Figure 3A,C). Based on these observations, LC3B, either directly or indirectly, seems to regulate intracellular proSP‐C content in response to AD treatment.
Figure 3.
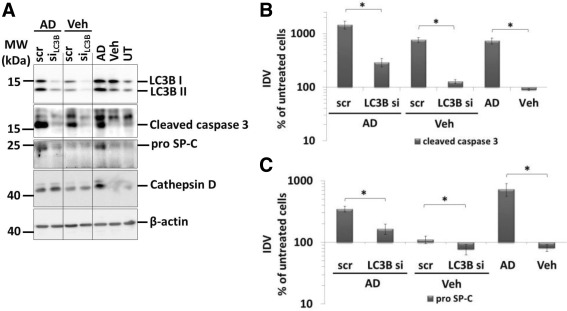
AD‐induced AECII apoptosis is mediated by LC3B. (A) Representative Western blot images for LC3B, cleaved caspase 3, proSP‐C, cathepsin D and β‐actin from MLE12 cells transfected with scrambled siRNA (scr) or siRNA for LC3B (LC3B si) and treated with AD or vehicle for 8 h. Cells treated only with AD, vehicle or untreated cells were included as controls. Different parts from the same Western blots are separated by vertical lines. (B and C) Densitometry analysis of cleaved caspase 3 (B) and proSP‐C (C) to β‐actin ratio was calculated and is given as a percentage of untreated cells. *p < 0.05.
AD induces macroautophagy and the fusion between autophagosome and lamellar bodies in AECII
In order to identify if AD induces macroautophagy in vivo, we performed intratracheal administration of AD every fifth day in mice and sacrificed the animals at days 7, 14, 21 and 28. Development of lung fibrosis was observed in AD‐treated mice from day 7 onwards 8. Analysis of lung homogenates for macroautophagy markers revealed a significant (∼10‐fold) increase in LC3BII as well as p62 levels (∼3‐fold) as compared to vehicle‐treated mice at all the time points analysed (Figure 4A,B, Supporting Information Figure S3a,b). The levels of Beclin1, another well‐known autophagy protein, remained unaltered (Figure 4A). Moreover, the overall level of LAMP2, a lysosomal marker, increased in AD treated lungs (Figure 4A,C), indicating a marked increase in the overall lysosomal content after AD treatment. Immunohistochemistry on serial lung sections revealed numerous LC3B positive AECII in AD‐treated mice as compared to vehicle‐treated controls (Figure 4D, Supporting Information Figure S3c). In order to further confirm this finding, we performed immunogold labelling for LC3B on lung tissues of AD and vehicle‐treated mice. This revealed preferential labelling of LC3B on the limiting membrane and the interior of lamellar bodies (LB) of AECII in AD as well as in vehicle‐treated mice (Figure 5A). After AD treatment, however, gold labelling appeared to be more intense compared to vehicle‐treated mice, confirming the results from our immunohistochemistry staining (Figure 5A). In addition, using EM tomography, we observed a connection between lamellar bodies and autophagosomes via membranes within the AECII of mice treated with AD (Figure 5B,C; Supporting Information video1).
Figure 4.
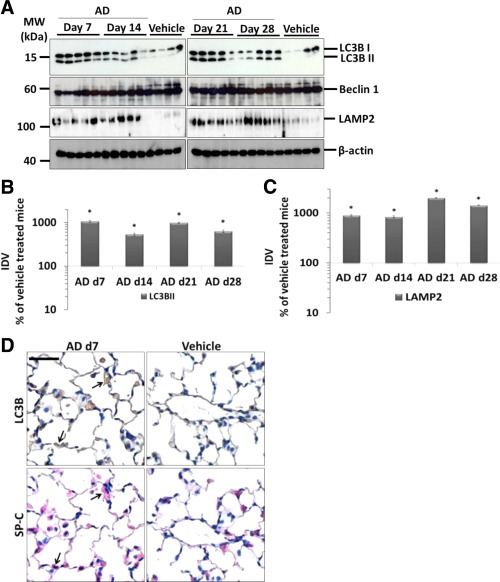
Increase in markers of autophagy in AECIIs from AD‐treated mice. (A) Western blot analysis of lung homogenates of (left) AD‐treated mice at days 7, 14 and vehicle at day 28 and (right) AD‐treated mice at days 21, 28 and vehicle at day 28 for LC3BII, Beclin 1, LAMP2 and β‐actin. (B and C) Graphical representation of densitometry analysis is shown for the LC3B and LAMP2 Western blots shown in (A). Representative blots and analysis from n = 5 mice. *p < 0.05. (D) Representative images of immunohistochemistry for LC3B and pro SP‐C performed on serial lung sections from day 7 AD and vehicle‐treated mice. Arrows indicate AECIIs positive for LC3B. Scale bar = 50 μm; Original magnification of pictomicrographs: x400.
Figure 5.
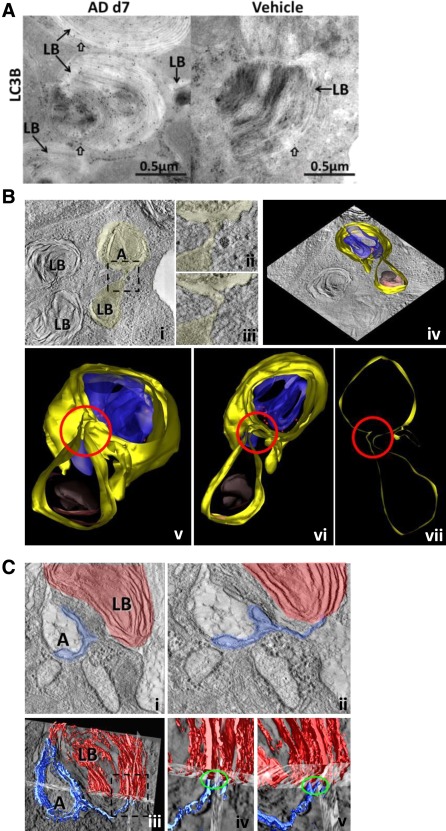
AD induces fusion between autophagosome and lamellar bodies in AECII. (A) Representative images from immunogold labelling for LC3B on lung sections of AD (day 7) and vehicle treated mice. Arrows indicate preferential binding of LC3B to lamellar bodies (LB) in AECII. Block arrows indicate LC3B‐bound gold particles in close proximity to the limiting membrane of LB of AECII in AD and vehicle‐treated mice. Scale bar=0.5 µm. (B) Single slice of EM tomogram showing a direct link between autophagosomes (A) and lamellar bodies (LB) via membranes (i); the boxed area is shown at higher magnification in (ii, iii). Different slices of the tomogram showing that the limiting membrane of the lamellar body and the autophagosome share the same membrane are shown adjacent to these images (iv). In the lower panel (v‐vii), different rotation views of the model of the connection between LB and the autophagosome are shown: the junction is highlighted by a red circle. Colour code: yellow, limiting membranes; blue, membranous content of the autophagosome; red, ribosomes; grey, core of the lamellar body; brown, lipid lamellae. (C) A second example obtained by means of EM tomography showing a direct link between autophagosomes (A) and lamellar bodies (LB) via membranes (i). The membranes of the LB also protrude and form a double membrane layer of an autophagosome which separates a compartment of low density within the autophagosome from the cytosol (ii). The lower row (iii‐v) represents a 3D reconstruction of the membranes of a LB (red) and the membranes of the autophagosome (blue).
Discussion
Our data indicate that AD induces enhanced fusion of autophagosomes to lysosomes as well as to lamellar bodies (which also belong to the lysosomal compartment), increased autophagy flux and markedly increased and LC3B‐dependent apoptosis of alveolar epithelial cells. We were also able to show localisation of LC3B to the lamellar bodies in AECIIs and report that the AD‐driven dysregulation of proSP‐C is dependent on LC3B.
AD is noted for its unusually long half‐life. It accumulates within the lysosomal compartments of various types of lung cells 2, 14, including AECIIs. Similarly, we also observed an increase in the size and number of lamellar bodies within AECIIs and altered intracellular surfactant homeostasis in AD‐treated mice. It was previously reported that the activity of Cathepsin D, a lysosomal enzyme, is increased after AD treatment 24 and we recently showed that Cathepsin D protein levels increase in AD‐treated mice lungs as well as in MLE cells 8. All these observations reflect altered lysosomal homeostasis following AD treatment. However, we did not observe Cathepsin D‐dependent apoptosis of MLE cells in response to AD treatment, indicating the involvement of other lysosome‐related pathways in driving apoptosis 8. Supporting this, we observed extensive vacuolisation and an increase in the autophagy markers when MLE12 cells were directly exposed to AD. This observation is in complete agreement with some previous studies, which reported AD‐induced LC3B positive vacuolar structures in different human cells in culture 25, 26. It may be logical to speculate that increased autophagy could be a mechanism to degrade the deposited drug from the lysosomal compartments. Supporting this concept, in response to AD, we observed a marked increase in LAMP2 protein levels (in mice and cells) and LC3B co‐localisation with LAMP2, a protein that has been shown to be pivotal for autophagosome maturation as well as for autophagosome‐lysosome fusion 27, 28. At first glance, it appears that AD treatment might lead to defective autophagy due to an increase in the autophagy substrate protein p62 following AD treatment. However, we observed up‐regulation of p62 transcript following AD treatment that might compensate for its protein level, questioning its use as a marker for autophagy flux. Further analysis of autophagy flux using inhibitors such as chloroquine and a combination of pepstatin and E64D (but not bafilomycin A1) revealed that AD actually increases autophagy flux in MLE12 cells. While this manuscript was in preparation, Lee et al 17 reported similar observations in other cell types, surprisingly using bafilomycin A1 as the inhibitor for flux studies, in spite of previous reports which impressively showed that bafilomycin A1 suppresses the uptake of AD 15.
AD is metabolised to its chief metabolite, desethylamiodarone (DES), by the P450 enzyme system 29. DES shares some toxicological and pharmacological characters of AD and some studies have documented that DES is much more cytotoxic than AD, causing cell death even at low doses in many cell types 30. Disruption of mitochondrial function, including production of reactive oxygen species (ROS), decrease in mitochondrial membrane potential and cytochrome c release, are well‐documented effects of both AD and DES 31, 32, 33, 34. Since we and others have shown that AD induces autophagy in many cell types, it may not be surprising to note that DES also exerts similar effects. Accumulation of DES in tissues due to prolonged AD treatment may also contribute in part to the dysregulated autophagy pathway and thereby apoptosis of AECIIs in mice in response to AD treatment. Basal autophagy is an extremely important housekeeping process that prevents accumulation of unwanted proteins within the cytoplasm 18, 19, 20. Under certain conditions such as cancer, it is believed to be potentially maladaptive 35 or under certain settings of uncontrolled increase in autophagy as in COPD, it might lead to cell death 19, 36. Hence, it appears important that the autophagy process is well‐balanced. The interplay between autophagy and apoptosis is a matter under continuous discussion and the intricate molecular mechanisms between these two pathways are extremely complex 19, 35. Nevertheless, there exists some evidence that molecules of the autophagy pathway do play a role in executing apoptosis in certain settings. For example, knockdown of LC3 rescued p53‐deficient HCT116 cells from apoptosis 37 and LC3B‐/‐ mice were shown to be resistant to apoptosis after cigarette smoke exposure 38. In line with these findings, we now show that AD‐induced cleaved caspase 3 levels are decreased after LC3B knockdown in MLE12 cells, indicating a role for LC3B in mediating their apoptosis and, probably, also lung fibrosis.
Under physiological conditions, the autophagy pathway orchestrates with other vital cellular pathways. It is hence not surprising that autophagy influences other pivotal cellular mechanisms or vice versa under certain pathological conditions 39. For example, in a previously described rat model of AD‐induced pulmonary toxicity, blockade of angiotensin formation inhibited the development of lung fibrosis. In fact, the angiotensin (AT) 1 receptor antagonist losartan that was used to inhibit AECII apoptosis and thereby lung fibrosis 40 is actually also an inhibitor of autophagy 41, 42.
Another intriguing observation that stems from the current work is the presence of LC3B at the vicinity of the limiting membrane of lamellar bodies within the AECIIs of mouse lungs. Sometimes, especially in response to AD, membranes of these lamellar bodies were observed to be in close connection with phagophores/ autophagosomal structures. It has been shown that an autophagic process is involved in the biogenesis of multilamellar bodies 43. However, proteomic analysis of lung lamellar bodies from rats did not reveal the presence of LC3B either on the limiting membrane or in the lumen of lamellar bodies 44. We nevertheless could convincingly show gold‐labelled LC3B particles both in the lumen as well as on the limiting membrane of lamellar bodies. Based on these observations, it may be reasonable to speculate on the role of LC3B in surfactant homeostasis in normal and, even more, in AD‐injured AECIIs. In complete agreement with this, we could now show that knockdown of LC3B impressively decreased the levels of AD‐induced accumulation of SP‐C in MLE12 cells.
In conclusion, our current study demonstrates a critical role for macroautophagy, especially the marker LC3B, in regulating AECII cell death in response to AD. Although not addressed experimentally here, it appears reasonable to speculate that the magnitude of autophagy flux is also linked to the extent of lung fibrosis. This study enhances our understanding of the molecular mechanisms underlying AD‐induced lung fibrosis. Based on our data, it appears reasonable to target autophagy pathways for improving epithelial survival and minimise AD‐induced lung fibrosis. However, a careful and critical analysis is warranted when targeting the autophagy pathway as too little or too much autophagy is deleterious for the cells.
Supporting information
Supporting Information
Figure S1: (a) Representative Western blot images from cell lysates from AD (8, 16 & 24 hours) or vehicle treated (24 hours) or untreated (UT) MLE12 cells for p62 and ß‐actin. (b). Graphical representation depicting quantification of p62 mRNA using q‐PCR in MLE 12 cells upon AD treatment for the indicated time points, vehicle or untreated cells. Below, a representative agarose gel image from semi‐quantitative RT‐PCR for p62 is shown. β‐actin was used as house‐keeping gene.
Figure S2: (a) Representative phase contrast images of MLE12 cells pre‐treated with bafilomycin A1, AD and both bafilomycin A1 and AD. Scale bar=100 µm. (b) Representative Western blot images for LC3B and β‐actin after pre‐incubation of MLE12 cells with vehicle (DMSO) or pepstatin + E64D (10 µg/mL each) for 1hour followed by AD or vehicle treatments for 8 hours.
Figure S3. (a) Western blot analysis of lung homogenates of (left) AD treated mice at days 7, 14 and vehicle at day 28 and (right) AD treated mice at days 21, 28 and vehicle at day 28 for p62 & β‐actin in AD treated mice. (b) Graphical representation of densitometry analysis is shown for p62 Western blots shown in (a). Representative blots and analysis from n=5 mice. *p < 0.05. (c). Immunohistochemistry performed on serial lung sections from mice treated with AD at 14, 21, 28 days and vehicle at day 28 for LC3B and pro SP‐C showing numerous LC3B positive AECIIs (indicated with arrows) in AD treated mice. Scale bar in all the images=50 μm; Original magnification of pictomicrographs: 400x.
Video1. Transmission electron tomography and modelling of an autophagosome connected to a lamellar body in a type II alveolar epithelial cell. Reconstructed volume is used for modelling of an autophagosome and a lamellar body (grey, core of the lamellar body; brown, lipid lamellae), sharing the same limiting membrane (yellow). The autophagosome exhibits membranous content (blue) and ribosomes (red). The 3D model reveals a junction between the lamellar body and the autophagosome and a remaining opening of the structure indicating a late phagophore/early autophagosome.
Acknowledgements
The authors thank Moritz Wattenbach, Susanne Fassbender and Sabine Fiedler for their superb technical assistance. Part of this work has been undertaken with the enormous support of the European Commission (European IPF Network, funded from 2008 to 2012 through the frame program 7).
Author contributions PM, AG conceived and designed the study. PM performed the experimental work and AG supervised the research. IH, SV, SA, SC, CR, MK participated in the experimental work. LK, JH, CW, MO performed and analysed electron microscopy and tomography.WS participated in the study design. PM, AG composed and finalised the manuscript. All authors were involved in writing the paper and had final approval of the submitted version.
The authors have no conflicts of interest to declare.
References
- 1. Chang SN, Hwang JJ, Hsu KL, et al Amiodarone‐related pneumonitis. J Formosan Med Assoc (Taiwan yi zhi) 2007;106:411–417. [DOI] [PubMed] [Google Scholar]
- 2. Okayasu K, Takeda Y, Kojima J, et al Amiodarone pulmonary toxicity: a patient with three recurrences of pulmonary toxicity and consideration of the probable risk for relapse. Intern Med 2006;45:1303–1307. [DOI] [PubMed] [Google Scholar]
- 3. Ott MC, Khoor A, Leventhal JP, et al Pulmonary toxicity in patients receiving low‐dose amiodarone. Chest 2003;123:646–651. [DOI] [PubMed] [Google Scholar]
- 4. Charles PE, Doise JM, Quenot JP, et al Amiodarone‐related acute respiratory distress syndrome following sudden withdrawal of steroids. Respiration 2006;73:248–249. [DOI] [PubMed] [Google Scholar]
- 5. January CT, Wann LS, Alpert JS, et al 2014. AHA/ACC/HRS Guideline for the Management of Patients With Atrial Fibrillation: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. Circulation 2014;130:2071–2104. [DOI] [PubMed] [Google Scholar]
- 6. Chiovato L, Martino E, Tonacchera M, et al Studies on the in vitro cytotoxic effect of amiodarone. Endocrinology 1994;134:2277–2282. [DOI] [PubMed] [Google Scholar]
- 7. Bargout R, Jankov A, Dincer E, et al Amiodarone induces apoptosis of human and rat alveolar epithelial cells in vitro. Am J Physiol 2000;278:L1039–L1044. [DOI] [PubMed] [Google Scholar]
- 8. Mahavadi P, Henneke I, Ruppert C, et al Altered surfactant homeostasis and alveolar epithelial cell stress in amiodarone‐induced lung fibrosis. Toxicol. Sci. 2014;142:285–297. [DOI] [PubMed] [Google Scholar]
- 9. Martin WJ, II , Kachel DL, Vilen T, et al Mechanism of phospholipidosis in amiodarone pulmonary toxicity. J Pharmacol Exp Ther 1989;251:272–278. [PubMed] [Google Scholar]
- 10. Ashrafian H, Davey P. Is amiodarone an underrecognized cause of acute respiratory failure in the ICU? Chest 2001;120:275–282. [DOI] [PubMed] [Google Scholar]
- 11. Sarma JS, Pei H, Venkataraman K. Role of oxidative stress in amiodarone‐induced toxicity. J Cardiov Pharmacol Ther 1997;2:53–60. [DOI] [PubMed] [Google Scholar]
- 12. Uhal BD, Zhang H, Abdul‐Hafez A, et al Amiodarone induces angiotensinogen gene expression in lung alveolar epithelial cells through activation protein‐1. Basic Clin Pharmacol. Toxicol 2007;100:59–66. [DOI] [PubMed] [Google Scholar]
- 13. Nagata N, Suematsu R, Yoshii C, et al Characterization of amiodarone pneumonitis as related to inflammatory cells and surfactant apoprotein. Chest 1997;112:1068–1074. [DOI] [PubMed] [Google Scholar]
- 14. Somani P, Bandyopadhyay S, Gross SA, et al Amiodarone and multilamellar inclusion bodies. Br J Clin Pharmacol 1987;24:237–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morissette G, Ammoury A, Rusu D, et al Intracellular sequestration of amiodarone: role of vacuolar ATPase and macroautophagic transition of the resulting vacuolar cytopathology. Br J Pharmacol 2009;157:1531–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Piccoli E, Nadai M, Caretta CM, et al Amiodarone impairs trafficking through late endosomes inducing a Niemann‐Pick C‐like phenotype. Biochem Pharmacol 2011;82:1234–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee KY, Oh S, Choi YJ, et al Activation of autophagy rescues amiodarone‐induced apoptosis of lung epithelial cells and pulmonary toxicity in rats. Toxicol Sci 2013;136:193–204. [DOI] [PubMed] [Google Scholar]
- 18. Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000;290:1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mizushima N, Levine B, Cuervo AM, et al Autophagy fights disease through cellular self‐digestion. Nature 2008;451:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. New Engl J Med 2013;368:1845–1846. [DOI] [PubMed] [Google Scholar]
- 21. Mahavadi P, Korfei M, Henneke I, et al Epithelial stress and apoptosis underlie Hermansky‐Pudlak syndrome‐associated interstitial pneumonia. Am J Respir Crit Care Med 2010;182:207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang XJ, Chen S, Huang KX, et al Why should autophagic flux be assessed? Acta Pharmacol Sin 2013;34:595–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Klionsky DJ, Abdalla FC, Abeliovich H, et al Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012;8:445–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li X, Rayford H, Shu R, et al Essential role for cathepsin D in bleomycin‐induced apoptosis of alveolar epithelial cells. Am J Physiol Lung Cell Molec Physiol 2004;287:L46–L51. [DOI] [PubMed] [Google Scholar]
- 25. Renna M, Jimenez‐Sanchez M, Sarkar S, et al Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases. J Biol Chem 2010;285:11061–11067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang L, Yu J, Pan H, et al Small molecule regulators of autophagy identified by an image‐based high‐throughput screen. Proc Natl Acad Sci USA 2007;104:19023–19028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gonzalez‐Polo RA, Boya P, Pauleau AL, et al The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci 2005;118:3091–3102. [DOI] [PubMed] [Google Scholar]
- 28. Saftig P, Beertsen W, Eskelinen EL. LAMP‐2: a control step for phagosome and autophagosome maturation. Autophagy 2008;4:510–512. [DOI] [PubMed] [Google Scholar]
- 29. Shayeganpour A, El‐Kadi AO, Brocks DR. Determination of the enzyme(s) involved in the metabolism of amiodarone in liver and intestine of rat: the contribution of cytochrome P450 3A isoforms. Drug Metab Dispos 2006;34:43–50. [DOI] [PubMed] [Google Scholar]
- 30. Mulder JE, Brien JF, Racz WJ, et al Mechanisms of amiodarone and desethylamiodarone cytotoxicity in nontransformed human peripheral lung epithelial cells. J Pharmacol Exp Ther 2011;336:551–559. [DOI] [PubMed] [Google Scholar]
- 31. Bolt MW, Card JW, Racz WJ, et al Disruption of mitochondrial function and cellular ATP levels by amiodarone and N‐desethylamiodarone in initiation of amiodarone‐induced pulmonary cytotoxicity. J Pharmacol Exp Ther 2001;298:1280–1289. [PubMed] [Google Scholar]
- 32. Varbiro G, Toth A, Tapodi A, et al Concentration dependent mitochondrial effect of amiodarone. Biochem Pharmacol 2003;65:1115–1128. [DOI] [PubMed] [Google Scholar]
- 33. Varbiro G, Toth A, Tapodi A, et al Protective effect of amiodarone but not N‐desethylamiodarone on postischemic hearts through the inhibition of mitochondrial permeability transition. J Pharmacol Exp Ther 2003;307:615–625. [DOI] [PubMed] [Google Scholar]
- 34. Zahno A, Brecht K, Morand R, et al The role of CYP3A4 in amiodarone‐associated toxicity on HepG2 cells. Biochem Pharmacol 2011;81:432–441. [DOI] [PubMed] [Google Scholar]
- 35. Corcelle EA, Puustinen P, Jaattela M. Apoptosis and autophagy: targeting autophagy signalling in cancer cells ‐‘trick or treats’? FEBS J 2009;276:6084–6096. [DOI] [PubMed] [Google Scholar]
- 36. Ryter SW, Lee SJ, Choi AM. Autophagy in cigarette smoke‐induced chronic obstructive pulmonary disease. Expert Rev Respir Med 2010;4:573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Scherz‐Shouval R, Weidberg H, Gonen C, et al p53‐dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci USA 2010;107:18511–18516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen ZH, Lam HC, Jin Y, et al Autophagy protein microtubule‐associated protein 1 light chain‐3B (LC3B) activates extrinsic apoptosis during cigarette smoke‐induced emphysema. Proc Natl Acad Sci USA 2010;107:18880–18885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang Y, Qin ZH. Coordination of autophagy with other cellular activities. Acta Pharmacol Sin 2013;34:585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Uhal BD, Wang R, Laukka J, et al Inhibition of amiodarone‐induced lung fibrosis but not alveolitis by angiotensin system antagonists. Pharmacol Toxicol 2003;92:81–87. [DOI] [PubMed] [Google Scholar]
- 41. Xiao R, Teng M, Zhang Q, et al Myocardial autophagy after severe burn in rats. PloS one 2012;7:e39488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lin L, Tang C, Xu J, et al Mechanical stress triggers cardiomyocyte autophagy through angiotensin II type 1 receptor‐mediated p38MAP kinase independently of angiotensin II. PloS one 2014;9:e89629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hariri M, Millane G, Guimond MP, et al Biogenesis of multilamellar bodies via autophagy. Molec Biol Cell 2000;11:255–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ridsdale R, Na CL, Xu Y, et al Comparative proteomic analysis of lung lamellar bodies and lysosome‐related organelles. PloS one 2011;6:e16482. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Figure S1: (a) Representative Western blot images from cell lysates from AD (8, 16 & 24 hours) or vehicle treated (24 hours) or untreated (UT) MLE12 cells for p62 and ß‐actin. (b). Graphical representation depicting quantification of p62 mRNA using q‐PCR in MLE 12 cells upon AD treatment for the indicated time points, vehicle or untreated cells. Below, a representative agarose gel image from semi‐quantitative RT‐PCR for p62 is shown. β‐actin was used as house‐keeping gene.
Figure S2: (a) Representative phase contrast images of MLE12 cells pre‐treated with bafilomycin A1, AD and both bafilomycin A1 and AD. Scale bar=100 µm. (b) Representative Western blot images for LC3B and β‐actin after pre‐incubation of MLE12 cells with vehicle (DMSO) or pepstatin + E64D (10 µg/mL each) for 1hour followed by AD or vehicle treatments for 8 hours.
Figure S3. (a) Western blot analysis of lung homogenates of (left) AD treated mice at days 7, 14 and vehicle at day 28 and (right) AD treated mice at days 21, 28 and vehicle at day 28 for p62 & β‐actin in AD treated mice. (b) Graphical representation of densitometry analysis is shown for p62 Western blots shown in (a). Representative blots and analysis from n=5 mice. *p < 0.05. (c). Immunohistochemistry performed on serial lung sections from mice treated with AD at 14, 21, 28 days and vehicle at day 28 for LC3B and pro SP‐C showing numerous LC3B positive AECIIs (indicated with arrows) in AD treated mice. Scale bar in all the images=50 μm; Original magnification of pictomicrographs: 400x.
Video1. Transmission electron tomography and modelling of an autophagosome connected to a lamellar body in a type II alveolar epithelial cell. Reconstructed volume is used for modelling of an autophagosome and a lamellar body (grey, core of the lamellar body; brown, lipid lamellae), sharing the same limiting membrane (yellow). The autophagosome exhibits membranous content (blue) and ribosomes (red). The 3D model reveals a junction between the lamellar body and the autophagosome and a remaining opening of the structure indicating a late phagophore/early autophagosome.