

Abstract
Besides tight and specific ligand-receptor interactions, the rate regulation of the formation of molecular assemblies is one of fundamental features of cells. But the latter receives little exploration for developing anticancer therapeutics. Here we show that a simple molecular design of the substrates of phosphatases—tailoring the number of phosphates on peptidic substrates—is able to regulate the rate of molecular self-assembly of the enzyme reaction product. Such a rate regulation allows selective inhibition of osteosarcoma cells over hepatocytes, which promises to target cancer cells in a specific organ. Moreover, our result reveals that the direct measurement of the rate of the self-assembly in a cell-based assay provides precise assessment of the cell targeting capability of self-assembly. This work, as the first report establishing rate regulation of a multiple-step process to inhibit cells selectively, illustrates a fundamentally new approach for controlling the fate of cells.
Keywords: self-assembly, enzyme, rate, selective, cancer
TOC image
Tailoring the number of phosphates on peptidic substrate is able to regulate the rate of self-assembly of the enzyme reaction product. Such a rate regulation allows selective inhibition of osteosarcoma cells over hepatocytes, which promises to target cancer cells in a specific organ. As the first report of using rate regulation of a multiple-step process to inhibit cells selectively, this work illustrates a fundamentally new approach to control the fate of cells.
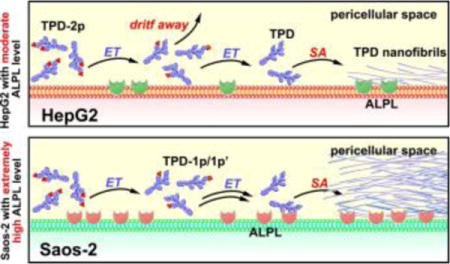
This communication reports targeting cancer cells by the control of the rate of the formation of supramolecular assemblies. Molecular-targeted therapeutics, which mostly relies on tight ligand-receptor interaction or enzyme inhibition, has been a key strategy for developing cancer drugs. However, recent advances in cancer biology have revealed the great complexity of cancers, such as redundant signaling pathways, adaptive drug resistance, genomic instability, intratumoral heterogeneity, and tumor microenvironment.[1] These conceptual advances not only elucidate that the major root of drug resistance in current cancer therapy is the reliance on specific, tight ligand-receptor binding of only one or two molecular targets (e.g., enzymes, receptors, or transcription factors), but also underscore an urgent need of new approaches for cancer therapy. Recognizing that cancer immunotherapy essentially is a form of spatiotemporal controlled apoptosis in human body and enzyme-instructed self-assembly (EISA) is an inherent feature of apoptosis,[2] we have departed from the dogma of specific ligand-receptor binding and are focusing on integrating enzyme transformation (ET) and self-assembly (SA)[3] to generate the fibrils of small molecules as potential anticancer therapeutics.[4]
Based on the development of enzymatic self-assembly and hydrogelation/aggregation of small molecules,[5] we have employed alkaline phosphatases (ALP) to catalyse the formation of nanofibrils of small peptidic hydrogelators in pericellular space of cancer cells, thus achieved selective inhibition of cancer cells, including drug-resistant ones.[4c] This approach turns out to be general since the use of a phosphorylated carbohydrate[6] or phosphorylated nanoparticles[7] as the substrates of ALP for EISA also selectively inhibit cancer cells. These results, undoubtedly, establish that EISA is able to utilize “undruggable” enzymes, such as ALP,[8] for generating molecular nanofibrils to inhibit cancer cells. Despite the promises of the ALP-based EISA for potential cancer therapy, the ubiquitous presence of ALPs in human body presents a unique challenge. As revealed by the studies of mammalian ALPs,[9] being a type of ectoenzymes and present in all tissues throughout the entire body for important biological functions, ALPs exist in four typical isozymes–placental alkaline phosphatase (ALPP), germ cell alkaline phosphatase (ALPP2), intestinal alkaline phosphatase (ALPI), and tissue non-specific alkaline phosphatase (ALPL). Although ALPP overexpresses only on certain cancer cells and not on normal cells, ALPL expresses on normal cells.[10] While the apparent solution for selectivity is to develop a substrate being dephosphorylated by ALPP only, it would not be an easy task for two reasons. First, the inability to develop a highly selective inhibitor for ALPP over ALPL implies that ALPP and ALPL are indiscriminate to their substrates. Second, there is little structural information of ALPL, which makes the design of specific substrates difficult, if not impossible. In addition, a single cell can co-express different isozymes of ALP, albeit at different levels, further complicating the situation. These facts demand a new strategy for precisely targeting cancer cells in a desired organ or tissue, particularly in the case of cancer cells overexpresses ALPL.
To meet the above need, we choose to use the rate of molecular self-assembly to amplify the difference of the expression level of ALPs in cancer and normal cells for targeting cancer cells. As a demonstration of concept, we select two cell lines (i.e., HepG2 (liver hepatocellular carcinoma) and Saos-2 (osteosarcoma)) known to express ALPL and design two kinds of substrates (i.e., monophosphorylated and diphosphorylated peptides, Figure 1) of ALPL for regulating the rate of self-assembly. We choose HepG2 and Saos-2 because the former acts as the model cell of hepatocytes[11] and the latter is known to overexpress ALPL on membrane.[6] Our antibody staining reveals that HepG2 cells, indeed, express less ALPLs than Saos-2 does. Tailoring the rate of self-assembly is able to amplify such a subtle difference in the expression of ALPL. As illustrated in Scheme 1, the rate for generating the self-assembling peptide (i.e., TPD) should be slower from the diphosphorylated substrates (i.e., TPD-2p) than from the monophosphorylated substrates (i.e., TPD-1p) because the former requires dephosphorylation twice. Cell viability assays indicate that, while TPD-2p and TPD-1p exhibit similar inhibitory activities against Saos-2 cells, TPD-2p is more cell compatible than TPD-1p towards HepG2 cells. Moreover, the fluorescent analogues of TPD-2p and TPD-1p (i.e., fTPD-2p and fTPD-1p) confirm that the diphosphorylated substrate results in little molecular assemblies on HepG2 cells, but monophosphorylated substrate affords significant amount of molecular assemblies on HepG2 cells. These results establish that it is feasible to achieve excellent targeting selectivity by using the rate of molecular self-assembly to amplify the difference of ectoenzyme expressions on different cells/tissues. Besides being the first report of using rate regulation of a multiple-step process to inhibit cells selectively, this work illustrates a novel approach for developing therapeutics that may pass by liver and target the tumor site (e.g., sites of osteosarcoma) for selectively killing cancer cells.
Figure 1.
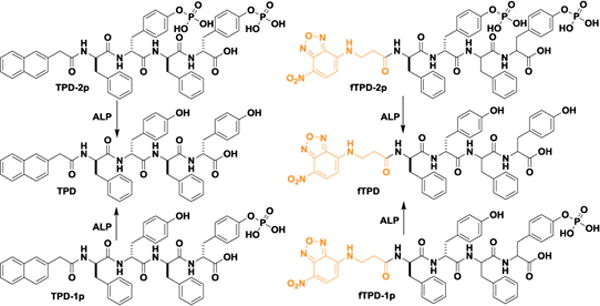
Molecular structures of the tetrapeptide derivatives/precursors with two or one phosphotyrosines (TPD-2p and TPD-1p) and their fluorescent analogs (fTPD-2p and fTPD-1p). These precursors turn into the self-assembling D-peptidic hydrogelators (i.e., TPD and fTPD) after enzymatic dephosphorylation by alkaline phosphatase (ALP).
Scheme 1.
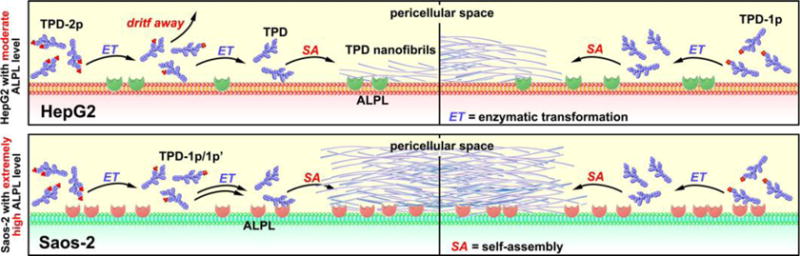
Schematic illustration of the use of the rate of molecular self-assembly (controlled by numbers of enzymatic site) to amplify the difference of the expression level of ALPs in different cell lines.
We have validated that a small D-tripeptide derivative, Nap-D-Phe-D-Phe-D-pTyr, with the tyrosine phosphorylated, selectively inhibits cancer cells due to the different expression of ALPs between cancer (e.g., HeLa) and normal (e.g., HS-5) cells.[4c, 4d, 10b] Based on this result and that aromatic ring of tyrosine promotes self-assembly[4e], we design a D-tetrapeptide derivative (TPD-1p, Figure 1) by inserting a tyrosine between two phenylalanine residues of that D-tripeptide derivative. Besides potentially retaining or even enhancing the self-assembly ability of the D-tetrapeptide, the additional tyrosine residue provides another site for phosphorylation, which affords a diphosphorylated substrate (TPD-2p) for ALP. We expect the ALP-catalysed formation of TPD would be slower from TPD-2p than from TPD-1P. To directly visualize the self-assembly behaviour of TPD generated from these two types of precursors, we design fTPD-2p and fTPD-1p (Figure 1) as the fluorescent analogues of TPD-2p and TPD-1p by replacing the Nap motif with nitrobenzoxadiazole (NBD), a fluorophore known to fluoresce brightly in assembled state.[4e] After preparing the non-commercial starting materials (e.g., Fmoc-phosphotyrosine and NBD-COOH (Figure S1)) prior to solid phase peptide synthesis (SPPS),[12] we used SPPS for the synthesis and high-performance liquid chromatography (HPLC) for the purification and obtained the molecules shown in Figure 1 in good yields (60–80%). LC-MS and 1H NMR confirm the purity and structures of these designed molecules (SI)
Both TPD-2p and TPD-1p dissolve well in water to make transparent solutions at physiological pH 7.4 (Figure S2A). The solubility of TPD-2p is better than that of TPD-1p due to an additional phosphate. In addition, the transmission electron microscopy (TEM) images of their solutions after water evaporation show that TPD-2p forms amorphous aggregates, while TPD-1p self-assembles to make uniform nanofibrils (d = 7±2 nm) (Figure 2). Having the same peptide backbone, either TPD-2p or TPD-1p results in the same self-assembling molecule TPD after its complete dephosphorylation catalyzed by ALP. This feature explains that treating the above solutions with ALP (1 U/mL) leads to the same hydrogels (Figure S2A) with same nanofibrils (d = 5±2 nm) in the hydrogels (Figure 2). TPD-2p takes longer time (Figure S2B) to form a solid hydrogel (1.5 h) than TPD-1p does (0.5 h) due to the need of the second dephosphorylation. fTPD-2p (or fTPD-1p) exhibits similar behavior as that of TPD-2p (or TPD-1p), except that replacing the Nap motif by NBD group slightly decreases the self-assembly ability. As is shown in Figure 2, the solution of fTPD-1p contains both aggregates and nanofibrils (d = 8±2 nm). Upon the treatment of ALP, both the solutions of fTPD-2p and fTPD-1p turn into hydrogels that fluoresce significantly. TEM images reveal similar nanofibrils (d = 4±2 nm) in the networks of the hydrogels (Figure 2).
Figure 2.
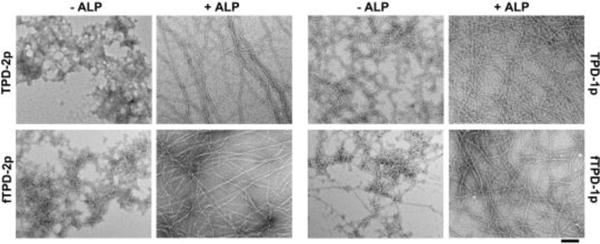
TEM images of aggregates/nanofibrils in the solutions of different precursors (TPD-2p, TPD-1p and fTPD-2p, fTPD-1p) or nanofibrils in the hydrogels formed by treating the solutions of the precursors with alkaline phosphatase (ALP). C = 0.5 wt%, pH = 7.4, [ALP] = 1 U/ml. The scale bar is 100 nm.
Despite that Western blot[13] indicates a significantly higher ALPL expression in Saos-2, it represents the enzyme amount in cell lysate, mainly from the cytosol. We thus use antibody staining/immunofluorescence to confirm the different ALPL levels on the cell membranes of HepG2 and Saos-2 cells. According to the results in Figure 3, Saos-2 cells, indeed, express extremely high level of ALPL, as the whole cell membrane emits bright green fluorescence visualized by confocal fluorescence microscopy. The expression level of ALPLs on HepG2 cell surface is moderate, compared with that of Saos-2 cells. This result justifies our choices of HepG2 and Saos-2 cells for proving the concept outlined in Scheme 1.
Figure 3.
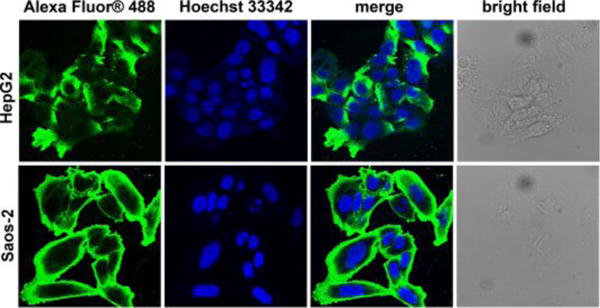
Confocal microscopy images of HepG2 and Saos-2 cells after ALPL antibody staining. Nuclei are stained by Hoechst 33342.
To verify that TPD-2p amplifies the difference of the ALP expression levels in different cell lines for targeting cancer cells, we use TPD-2p to treat HepG2 and Saos-2 cells, and use TPD-1p as a control. After treating HepG2 and Saos-2 cells with TPD-2p or TPD-1p at different concentrations, the cell viabilities at different time (i.e., 24, 48, and 72 hr) (Figure 4 and Figure S3) show that TPD-2p and TPD-1p exhibit similar cytotoxicity on Saos-2 cell, with the 48hr-IC50 values of 130, 150 μM, respectively, but TPD-2p is less cytotoxic than TPD-1p on HepG2 cells (with the 48hr-IC50 value of 300, 180 μM, respectively). More specifically, as shown in Figure 4A, by adding a second phosphate on the molecule of TPD-1p to obtain TPD-2p, we successfully enhance the selectivity for targeting Saos-2 cells over HepG2 cells, from 30% cell viability difference to 48% cell viability difference, 1.6-fold enhancement, at the concentration of 200 μM, and 1.56, 1.82, 1.90 fold enhancement at the concentration of 300, 400, 500 μM, respectively.
Figure 4.
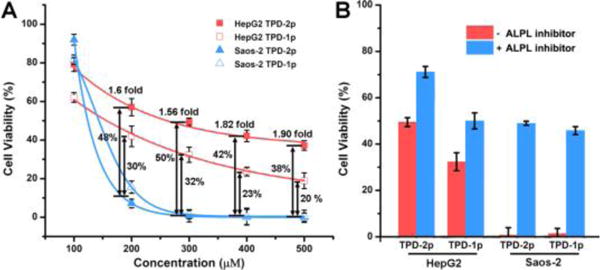
(A) 48-hour cytotoxicity of TPD-2p and TPD-1p on HepG2 and Saos-2 cells at different concentration. The viability differences between the two cell lines are labelled as well. (B) 48-hour cell viability of HepG2 and Saos-2 cells incubated with TPD-2p and TPD-1p (300 μM) with or without ALPL inhibitors (DQB, 2 μM) for 48 h. The initial cell number is 1 × 104 cells/well.
The above results agree with the significantly higher ALPL expression on Saos-2 cells (Figure 3) and support the concept illustrated in Scheme 1. That is, the dephosphorylation of TPD-1p generates TPD for immediate self-assembly, but the dephosphorylation of TPD-2p first results in intermediates (TPD-1p and TPD-1p′, SI), which are more soluble and diffuse more freely than TPD, before the second dephosphorylation to form TPD. High ALPL level on Saos-2 cells warrants rapidly dephosphorylate TPD-2p to TPD before the intermediates diffuse away. So TPD-2p and TDP-1p exhibit almost same inhibitory activities to Saos-2 cells. However, in the case of less ALPL on HepG2, dephosphorylation of TPD-1p forms TPD that self-assembles immediately to result in the nanofibrils on cell surface to cause cell death, but dephosphorylation the TPD-2p firstly generates the intermediates (TPD-1p and TPD-1p′) (Figure S4), which can drift away due to their good solubility before continuously interacting with ALPL to lose another phosphate and then self-assemble. So TPD-2p is more cell compatible than TDP-1p.
To further confirm the ALPL-catalyzed dephosphorylation is responsible for the observed cell inhibition, we incubate HepG2 and Saos-2 cell by TPD-2p and TPD-1p together with ALPL inhibitor[14] (2,5-dimethoxy-N-(quinolin-3-yl)benzenesulfonamide, denoted as DQB and at 2 μM). As shown in Figure 4B and Figure S3, the presence of ALPL inhibitor, DQB, significantly reduces the inhibition of Saos-2 and HepG2 cells. Obviously, DQB rescues more Saos-2 cells than HepG2 cells (e.g., 50% vs 20%) for both TPD-2p and TPD-1p. These results not only validate the critical role of ALPLs for instructing the self-assembly of small D-peptide derivatives to inhibit cancer cells, but also agree with the different ALPL expression levels on Saos2 and HepG2 cells.
The replacement of N-terminal-capped motifs results in reduced self-assembly ability and recued cytotoxicity (Figure S5), but still allows their corresponding self-assembling molecules fTPD to self-assemble and form nanofibrils in pericellular space of cells. According to Figure 5, treating HepG2 cells with fTPD-2p (500 μM) for 12 hours only leads to faint yellow fluorescence in the cells, likely due to endocytosis, while the addition of fTPD-1p into HepG2 cell culture results in strong fluorescence on the surface of the cells. Contrasting to the case of HepG2 cells, the addition of fTPD-2p or fTPD-1p in Saos-2 cell culture leads to the similar result—significant fluorescence on cell surface (Figure 5), which is more fluorescent than that on HepG2 cell treated by fTPD-1p. Because NBD-modified peptides fluoresce intensely in self-assembled nanofibrils,[4e] the yellow fluorescence reflects the amount of nanofibrils formed by EISA of fTPD-2p or fTPD-1p. Moreover, treating the Saos-2 and HepG2 cells with fTPD-2p or fTPD-1p together with ALPL inhibitor DQB (10 μM) for 4 hours prior to imaging, we observe obvious difference between the cells with and without DQB even by naked eyes (Figure S6). Confocal fluorescence images further confirm the inhibitory effect (Figure S7). These results indicate that ALPL catalysed self-assembly to generate the strong fluorescence, agreeing with the cell viability data discussed above.
Figure 5.
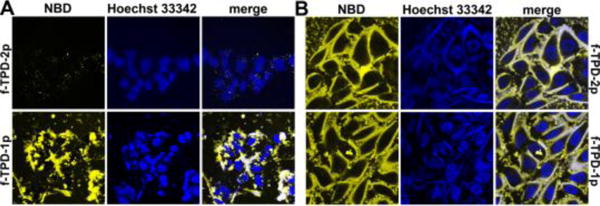
Confocal microscopy images of (A) HepG2 and (B) Saos-2 cells treated with fTPD-2p and fTPD-1p at the concentration of 500 μM for 12 hours. Nuclei are stained by Hoechst 33342.
The direct measurement of the formation rate of the self-assembling molecules in a cell-based assay (SI) provides more accurate assessment of the cell targeting capability of self-assembly. Specifically, we treat the solution of TPD-2p and TPD-1p (500 μM in PBS buffer, pH 7.4) with the cell lysate of HepG2 or Saos-2 (~500 μg total protein) and examine the amount (%) of each component at different time points by analytical HPLC. As shown in Figure 6 and Figure S8, in the first hour, the lysates of Saos-2 and HepG2, respectively, dephosphorylate more than 40 % and less than 5% of TPD-1p. The conversion of TPD-2p follows the similar trend as that of TPD-1p. Moreover, Saos-2 lysate dephosphorylates 50% of TPD-2p or TPD-1p within 2 hours, while HepG2 lysate takes more than 15 hours to consume half of TPD-2p and more than 6 hours to dephosphorylate half of TPD-1p. This significant difference, agreeing with the antibody staining, further supports the cell viability data and fluorescence imaging.
Figure 6.
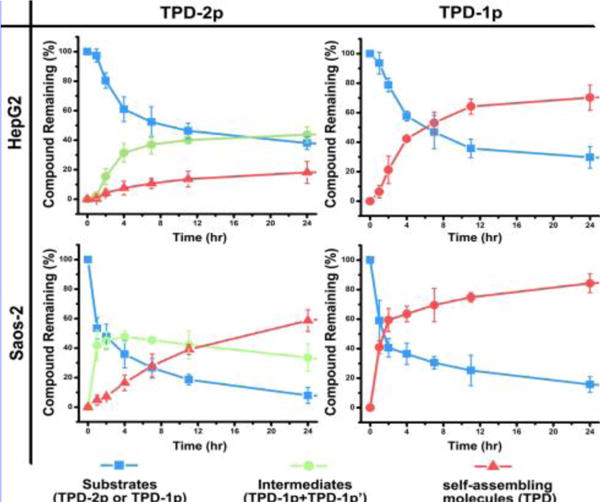
The time-dependent curves for dephosphorylation process of TPD-2p and TPD-1p (500 μM) after incubation with the cell lysate of HepG2 and Saos-2 at 37 °C in PBS buffer.
In conclusion, this work demonstrates a simple strategy to regulate the rate of molecular self-assembly of the enzyme reaction product by tailoring the number of phosphates on peptidic substrates. Our results verify the feasibility to achieve better target selectivity by using the self-assembling rate to amplify the difference in enzyme expressions on different cells/tissues and using molecular engineering to reduce side effects. For example, comparing to TPD-1p, TDP-2p would be a better choice for targeting osteosarcoma (e.g., Saos-2) by intravenous injection because TPD-2p would be more likely than TPD-1p to pass by liver (i.e. hepatocytes like HepG2), which expresses ALPL, to finally reach the tumor site (e.g., Saos-2) (Scheme 1). Although this work focuses on the same isoenzyme, the principle established would be applicable to different isoenzymes that exhibit different reactivity, which promises a new way for controlling dosage in precision medicine.
Supplementary Material
Acknowledgments
This work was partially supported by NIH (R01CA142746). We thank Brandeis EM and Optical Imaging facilities for TEM. JZ is a HHMI international student fellow.
Footnotes
Supporting information for this article is available on the WWW under http://
References
- 1.a) Hanahan D, Weinberg RA. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]; b) Hanahan D, Weinberg RA. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Zhou J, Xu B. Bioconjugate Chem. 2015;26:987–999. doi: 10.1021/acs.bioconjchem.5b00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Yamanaka M, Haraya N, Yamamichi S. Chem Asian J. 2011;6:1022–1025. doi: 10.1002/asia.201000791. [DOI] [PubMed] [Google Scholar]; b) Estroff LA, Hamilton AD. Chem Rev (Washington, DC, U S) 2004;104:1201–1217. doi: 10.1021/cr0302049. [DOI] [PubMed] [Google Scholar]; c) Noguchi T, Roy B, Yoshihara D, Tsuchiya Y, Yamamoto T, Shinkai S. Chem Sci. 2015;6:3863–3867. doi: 10.1039/c5sc00863h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Yang ZM, Xu KM, Guo ZF, Guo ZH, Xu B. Adv Mater. 2007;19:3152–3156. [Google Scholar]; b) Li J, Kuang Y, Shi J, Zhou J, Medina JE, Zhou R, Yuan D, Yang C, Wang H, Yang Z, Liu J, Dinulescu DM, Xu B. Angew Chem Int Edit. 2015;54:13307–13311. doi: 10.1002/anie.201507157. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kuang Y, Shi J, Li J, Yuan D, Alberti KA, Xu Q, Xu B. Angew Chem Int Edit. 2014;53:8104–8107. doi: 10.1002/anie.201402216. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Shi J, Du X, Yuan D, Zhou J, Zhou N, Huang Y, Xu B. Biomacromolecules. 2014;15:3559–3568. doi: 10.1021/bm5010355. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhou J, Du X, Li J, Yamagata N, Xu B. J Am Chem Soc. 2015;137:10040–10043. doi: 10.1021/jacs.5b06181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Yang Z, Gu H, Fu D, Gao P, Lam JK, Xu B. Adv Mater. 2004;16:1440–1444. [Google Scholar]; b) Toledano S, Williams RJ, Jayawarna V, Ulijn RV. J Am Chem Soc. 2006;128:1070–1071. doi: 10.1021/ja056549l. [DOI] [PubMed] [Google Scholar]; c) Yang Z, Liang G, Xu B. Acc Chem Res. 2008;41:315–326. doi: 10.1021/ar7001914. [DOI] [PubMed] [Google Scholar]; d) Miao X, Cao W, Zheng W, Wang J, Zhang X, Gao J, Yang C, Kong D, Xu H, Wang L, Yang Z. Angew Chem, Int Ed. 2013;52:7781–7785. doi: 10.1002/anie.201303199. [DOI] [PubMed] [Google Scholar]; e) Zelzer M, Todd SJ, Hirst AR, McDonald TO, Ulijn RV. Biomater Sci. 2013;1:11–39. doi: 10.1039/c2bm00041e. [DOI] [PubMed] [Google Scholar]; f) Cheetham AG, Keith D, Zhang P, Lin R, Su H, Cui H. Curr Cancer Drug Targets. 2015 doi: 10.2174/1568009616666151130214646. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Cui L, Tokarz D, Cisek R, Ng KK, Wang F, Chen J, Barzda V, Zheng G. Angew Chem, Int Ed. 2015;54:13928–13932. doi: 10.1002/anie.201506171. [DOI] [PubMed] [Google Scholar]
- 6.Pires RA, Abul-Haija YM, Costa DS, Novoa-Carballal R, Reis RL, Ulijn RV, Pashkuleva I. J Am Chem Soc. 2015;137:576–579. doi: 10.1021/ja5111893. [DOI] [PubMed] [Google Scholar]
- 7.Du X, Zhou J, Wu L, Sun S, Xu B. Bioconjugate Chem. 2014;25:2129–2133. doi: 10.1021/bc500516g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blaskovich MAT. Curr Med Chem. 2009;16:2095–2176. doi: 10.2174/092986709788612693. [DOI] [PubMed] [Google Scholar]
- 9.Millan JL. Mammalian Alkaline Phosphatases: From Biology to Applications in Medicine and Biotechnology. WILEY-VCH Verlag GmbH & Co. KGaA; Weinheim: 2006. [Google Scholar]
- 10.a) Fishman WH, Inglis NR, Green S, Anstiss CL, Gosh NK, Reif AE, Rustigian R, Krant MJ, Stolbach LL. Nature. 1968;219:697–699. doi: 10.1038/219697a0. [DOI] [PubMed] [Google Scholar]; b) Uhlen M, Fagerberg L, Hallstroem BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjoestedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CAK, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F. Science. 2015;347:1260419-1260411–1260419-1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 11.Schuetz EG, Schuetz JD, Strom SC, Thompson MT, Fisher RA, Molowa DT, Li D, Guzelian PS. Hepatology. 1993;18:1254–1262. [PubMed] [Google Scholar]
- 12.Chan WC, White PD. Fmoc Solid Phase Peptide Synthesis: A Practical Approach. Oxford Univ. Press; 2000. [Google Scholar]
- 13.http://www.abcam.com/Alkaline-Phosphatase-Tissue-Non-Specific-antibody-EPR4477-ab108337-references.html.
- 14.Dahl R, Sergienko EA, Su Y, Mostofi YS, Yang L, Simao AM, Narisawa S, Brown B, Mangravita-Novo A, Vicchiarelli M, Smith LH, O’Neill WC, Millan JL, Cosford NDP. J Med Chem. 2009;52:6919–6925. doi: 10.1021/jm900383s. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.