

ABSTRACT
Histone deacetylase inhibitors (HDACIs) like valproic acid (VPA) display activity in leukemia models and induce tumor-selective cytotoxicity against acute myeloid leukemia (AML) blasts. As there are limited data on HDACIs effects, we aimed to dissect VPA effects in vitro using myeloid cell lines with the idea to integrate findings with in vivo data from AML patients treated with VPA additionally to intensive chemotherapy (n = 12). By gene expression profiling we identified an in vitro VPA response signature enriched for genes/pathways known to be implicated in cell cycle arrest, apoptosis, and DNA repair. Following VPA treatment in vivo, gene expression changes in AML patients showed concordant results with the in vitro VPA response despite concomitant intensive chemotherapy. Comparative miRNA profiling revealed VPA-associated miRNA expression changes likely contributing to a VPA-induced reversion of deregulated gene expression. In addition, we were able to define markers predicting VPA response in vivo such as CXCR4 and LBH. These could be validated in an independent cohort of VPA and intensive chemotherapy treated AML patients (n = 114) in which they were inversely correlated with relapse-free survival. In summary, our data provide new insights into the molecular mechanisms of VPA in myeloid blasts, which might be useful in further advancing HDAC inhibition based treatment approaches in AML.
KEYWORDS: Acute myeloid leukemia (AML), gene expression, histone deacetylase inhibitor (HDACI), miRNA expression, prognostic marker, valproic acid (VPA)
Abbreviations
- AML
acute myeloid leukemia
- AMLSG
German-Austrian AML Study Group
- BM
bone marrow
- GEP
gene expression profiling
- GSEA
gene set enrichment analysis
- HDAC
histone deacetylase
- HDACI
histone deacetylase inhibitor
- ICE
idarubicin, cytarabine, and etoposide (standard induction chemotherapy)
- LDH
lactate dehydrogenase
- MDS
myelodysplastic syndrome
- RFS
relapse free survival
- RMA
robust multichip average
- VPA
valproic acid.
Introduction
Acute myeloid leukemia (AML) is a genetically heterogeneous clonal stem cell disorder characterized by the accumulation of genetic alterations that lead to aberrant proliferation and differentiation.1 In addition to chromosomal abnormalities and gene mutations, altered gene expression and/or differentially expressed microRNAs (miRNAs) could be identified to be of pathogenic and prognostic relevance.2 Moreover, DNA methylation and histone deacetylation, i.e., epigenetic mechanisms leading to transcriptional gene expression alteration, have been implicated in the pathogenesis of AML recently. In accordance, leukemia-associated epigenetic changes in part explain the deregulated gene expression contributing to leukemogenesis.3
In general, malignant cells including AML blasts show aberrant DNA methylation and/or histone methylation and acetylation patterns, as leukemia specific fusion genes, such as PML-RARA, KMT2A-MLL3T (MLL-AF9), and RUNX1-RUNX1T1, affect epigenetic modifications. Moreover, mutations in epigenetic modifiers, such as TET2, IDH1, IDH2, DNMT3A, ASXL1, and EZH2, were recently identified.4,5 Functional studies could confirm the important pathogenic role of these genes in the deregulation of aberrant epigenetic programs in hematologic malignancies including AML.6
Notably, DNA methylation and histone acetylation are not only of pathogenic relevance, but also of therapeutic interest, since they are reversible, in contrast to genetic alterations. Novel treatment strategies using DNA hypomethylating agents or histone deacetylase inhibitors (HDACIs) have been demonstrated to restore altered gene expression and to be clinically active in a subset of patients with myelodysplastic syndrome (MDS) and AML.7 Notably, only nanomolar doses of demethylating drugs can have antitumor effects on leukemic cells that are accompanied by sustained, genome-wide changes in promoter DNA methylation and gene expression.8 Similarly, HDACIs reverse tumor-associated aberrant epigenetic states and display activity in several malignant cells including myeloid blasts.9,10
Valproic acid (VPA), a well-known drug from the treatment of epilepsy for 3 decades, causes hyperacetylation of the N-terminal tails of H3 and H4 histones in vitro and in vivo by inhibiting the catalytic activity of class I HDACs and by inducing proteasomal degradation of HDAC2.11 It inhibits HDAC activity at concentrations of 0.3–1.0 mM, thereby inducing differentiation and/or apoptosis in murine PML-RARA- and RUNX1-RUNX1T1-driven leukemias, both in AML cell lines as well as in primary AML blasts. While recently several studies examined the underlying VPA-mediated mechanisms in vitro,12,13 the global VPA treatment effects on gene expression in vivo given in combination with conventional chemotherapy in first-line AML patient management have not been extensively studied yet.
Therefore, to gain additional insight into the molecular pathway of VPA treatment in AML, we performed global gene expression and miRNA profiling and integrated in vitro results from myeloid cell line experiments with in vivo data derived from molecularly well-characterized primary AML samples (Table 1) followed by the correlation of our findings with clinical data (Fig. 1).
Table 1.
Detailed clinical characteristics of patients including cytogenetic and molecular genetic information.
| Training Set (n = 18) | Test Set (n = 114) | |
|---|---|---|
| Clinical data | ||
| Gender (male/female) | 12 (66%) / 6 (33%) | 53 (46%) / 61 (54%) |
| Age (y, median) | 50 | 44 |
| AML history | ||
| De novo | 15 (83%) | 100 (88%) |
| Secondary | 1 (6%) | 4 (4%) |
| Therapy related | 2 (11%) | 10 (8%) |
| WBC count (106/L, median) | 35 | 16 |
| Platelet count (106/L, median) | 45 | 64 |
| PB blast count (%, median) | 71 | 32 |
| BM blast count (%, median) | 80 | 75 |
| LDH count (U/L, median) | 562 | 442 |
| Response to induction therapy | ||
| Complete remission | 12 (66%) | 85 (75%) |
| Refractory disease | 6 (33%) | 24 (21%) |
| Early death | 0 (0%) | 5 (4%) |
| Outcome | ||
| OS (days, median) | 448 | 594 |
| EFS (days, median) | 234 | 224 |
| RFS (days, median) | 574 | 391 |
| Cytogenetic risk groups (ELN 2010) | ||
| Favorable | 9 (50%) | 38 (33%) |
| Intermediate-I | 3 (17%) | 37 (32%) |
| Intermediate-II | 3 (17%) | 17 (15%) |
| Unfavorable | 3 (17%) | 17 (15%) |
| NA | 0 (0%) | 5 (4%) |
Abbreviations: WBC, white blood cell count; BM, bone marrow; PB, peripheral blood; LDH, lactate dehydrogenase; OS, overall survival; EFS, event free survival; RFS, relapse free survival; NA, not available
Figure 1.

Illustration of the experimental set up. First, VPA effects were investigated in vitro using myeloid cell lines (n = 5) and in vivo using primary AML samples from patients receiving intensive chemotherapy in combination with VPA (n = 12) or without VPA (n = 6). Based on global gene expression and miRNA profiling, a VPA response signature was generated. This data was intersected with a VPA response predictor generated in 88 diagnostic AML cases who have been also intensively treated in combination with VPA (n = 40) or without VPA (n = 48). Finally, the impact of the refined VPA response predictor genes was validated in an independent cohort of 114 primary AML samples treated intensively in combination with VPA.
Results
Effects of VPA treatment in vitro
To verify our in vitro VPA treatment cell line models, we looked for protein expression changes of HDAC2, UBC8, and acetylated histone H4, known to be influenced by VPA, thereby serving as positive controls for VPA function. As expected, Western Blot analysis showed a dosage dependent downregulation of HDAC2 and upregulation of UBC8 and acetylated histone H4 protein levels following 48 h (data not shown) and 72 h VPA treatment at doses ranging from 10 µM to 10 mM (Fig. 2A), thereby confirming previous reports.
Figure 2.

Protein and gene expression changes related to VPA treatment. (A) Compared to untreated controls, leukemia cell lines treated with VPA (final concentration 1 mM) for 48 h showed a dosage dependent downregulation of HDAC2 and upregulation of UBC8 and acetylated histone H4 protein levels. (B) and (C) Comparative gene expression profiling (comparison of VPA treated vs. untreated cases) revealed an VPA response signature enriched for genes/pathways known to be implicated in cell cycle arrest, apoptosis, and DNA repair (e.g., cell cycle G1/S checkpoint, ATM signaling pathway, and RB tumor suppressor signaling in response to DNA damage). (D) Gene expression changes of primary leukemia patients following in vivo VPA treatment showed results concordant with the in vitro VPA response signature, which was also significantly enriched despite concomitant chemotherapy (P < 0.0001). (E) and (F) Comparative miRNA profiling in cell lines revealed VPA-associated miRNA expression changes likely contributing to a VPA-induced reversion of deregulated gene expression with miR-150 and miR-106b being the most significantly down- (2.7-fold) and upregulated (2.24-fold) miRNAs, respectively.
To further characterize key genes and molecular pathways affected by VPA treatment in vitro, we performed comparative gene expression profiling (GEP) in 5 myeloid cell lines [VPA treated (1 mM for 48 h) vs. untreated]. Class comparison analysis revealed 4876 clones to be significantly differentially expressed among groups at a nominal P-value < 0.05 (Supplementary Table 1). One of the most differentially expressed genes was the cell cycle regulator CDKN1A (p21), known to be influenced by VPA, with a significantly higher expression in VPA-treated cell lines (P < 0.0001; Fig. 2B). Pathway class comparison analysis showed the VPA signature to be enriched for 67/295 investigated Biocarta gene sets (P < 0.05; Supplementary Table 2). These included pathways implicated in cell cycle arrest, apoptosis, and tumor suppression (e.g., cell cycle G2/M checkpoint, cell cycle G1/S checkpoint, ATM signaling pathway, and RB tumor suppressor/checkpoint signaling in response to DNA damage), but, interestingly, also pathways implicated in epigenetic modification (e.g., basic mechanisms of SUMOylation and the PRC2 complex sets long-term gene silencing through modification of histone tails). In accordance, a gene set enrichment analysis (GSEA) revealed similar findings, especially with regard to cell cycle genes, the RB pathway/DNA damage pathway, which were positively correlated with untreated cell lines, whereas genes involved in haematopoietic differentiation and genes reported to be upregulated following HDAC treatment were significantly positively correlated with the VPA-treated cell lines (P < 0.001 for all plots; Fig. 2C).
Effects of VPA treatment in vivo
To correlate our in vitro cell line data with in vivo VPA treatment derived AML data, we performed comparative GEP in 12 primary AML samples with high peripheral blast counts and VPA serum levels [median serum level 59 mg/L (0.41 mM), range: 44–97 mg/L (0.31–0.68 mM)] and compared findings with 6 control cases receiving chemotherapy only (samples were derived following 48 h of chemotherapy and/or VPA treatment and compared with pretreatment samples). Class comparison analysis revealed 2183 clones to be significantly differentially expressed among groups at a nominal P-value < 0.05 (Supplementary Table 3). In accordance with in vitro data, one of the most differentially expressed genes was CDKN1A with a significantly higher expression in VPA-treated AML samples (P = 0.001; Fig. 2D and Supplementary Fig. 1). Notably, the in vitro and in vivo gene VPA response signatures showed a significant overlap of 599 differentially expressed genes with 364 genes being upregulated and 235 genes being downregulated following VPA treatment (P < 0.0001). In addition to CDKN1A, we found a significant upregulation for genes playing a role in differentiation or apoptosis, such as FOXO1A, CD9, STAT4, and TNFRSF21. Several of the most significantly downregulated genes play a role in purine synthesis, such as DHFR and MTHFD1, in histone acetylation, such as HAT1, or DNA damage and DNA repair, such as RAD51, RBBP8, and BRCA1.
Pathway class comparison analysis showed the in vivo VPA signature to be enriched for 93/256 of the investigated Biocarta gene sets (P < 0.05; Supplementary Table 4). These included many pathways also seen in our in vitro data, namely pathways implicated in cell cycle arrest, apoptosis, or tumor suppression (e.g., cell cycle G2/M checkpoint, cell cycle G1/S checkpoint, ATM signaling pathway, or RB tumor suppressor/checkpoint signaling in response to DNA damage); however, the in vivo signature showed no enrichment for pathways implicated in epigenetic modifications (e.g., basic mechanisms of SUMOylation and the PRC2 complex sets long-term gene silencing through modification of histone tails), but similar to our in vitro signature VPA response signature, we found significantly decreased EZH2 expression, a gene playing a central role in the PRC2 complex. Pathways identified in only primary AML samples included the PTEN and the CXCR4 signaling pathways, both known to be deregulated in AML (Supplementary Table 4).
VPA-associated miRNA profiles and delineation of potential target genes
To address the potential impact of miRNAs we additionally performed comparative miRNA expression profiling in the myeloid cell lines. Supervised analysis identified 20 differentially expressed miRNAs correlated with VPA treatment (Supplementary Table 5). Of these, miR-150 and miR-106b were the most significantly down- (2.70-fold) and up-regulated (2.24-fold) miRNAs, respectively (P < 0.05; Fig. 2E). To identify putative target genes of VPA-regulated miRNAs, we performed an integrative analysis comparing predicted target genes of VPA associated miRNAs with our in vitro VPA gene expression signature (see above), as it was shown that mammalian miRNAs predominantly act to decrease target mRNA levels.14 Using different prediction algorithms [MicroCosm Targets (formerly miRBase Targets), miRWalk, DIANA - microT v3.0, TargetScan, and PicTar], we identified mRNAs potentially targeted by one of the VPA regulated miRNAs (miRNA-mRNA interactions were included if they were predicted by at least one of the algorithms). Among the genes significantly downregulated by VPA treatment, e.g., CHEK1 and RAD51, members of the ATM signaling pathway, as well as TYMS were identified as putative targets of miR-15a and miR-16, which were upregulated by VPA. Similarly, of the genes upregulated by VPA treatment HOXA1 and HPSE were identified as potential target genes of miR-99a/b and miR-516-3p, which are downregulated by VPA. Furthermore, GSEA also showed a significant deregulation of miR-106b target genes (Fig. 2F; P < 0.001).
Prediction for VPA response
Next, we wanted to see whether the subset of AML patients in whom the VPA treatment effects were not only short lasting, but had longer lasting impact improving outcome could be predicted in order to better guide treatment decisions in the future. Using in vitro derived GEP VPA response data, we were able to devise a VPA response predictor consisting of 9 genes (ASNS, CXCR4, EGLN3, GARS, GDF15, LBH, SEL1L3, STOM, TNFAIP3) represented by 11 probes sets (Supplementary Table 6). When this predictor was combined with predictors for idarubicine, cytarabine, and etoposide (ICE), it was able to independently predict response to therapy as well as relapse-free survival even when all other prognostic and predictive factors are taken into account in a multivariate analysis (Table 2). Note that because it is based on small sample numbers this analysis is limited and, thus, has to be cautiously interpreted. The predictors were applied to diagnostic gene expression data available from a previous GEP study performed on AMLSG 07–04 patient samples.15
Table 2.
Multivariate analysis of in vitro-derived predictors of response to therapy in a cohort of 88 normal karyotype AML patients.
| RFS (P-value) | RFS (P-value) | RD cycle1 | RD cycle1 | |
|---|---|---|---|---|
| ICE | 0.07 | 0.74 | ||
| V-ICE | 0.005 | 0.06 | ||
| A-ICE | 0.32 | 0.30 | ||
| AV-ICE | 0.72 | 0.47 | ||
| DA | 0.79 | 0.59 | ||
| Combined | 0.048 | 0.02 | ||
| BM Blasts | 0.55 | 0.72 | 0.29 | 0.13 |
| platelets | 0.06 | 0.07 | 0.21 | 0.22 |
| LDH | 0.09 | 0.09 | 0.24 | 0.32 |
| WBC | 0.15 | 0.047 | 0.12 | 0.16 |
| FLT3 ITD | 0.02 | 0.048 | 0.31 | 0.49 |
| FLT3 TKD | 0.22 | 0.20 | 0.55 | 0.62 |
| CEBPA | 0.85 | 0.85 | 0.08 | 0.15 |
| NPM1 | 0.72 | 0.91 | 0.0002 | 0.0002 |
| Total model | 0.002 | 0.002 | 0.045 | 0.018 |
Twenty-one patients had missing values for one or more of the clinical covariates or outcome measures used in the table. Patients were randomized to treatment with ICE, V-ICE, A-ICE or VA-ICE, and the predicted sensitivity to the treatment given was compared to clinical response after first treatment cycle, as well to relapse-free survival (RFS). P-values of association between predictors and outcome are shown. Combined uses a single score for each patient equal to the prediction score for the treatment given. Both V-ICE and the combined score are independent predictors of response, even when all other relevant factors are taken into account.
Evaluation of the VPA response predictor genes in an independent AML cohort
Based on the potential functional relevance of the VPA response associated genes in leukemogenesis, we subsequently investigated the pre-treatment expression of LBH and CXCR4 as VPA response surrogates, as LBH was found to be upregulated both in cell line and primary AML samples and CXCR4 signaling was significantly affected following VPA treatment in vivo. Using qRT-PCR in an independent cohort of 114 VPA treated younger AML cases enrolled in the AMLSG 07–04 treatment trial [median age, 47 y (range 25–60)] the mean-centered normalized log2 transformed expression values for CXCR4 and LBH ranged from −6.30 to 4.14 and −3.81 to 4.58, respectively. Correlation with clinical characteristics and outcome was performed based on: (i) data dichotomized at the median CXCR4 and LBH expression, and (ii) data grouped according to the quartiles of CXCR4 and LBH expression, respectively. Patients with low LBH expression (below median expression of the entire cohort) showed higher LDH serum levels, white blood cell and bone marrow blast counts (median, 514 U/l vs. 354 U/l, P = 0.04; 24.5 G/L vs. 9.1 G/L, P = 0.01; and 80% vs. 60%, P < 0.001). There was no further significant correlation for any of the other clinical characteristics such as age, LDH serum level, peripheral blasts, cytogenetic risk groups, or gene mutations, for either CXCR4 or LBH expression.
For neither CXCR4 nor LBH expression an impact on response to chemotherapy could be identified. However, AML cases with high CXCR4 expression levels (4th quartile) were associated with a significantly longer relapse free survival (RFS; median survival not reached vs. 339 d, P = 0.0496) (Fig. 3A), as opposed to low LBH expression (1st quartile) cases, who showed an inferior RFS (median survival 261 vs. 474 d, P = 0.0464) (Fig. 3B). Notably, there was no overlap between prognostically favorable 4th quartile CXCR4 high expression and LBH 1st quartile low expression cases. In accordance, a comparison of samples based on these respective quartiles showed also a significant difference in RFS (median survival not reached vs. 261 d, P = 0.0182) (Fig. 3C). In contrast, no difference in outcome was seen for a respective LBH and CXCR4 analysis in a previously published cohort of intensively treated AML cases that did not receive VPA16 (Supplementary Table 2).
Figure 3.
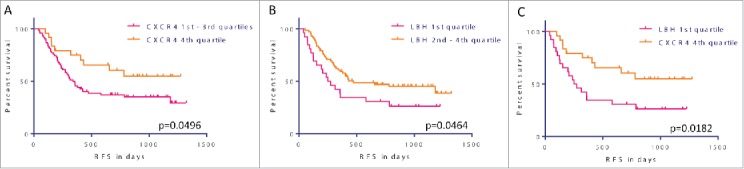
VPA response and outcome prediction. (A) - (C) Kaplan-Meier survival estimates according to CXCR4 and LBH expression. AML cases with high CXCR4 expression levels (4th quartile) were associated with a significantly longer relapse free survival (RFS, median survival not reached vs. 339 d, P = 0.0496) (A), as opposed to low LBH expression (1st quartile) cases, who showed an inferior RFS (median survival 261 vs. 474 d, P = 0.0464) (B). Notably, there was no overlap between prognostically favorable 4th quartile CXCR4 high expression and LBH 1st quartile low expression cases. In accordance, a comparison of samples based on these respective quartiles showed also a significant difference in RFS (median survival not reached vs. 261 d, P = 0.0182) (C).
Discussion
Epigenetic modifications such as DNA methylation and histone acetylation are not only of pathogenic relevance in tumorigenesis and leukemogenesis, but also of rising therapeutic interest, since they are reversible (in contrast to genetic alterations, such as chromosomal aberrations or gene mutations). Novel treatment strategies using DNA hypomethylating agents or HDACIs have been demonstrated to restore altered gene expression and to be clinically active in a subset of patients with myelodysplastic syndrome (MDS) and AML.17
For example, the HDACI VPA causes hyperacetylation of the N-terminal tails of H3 and H4 histones in vitro and in vivo by inhibiting the catalytic activity of class I HDACs and inducing proteasomal degradation of HDAC2, thereby inducing differentiation and/or apoptosis in cell lines as well as in fresh myeloid leukemic blasts.11 Here, we could validate the dosage dependent downregulation of HDAC2 and upregulation of acetylated histone H4 protein levels by VPA treatment at doses that can be achieved in primary patients without considerable toxicity. As previously described, we also could confirm that the downregulation of HDAC2 depends on the E2 ubiquitin conjugase UBC8,18 for which protein levels were upregulated upon VPA treatment. These data suggest a clinical efficacy of VPA treatment at dosages of 0.3 to 1.0 mM.
Next, we delineated miRNA and mRNA profiles associated with VPA treatment and dissected molecular mechanisms of the VPA effect in AML. One of the most upregulated genes by VPA treatment in vitro and in vivo was the cell cycle regulator CDKN1A (p21), previously reported to be induced by VPA.19 While it remains to be shown that VPA directly impacts CDKN1A expression via restoring an activating histone acetylation pattern, for the HDACI suberoylanilide hydroxamic acid (SAHA) a direct CDKN1A upregulation by acetylation of histone H3K9 (as well as histones H3K14, H4K5, H4K8, and H4K12) has been already shown.20 Since four of the 5 analyzed cell lines exhibit TP53 alteration (CMK and HEL monoallelic; NB-4 and HL-60 biallelic), a CDKN1A induction via VPA induced TP53 hyperacetylation, as recently described,21 seems to be unlikely. Further support for a direct regulation of CDKN1A expression stems from our observation that mediators of TGFβ signaling and DNA damage response (such as, SMAD3, SMAD4, and TP53), pathways known to transcriptionally activate CDKN1A, were found downregulated after VPA treatment.13 Alternatively, the induction of expression could also be in part indirectly caused by KLF6,22 which was upregulated by VPA treatment.
Similarly, the results of our pathway class comparison analysis provide further evidence that the major impact of VPA treatment might result from the restoration of deregulated cell cycle regulation. ‘Sustaining proliferative signaling’ has been demonstrated to be a classical hallmark of cancer;23 thus, cell cycle deregulation that sustains chronic proliferation is definitely one of the most fundamental traits of cancer, including leukemia. In normal hematopoiesis, growth-promoting signals that instruct entry into and progression through the cell growth-and-division cycle are tightly controlled by epigenetic mechanisms, thereby ensuring a homeostasis of normal tissue architecture and function. As leukemic blasts become masters of their own destinies by deregulation of these signals, the use of VPA seems to restore at least in part the non-malignant epigenetic profile preventing ongoing proliferation in cells. In accordance, epigenetic deregulation might also contribute to other hallmarks of cancer, such as apoptosis and tumor suppression. These, in turn, influence genomic stability, which seems impacted by VPA treatment, as the VPA signature was also found enriched for pathways implicated in the “ATM signaling pathway” and “RB tumor suppressor/checkpoint signaling in response to DNA damage.” Finally, in line with the hypothesis that the VPA effects are mainly caused by reversing aberrant histone acetylation patterns, we also found the VPA signature enriched for pathways implicated in epigenetic modification (“basic mechanisms of SUMOylation” and “the PRC2 complex sets long-term gene silencing through modification of histone tails”). This observation is in line with previous data demonstrating a positive correlation for sumoylation and HDAC2 activity,24 and an interaction of PRC2 with HDAC1 and HDAC2, respectively.25,26
In accordance with in vitro data, one of the most differentially expressed genes in vivo was also CDKN1A with a significantly higher expression in combined VPA and chemotherapy treated AML samples (P = 0.001), excluding CDKN1A induction as in vitro effect only. This is further underlined by previous observations demonstrating a synergistic effect of VPA and idarubicin or cytarabine.27 Notably, the in vitro and in vivo VPA gene response signatures showed a significant overlap of differentially expressed genes following VPA treatment; among the 364 upregulated genes, we found candidates playing a role in differentiation and apoptosis, such as FOXO1A, CD9, STAT4, and TNFRSF21. Among the 235 downregulated genes, many are associated with purine synthesis (DHFR and MTHFD1), histone acetylation (HAT1), or DNA damage and DNA repair (RAD51, RBBP8, and BRCA1). With regard to MTHFD1 and DHFR, which are also downregulated upon HDACIs in all cell lines,28 it remains elusive if the downregulation is a major result of VPA treatment due to cell cycle arrest or a passenger observation due to reduced proliferation. However, given the fact that the DHFR and MTHFD1 loci can exhibit hyperacetylation of H3K9 and H3K27, their downregulation is unlikely a direct VPA effect. Likewise, the downregulation of RAD51, RBBP8, BRCA1, CHEK1, and TYMS (all carrying hyperacetylated H3K9 and H3K27 marks too) suggests an indirect effect by, e.g., altered miRs, as supposed for CHEK1, RAD51, and TYMS (miR-15a and miR-16) rather than a direct VPA effect. Furthermore, the downregulation of HAT1 by VPA treatment assumes a “negative feedback mechanism” between histone acetylation and histone deacetylation inhibition.
Importantly, pathway class comparison analysis showed the in vivo VPA signature enriched for the same pathways also seen in our in vitro experiments, namely pathways implicated in cell cycle arrest, apoptosis, or tumor suppression (e.g., cell cycle G2/M checkpoint, cell cycle G1/S checkpoint, ATM signaling pathway, or RB tumor suppressor/checkpoint signaling in response to DNA damage). This demonstrates that in vivo VPA patient and in vitro cell line treatment affects identical molecular mechanisms, including a decrease in EZH2 expression, a gene playing a central role in the PRC2 complex. This complex, consisting of EZH2, SUZ12, and EED, leads to transcriptional repressive marks and chromatin compaction, and overexpression of EZH2 is found in advanced and/or metastatic stages of many solid tumors. While activating EZH2 Y641 mutations have been described in lymphoma, in myeloid neoplasms inactivating mutations are associated with poor prognosis.29
Next, integrative analysis of mRNA and miRNA expression showed that part of the VPA effect on cell cycle and differentiation might be indirect by also restoring miRNA regulation networks. Previously, it has been shown that VPA induces the proteasomal degradation of DICER, a key protein in the generation of miRNAs,30 but the upregulation of several miRNAs after VPA treatment might be due to the upregulation of the respective hosting genes. In accordance, upregulation of miR-106b was correlated with a significant enrichment of known miR-106b target genes and recent studies suggest a tumor suppressor function for miR-106b.31,32
To identify patients who might achieve an enduring reversal of the aberrant epigenetic state and who would likely benefit from VPA treatment in addition to chemotherapy, we delineated a 9-gene VPA response predictor comprising genes directly affected by VPA in vitro and in vivo treatment. Of these, deregulated CXCR4 and LBH expression have been associated with solid tumors and leukemias,33,34 and therefore we investigated their expression and prognostic impact in an independent cohort of 114 VPA treated primary AMLs. The CXCR4 high expression group was associated with a significantly higher RFS rate than LBH low expression VPA treated cases. This observation is of note, because high CXCR4 expression has been associated with poor prognosis in AML 35 and, thus, our data suggest that this negative impact might be in part overcome by additional VPA treatment. In contrast, the transcriptional activator LBH has so far not been associated with AML, but recently LBH was linked to the Wnt signaling pathway;36 in addition, LBH was shown to be involved in inducing cell cycle arrest at the G1/S transition and in suppressing the growth of tumors in vivo.34
In summary, our data provide new insights into the molecular mechanisms of VPA effect in vitro and we, for the first time, provide evidence that similar effects are seen in primary AML patients treated with VPA in combination with intensive chemotherapy. Thus, synergistic effects of VPA and other HDAC inhibitors in addition to intensive chemotherapy might further improve RFS in AML, and first clinical trials evaluating the addition of VPA to AML therapy are promising.37,38 To further advance the impact of HDACI in AML treatment approaches, accurate prediction tools like the one presented in this study, which of course warrants further testing in independent studies, also in low-toxicity regimens (VPA in combination with low-dose cytarabine or azacitidine/decitabine), will contribute to a successful integration of VPA/HDACI treatment in individualized AML patient management.
Patients and methods
Cell lines and cell culture
Leukemia cell lines (CMK, HEL, K-562, NB-4, and HL-60) were purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ) and cultured according to their guidelines (www.dsmz.de/human_and_animal_cell_lines/) as previously reported.39
Primary AML samples
Adult AML patient samples (n = 18 and n = 114) were provided by the German-Austrian AML Study Group (AMLSG) with patient informed consent obtained in accordance with the Declaration of Helsinki and institutional review board approval from all participating centers. Samples were enriched for mononuclear cells by Ficoll gradient purification from diagnostic (ipre-treatment) peripheral blood and/or bone marrow. Following enrichment, the percentage of leukemic cells/blasts was at least 80%. All patients were enrolled into the AMLSG treatment protocols AMLSG 07–04 (NCT00151242) for younger patients (<60 years) or AMLSG 06–04 (NCT00151255) for older patients (>60 years). The backbone of both studies was an age-adjusted intensive induction chemotherapy, as previously described for the AMLSG 06–04 protocol.38 Within the AMLSG 07–04 protocol, patients were randomly assigned to receive double induction chemotherapy consisting of 2 cycles of ICE (idarubicin, 12 mg/m2 intravenously, days 1, 3, and 5; cytarabine, 100 mg/m2 continuously intravenously, days 1 to 7; etoposide, 100 mg/m2 intravenously, days 1 to 3) +/− ATRA [by mouth, 45 mg/m2, days 6 to 8, and 15 mg/m2, days 9 to 21 (A-ICE)] +/− valproic acid (VPA) [started at a dosage of 400 mg twice a day by mouth and then adapted according to the biweekly measured serum level, starting from day 3, to obtain a serum level of 100 mg/L (60–150 mg/L) continuously till end of second induction (V-ICE and VA-ICE, respectively)]. Patients achieving a CR or partial remission (PR) after the first induction received a second cycle identical to the first one, according to initial randomization. For consolidation therapy, patients with high-risk AML, defined either by high-risk cytogenetics or induction failure, were assigned to receive allogeneic haematopoietic stem cell transplantation (HSCT) from a matched related donor (MRD) or match unrelated donor (MUD). All other patients were assigned to 3 cycles of high-dose cytarabine (HiDAC; 18 g/m2 per cycle) +/− ATRA [by mouth, 15 mg/m2, days 6 to 21 (A-C)] +/− VPA (V-C and VA-C, respectively) according to the initial randomization. If a MRD was available, an allogeneic HSCT was intended in all patients except those with core-binding factor AML. Detailed clinical characteristics of patients, as well as cytogenetic and molecular genetic analyses results are provided in Table 1.
VPA treatment
Leukemia cells lines (CMK, HEL, K-562, NB-4, and HL-60) were treated with VPA (final concentration 1 mM) for 48 h or left untreated. As scheduled in the protocols, patients were randomized to receive standard induction chemotherapy (idarubicin, cytarabine, and etoposide; ICE) with or without VPA at a dosage of 400 mg/bid i.v. followed by oral administration from day 3 achieving serum levels of 60–110 mg/L (0.42–0.76 mM). After treatment for 48 h, peripheral blood was collected and processed as aforementioned for further analyses. RNA isolation from cell lines and primary leukemic cells was performed using Trizol reagent (Invitrogen).
Immunoblot analysis
Total cell extracts were fractionated on 12% or 4–12% SDS polyacrylamide gels (NuPAGE Bis-Tris Gels, Invitrogen) and blotted to PVDF membranes (Immobilon-P, Millipore). Membranes were reacted with anti-HDAC2, anti-histone H4, anti-UBC8 (Santa Cruz biotechnology), followed by incubation with secondary HRP-linked antibodies (GE Healthcare). Immunoreactivity was determined using ECL Western Blotting detection reagents (GE Healthcare).
Gene expression profiling
Comparative gene expression profiling (GEP; VPA treated vs. untreated) of myeloid cell lines (n = 5) and primary AML samples (n = 12 and n = 6, respectively) was performed using microarray technology according to standard protocols reported elsewhere.40 Primary AML samples were selected based on the peripheral blast counts (median: 63%, after enrichment > 85%) and VPA serum levels [median: 59 mg/L (0.41 mM)].
miRNA expression profiling
To screen miRNA expression in leukemic cell lines we set up a microarray platform using a commercially available oligonucleotide probe set based on version 6.0 of the Sanger miRNA database (mirVana miRNA Probe Set, Ambion) and performed miRNA profiling as previously described.41,42 Normalized miRNA expression data are provided as supplementary information (Supplementary Table 7).
Data analysis of microarray based expression data sets
Supervised analyses to correlate miRNA and/or mRNA expression findings with molecular genetics were carried out using the class comparison features of the BRB-Array Tools Version 3.6.1 (http://linus.nci.nih.gov/BRB-ArrayTools.html) and R Version 2.13.2 (https://www.r-project.org/) as well as the GSEAPreranked module of the GenePattern 3.9.4 platform (http://genepattern.broadinstitute.org/). In order to not miss relevant VPA-associated gene expression changes, low stringency analyses were performed as indicated.
For the VPA response predictor, in vitro gene expression measurements were correlated to growth inhibition [-log(GI50)]. Genes with a Pearson correlation above 0.3 or below −0.3 were considered biomarkers of sensitivity and resistance, respectively, and retained as a response profile for the drug in question. To reduce the number of false positive markers passing the Pearson correlation cutoff, we applied a biological relevance filter that mapped subnetworks of markers known to interact, similar to the approach described by Chuang et al.43
To predict VPA sensitivity in clinical samples, VPA treated cases (n = 40) contained in a previously published microarray data set 15 were normalized using RMA and the expression of each gene in the response profile was used to predict sensitivity [Prediction score = mean (positively correlated genes) – mean (negatively correlated genes)]. Next, the prediction score was normalized to a scale from 0 to 100 by a linear transformation of the prediction score of all patient samples. For prediction of sensitivity to drug combinations, the genes for the individual drugs were joined before calculation of the prediction score, while omitting duplicate genes. Statistical significance of the prediction was calculated using a multivariate linear model on the prediction scores of patients with remission status after the first cycle, or a multivariate cox proportional hazards model on the prediction scores with relapse-free survival time.
Quantitative RT-PCR (qRT-PCR) for mRNA detection
Using the SuperScript III First-Strand Synthesis System (Invitrogen), cDNA synthesis was primed with random hexamer primers according to the manufacturer's protocol. Primers (CXCR4: sense 5′-GGAGGGGATCAGTATATACACTTCAG-3′; anti-sense 5′-GTAGATGGTGGGCAGGAAGA-3′; LBH: sense 5′-CCTACCAGATCTTCCCAGACC-3′, anti-sense 5′-TCGCTGTCTCTTCGCAGTTA-3′; ACTB: sense 5′-AGAGCTACGAGCTGCCTGAC-3′, anti-sense 5′-AGCACTGTGTTGGCGTACAG-3′) were tested by conventional PCR before qRT-PCR (annealing temperature 60°C). Analyses were carried out using the Fast SYBR Green Master Mix (Applied Biosystems) with a 7900HT Fast Real-Time PCR System (Applied Biosystems) in the fast mode and quantity mean values were calculated according to the standard curve method as previously reported.42
Statistical data analysis
Group wise comparisons of the distributions of clinical variables or experimental results were performed using Fisher's exact test, one-sample t test, or unpaired t test, as appropriate. Outcome analyses were performed by Kaplan Meier plot, and curve comparisons were performed using a Log-rank test. A Cox model was used to evaluate prognostic variables. The following variables were evaluated: BM blasts, white blood cell count, platelets, LDH, treatment randomization group, and FLT3, CEBPA and NPM1 mutational status. All tests were 2-tailed, and an effect was considered significant if the P-value was 0.05 or less. Data was visualized and analyzed using GraphPad Prism 5.0 (GraphPad Software) and the statistical software environment R, version 2.14.0, using the R packages rms, version 3.3–1, and cmprsk, version 2.2–2.
Supplementary Material
Disclosure of potential conflicts of interest
Steen Knudsen declares ownership interest in Medical Prognosis Institute.
Acknowledgements
The authors thank all members of the German-Austrian AML Study Group (AMLSG) for their participation in this study and for providing patient samples.
Funding
This study was supported in part by the Deutsche Krebshilfe (DKH 110530), and LB was supported in part by the Deutsche For-schungsgemeinschaft (Heisenberg-Professur BU 1339/8-1).
Availability of supporting data
The complete gene expression microarray dataset is available at the Gene Expression Omnibus data repository (accession number: GSE32240 available at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?accDGSE32240).
References
- 1.Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, et al.. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010; 115:453-74; PMID:19880497; http://dx.doi.org/ 10.1182/blood-2009-07-235358 [DOI] [PubMed] [Google Scholar]
- 2.Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013; 368:2059-74; PMID:23634996; http://dx.doi.org/ 10.1056/NEJMoa1301689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goodell MA, Godley LA. Perspectives and future directions for epigenetics in hematology. Blood 2013; 121:5131-7; PMID:23692857; http://dx.doi.org/ 10.1182/blood-2013-04-427724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel JP, Levine RL. How do novel molecular genetic markers influence treatment decisions in acute myeloid leukemia? Hematology Am Soc Hematol Educ Program 2012; 2012:28-34; PMID:23233557 [DOI] [PubMed] [Google Scholar]
- 5.Dohner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med 2015; 373:1136-52; PMID:26376137; http://dx.doi.org/ 10.1056/NEJMra1406184 [DOI] [PubMed] [Google Scholar]
- 6.Abdel-Wahab O, Levine RL. Mutations in epigenetic modifiers in the pathogenesis and therapy of acute myeloid leukemia. Blood 2013; 121:3563-72; PMID:23640996; http://dx.doi.org/ 10.1182/blood-2013-01-451781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santini V, Melnick A, Maciejewski JP, Duprez E, Nervi C, Cocco L, Ford KG, Mufti G. Epigenetics in focus: pathogenesis of myelodysplastic syndromes and the role of hypomethylating agents. Crit Rev Oncol Hematol 2013; 88:231-45; PMID:23838480; http://dx.doi.org/ 10.1016/j.critrevonc.2013.06.004 [DOI] [PubMed] [Google Scholar]
- 8.Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, Shin JJ, Harbom KM, Beaty R, Pappou E, et al.. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell 2012; 21:430-46; PMID:22439938; http://dx.doi.org/ 10.1016/j.ccr.2011.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell 2012; 150:12-27; PMID:22770212; http://dx.doi.org/ 10.1016/j.cell.2012.06.013 [DOI] [PubMed] [Google Scholar]
- 10.Popovic R, Licht JD. Emerging epigenetic targets and therapies in cancer medicine. Cancer Discov 2012; 2:405-13; PMID:22588878; http://dx.doi.org/ 10.1158/2159-8290.CD-12-0076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, et al.. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 2001; 20:6969-78; PMID:11742974; http://dx.doi.org/ 10.1093/emboj/20.24.6969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Batty N, Malouf GG, Issa JP. Histone deacetylase inhibitors as anti-neoplastic agents. Cancer Lett 2009; 280:192-200; PMID:19345475; http://dx.doi.org/ 10.1016/j.canlet.2009.03.013 [DOI] [PubMed] [Google Scholar]
- 13.Fredly H, Gjertsen BT, Bruserud O. Histone deacetylase inhibition in the treatment of acute myeloid leukemia: the effects of valproic acid on leukemic cells, and the clinical and experimental evidence for combining valproic acid with other antileukemic agents. Clin Epigenetics 2013; 5:12; PMID:23898968; http://dx.doi.org/ 10.1186/1868-7083-5-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009; 136:215-33; PMID:19167326; http://dx.doi.org/ 10.1016/j.cell.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kohlmann A, Bullinger L, Thiede C, Schaich M, Schnittger S, Dohner K, Dugas M, Klein HU, Dohner H, Ehninger G, et al.. Gene expression profiling in AML with normal karyotype can predict mutations for molecular markers and allows novel insights into perturbed biological pathways. Leukemia 2010; 24:1216-20; PMID:20428199; http://dx.doi.org/ 10.1038/leu.2010.73 [DOI] [PubMed] [Google Scholar]
- 16.Bullinger L, Dohner K, Bair E, Frohling S, Schlenk RF, Tibshirani R, Dohner H, Pollack JR. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med 2004; 350:1605-16; PMID:15084693; http://dx.doi.org/ 10.1056/NEJMoa031046 [DOI] [PubMed] [Google Scholar]
- 17.Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol 2009; 27:5459-68; PMID:19826124; http://dx.doi.org/ 10.1200/JCO.2009.22.1291 [DOI] [PubMed] [Google Scholar]
- 18.Kramer OH, Zhu P, Ostendorff HP, Golebiewski M, Tiefenbach J, Peters MA, Brill B, Groner B, Bach I, Heinzel T, et al.. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J 2003; 22:3411-20; PMID:12840003; http://dx.doi.org/ 10.1093/emboj/cdg315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng YC, Lin H, Huang MJ, Chow JM, Lin S, Liu HE. Downregulation of c-Myc is critical for valproic acid-induced growth arrest and myeloid differentiation of acute myeloid leukemia. Leuk Res 2007; 31:1403-11; PMID:17445886; http://dx.doi.org/ 10.1016/j.leukres.2007.03.012 [DOI] [PubMed] [Google Scholar]
- 20.Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci U S A 2000; 97:10014-9; PMID:10954755; http://dx.doi.org/ 10.1073/pnas.180316197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paradis FH, Hales BF. Valproic acid induces the hyperacetylation of P53, expression of P53 target genes, and markers of the intrinsic apoptotic pathway in midorganogenesis murine limbs. Birth Defects Res B Dev Peprod Toxicol 2015; 104:177-83; PMID:26305274; http://dx.doi.org/ 10.1002/bdrb.21149 [DOI] [PubMed] [Google Scholar]
- 22.Calderon MR, Verway M, An BS, DiFeo A, Bismar TA, Ann DK, Martignetti JA, Shalom-Barak T, White JH. Ligand-dependent corepressor (LCoR) recruitment by Kruppel-like factor 6 (KLF6) regulates expression of the cyclin-dependent kinase inhibitor CDKN1A gene. J Biol Chem 2012; 287:8662-74; PMID:22277651; http://dx.doi.org/ 10.1074/jbc.M111.311605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 24.Yang SH, Sharrocks AD. SUMO promotes HDAC-mediated transcriptional repression. Mol Cell 2004; 13:611-7; PMID:14992729; http://dx.doi.org/ 10.1016/S1097-2765(04)00060-7 [DOI] [PubMed] [Google Scholar]
- 25.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev 2002; 16:2893-905; PMID:12435631; http://dx.doi.org/ 10.1101/gad.1035902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van der Vlag J, Otte AP. Transcriptional repression mediated by the human polycomb-group protein EED involves histone deacetylation. Nat Genet 1999; 23:474-8; PMID:10581039; http://dx.doi.org/ 10.1038/70602 [DOI] [PubMed] [Google Scholar]
- 27.Sanchez-Gonzalez B, Yang H, Bueso-Ramos C, Hoshino K, Quintas-Cardama A, Richon VM, Garcia-Manero G. Antileukemia activity of the combination of an anthracycline with a histone deacetylase inhibitor. Blood 2006; 108:1174-82; PMID:16675713; http://dx.doi.org/ 10.1182/blood-2005-09-008086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leclerc GJ, Mou C, Leclerc GM, Mian AM, Barredo JC. Histone deacetylase inhibitors induce FPGS mRNA expression and intracellular accumulation of long-chain methotrexate polyglutamates in childhood acute lymphoblastic leukemia: implications for combination therapy. Leukemia 2010; 24:552-62; PMID:20072153; http://dx.doi.org/ 10.1038/leu.2009.282 [DOI] [PubMed] [Google Scholar]
- 29.Chase A, Cross NC. Aberrations of EZH2 in cancer. Clin Cancer Res 2011; 17:2613-8; PMID:21367748; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-2156 [DOI] [PubMed] [Google Scholar]
- 30.Zhang Z, Convertini P, Shen M, Xu X, Lemoine F, de la Grange P, Andres DA, Stamm S. Valproic acid causes proteasomal degradation of DICER and influences miRNA expression. PloS One 2013; 8:e82895; PMID:24358235; http://dx.doi.org/ 10.1371/journal.pone.0082895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lozano-Velasco E, Vallejo D, Esteban FJ, Doherty C, Hernandez-Torres F, Franco D, Aranega AE. A Pitx2-MicroRNA pathway modulates cell proliferation in myoblasts and skeletal-muscle satellite cells and promotes their commitment to a myogenic cell fate. Mol Cell Biol 2015; 35:2892-909; PMID:26055324; http://dx.doi.org/ 10.1128/MCB.00536-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen S, Chen X, Xiu YL, Sun KX, Zhao Y. Inhibition of ovarian epithelial carcinoma tumorigenesis and progression by microRNA 106b mediated through the RhoC pathway. PloS One 2015; 10:e0125714; PMID:25933027; http://dx.doi.org/ 10.1371/journal.pone.0125714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kremer KN, Peterson KL, Schneider PA, Meng XW, Dai H, Hess AD, Smith BD, Rodriguez-Ramirez C, Karp JE, Kaufmann SH, et al.. CXCR4 chemokine receptor signaling induces apoptosis in acute myeloid leukemia cells via regulation of the Bcl-2 family members Bcl-XL, Noxa, and Bak. J Biol Chem 2013; 288:22899-914; PMID:23798675; http://dx.doi.org/ 10.1074/jbc.M113.449926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Q, Guan X, Lv J, Li X, Wang Y, Li L. Limb-bud and Heart (LBH) functions as a tumor suppressor of nasopharyngeal carcinoma by inducing G1/S cell cycle arrest. Sci Rep 2015; 5:7626; PMID:25557837; http://dx.doi.org/ 10.1038/srep07626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spoo AC, Lubbert M, Wierda WG, Burger JA. CXCR4 is a prognostic marker in acute myelogenous leukemia. Blood 2007; 109:786-91; PMID:16888090; http://dx.doi.org/ 10.1182/blood-2006-05-024844 [DOI] [PubMed] [Google Scholar]
- 36.Rieger ME, Sims AH, Coats ER, Clarke RB, Briegel KJ. The embryonic transcription cofactor LBH is a direct target of the Wnt signaling pathway in epithelial development and in aggressive basal subtype breast cancers. Mol Cell Biol 2010; 30:4267-79; PMID:20606007; http://dx.doi.org/ 10.1128/MCB.01418-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grishina O, Schmoor C, Dohner K, Hackanson B, Lubrich B, May AM, Cieslik C, Muller MJ, Lubbert M. DECIDER: prospective randomized multicenter phase II trial of low-dose decitabine (DAC) administered alone or in combination with the histone deacetylase inhibitor valproic acid (VPA) and all-trans retinoic acid (ATRA) in patients >60 years with acute myeloid leukemia who are ineligible for induction chemotherapy. BMC Cancer 2015; 15:430; PMID:26008690; http://dx.doi.org/ 10.1186/s12885-015-1432-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tassara M, Dohner K, Brossart P, Held G, Gotze K, Horst HA, Ringhoffer M, Kohne CH, Kremers S, Raghavachar A, et al.. Valproic acid in combination with all-trans retinoic acid and intensive therapy for acute myeloid leukemia in older patients. Blood 2014; 123:4027-36; PMID:24797300; http://dx.doi.org/ 10.1182/blood-2013-12-546283 [DOI] [PubMed] [Google Scholar]
- 39.Rucker FG, Sander S, Dohner K, Dohner H, Pollack JR, Bullinger L. Molecular profiling reveals myeloid leukemia cell lines to be faithful model systems characterized by distinct genomic aberrations. Leukemia 2006; 20:994-1001; PMID:16721385; http://dx.doi.org/ 10.1038/sj.leu.2404235 [DOI] [PubMed] [Google Scholar]
- 40.Rucker FG, Bullinger L, Schwaenen C, Lipka DB, Wessendorf S, Frohling S, Bentz M, Miller S, Scholl C, Schlenk RF, et al.. Disclosure of candidate genes in acute myeloid leukemia with complex karyotypes using microarray-based molecular characterization. J Clin Oncol 2006; 24:3887-94; PMID:16864856; http://dx.doi.org/ 10.1200/JCO.2005.04.5450 [DOI] [PubMed] [Google Scholar]
- 41.Rucker FG, Russ AC, Cocciardi S, Kett H, Schlenk RF, Botzenhardt U, Langer C, Krauter J, Frohling S, Schlegelberger B, et al.. Altered miRNA and gene expression in acute myeloid leukemia with complex karyotype identify networks of prognostic relevance. Leukemia 2013; 27:353-61; PMID:22810507; http://dx.doi.org/ 10.1038/leu.2012.208 [DOI] [PubMed] [Google Scholar]
- 42.Russ AC, Sander S, Luck SC, Lang KM, Bauer M, Rucker FG, Kestler HA, Schlenk RF, Dohner H, Holzmann K, et al.. Integrative nucleophosmin mutation-associated microRNA and gene expression pattern analysis identifies novel microRNA - target gene interactions in acute myeloid leukemia. Haematologica 2011; 96:1783-91; PMID:21880628; http://dx.doi.org/ 10.3324/haematol.2011.046888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chuang HY, Lee E, Liu YT, Lee D, Ideker T. Network-based classification of breast cancer metastasis. Mol Syst Biol 2007; 3:140; PMID:17940530; http://dx.doi.org/ 10.1038/msb4100180 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.