

ABSTRACT
Nucleosomes are the basic unit of chromatin. Nucleosome positioning (NP) plays a key role in transcriptional regulation and other biological processes. To better understand NP we used MNase-seq to investigate changes that occur as human embryonic stem cells (hESCs) transition to nascent mesoderm and then to smooth muscle cells (SMCs). Compared to differentiated cell derivatives, nucleosome occupancy at promoters and other notable genic sites, such as exon/intron junctions and adjacent regions, in hESCs shows a stronger correlation with transcript abundance and is less influenced by sequence content. Upon hESC differentiation, genes being silenced, but not genes being activated, display a substantial change in nucleosome occupancy at their promoters. Genome-wide, we detected a shift of NP to regions of higher G+C content as hESCs differentiate to SMCs. Notably, genomic regions with higher nucleosome occupancy harbor twice as many G↔C changes but fewer than half A↔T changes, compared to regions with lower nucleosome occupancy. Finally, our analysis indicates that the hESC genome is not rearranged and has a sequence mutation rate resembling normal human genomes. Our study reveals another unique feature of hESC chromatin, and sheds light on the relationship between nucleosome occupancy and sequence G+C content.
KEYWORDS: G+C content, hESC differentiation, MNase-seq, nucleosome positioning, nucleosome occupancy, sequence mutation, transcription
Introduction
Human embryonic stem cells (hESCs) are pluripotent and have the capacity to differentiate into all adult cell types.1,2 If cultured under appropriate conditions, pluripotent stem cells maintain their genomic integrity, ensuring fidelity in transmission of genetic information from generation to generation.3,4 These unique features (pluripotency and genomic stability) could be attributed to epigenetic mechanisms, which are critical in chromatin remodeling, cell fate specification, and cell identity establishment.5-15 Indeed, studies have shown that hESCs bear unique chromatin composition compared to somatic cells, including prevalent bivalent histone modifications and DNA hydroxymethylation.16-23 Additionally, changes in chromatin architecture, DNA methylation, and histone modification occur frequently and extensively throughout the course of differentiation.5-8,13,17,18,21,22,24
Nucleosomes are the basic unit of chromatin. The canonical nucleosome is composed of 147 bp of DNA wrapped around a histone octamer core consisting of 2 copies each of histone H2A, H2B, H3, and H4.25-27 Nucleosome positioning (NP) along the DNA sequence, the most fundamental structure of chromatin, provides an essential level of epigenetic mechanism regulating biological processes, such as transcription and DNA repair.28,29 NP is influenced by DNA sequence composition in a passive and ATP-independent manner, as well as by the cellular chromatin remodeling machinery in an active and ATP-dependent fashion.
Current epigenetic research on hESC and its differentiation has greatly focused on histone modifications and DNA methylation. This leaves NP,30-37 an equally fundamental epigenetic mechanism, relatively understudied when compared to histone modifications or DNA methylation. For example, the NIH Roadmap Epigenomics project (www.roadmapepigenomics.org) has generated DNA methylation and various histone modification data for numerous human and mouse cell lines and types, including hESCs and their differentiated products. However, very few of them have been subjected to NP analyses.38-40 Although the DNA accessibility of these cell lines/types has been investigated by DNase-seq studies,41 these data do not readily provide genome-wide NP information.42
To more comprehensively understand hESC chromatin and its changes during differentiation, we investigated NP in a well-defined differentiation system, WA09 hESCs → ISL1+ nascent mesoderm (INM) → smooth muscle cells (SMCs), via paired-end sequencing of micrococcal nuclease (MNase)-digested mononucleosomal DNA fragments (MNase-seq). We have been using this system to investigate epigenetic changes of individual genes and genomic loci during hESC differentiation for some time.
Results
Expression analysis supports cell identity and homogeneity of hESC differentiation
We conducted expression microarray experiments to characterize WA09-hESC → INM → SMC differentiation. The principle component analysis (PCA) of the microarray data indicates a clear separation among the 3 cell types (Fig. 1A), supporting homogeneity for each of them. We then investigated established markers characteristic of each cell type and observed the corresponding changes. These include: 1) Silencing of pluripotent markers SOX2, OCT4, and NANOG upon WA09-hESC differentiation; 2) High level expression of INM markers ISL1 and HAND1 in INM; and 3) Significant activation of SMC markers, such as ACTA2, in SMCs (Table S1A).
Figure 1.
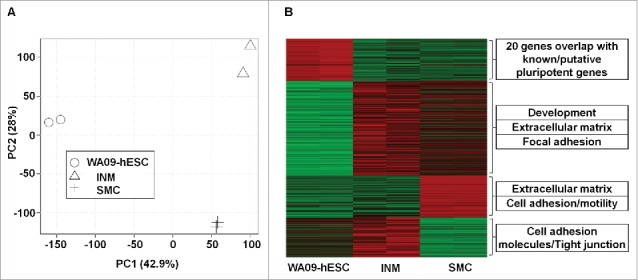
Gene expression analysis supports the cell identity and homogeneity of the WA09-human embryonic stem cell (hESC) → ISL1+ nascent mesoderm (INM) → smooth muscle cell (SMC) differentiation. (A) The principle component analysis (PCA) was performed with the entire transcript set (22,089 in total) included in the Affymetrix Human Gene 1.0 ST array. (B) Heat map indicates genes with significant changes (red: upregulated; green: downregulated) among the 3 cell types and their enriched functional groups (see also Table S1).
These individual marker observations are further corroborated by global analyses. Specifically, at false discovery rate (FDR) of < 0.05, we found a total of 753 genes showing altered expression during the first differentiation stage of WA09-hESC → INM. Among them, while known and putative pluripotent marker genes (20 in total)43 are being silenced (P < 2.2e-16), genes related to development, extracellular matrix (ECM), and focal adhesion are being activated (P < 3.0e-11) (Fig. 1B and Table S1A-B). The 2nd stage of differentiation, INM → SMC, is however characterized by downregulation of cell adhesion and tight junction genes (P < 1.0e-04) (Fig. 1B and Table S1A-B), as well as by upregulation of genes that are consistent with SMC properties, including 29 genes that are associated with smooth muscle, actin, or calponin (P = 1.1e-07) (Table S1A).
MNase-seq analysis for each cell type
To conduct MNase-seq, we treated the cells with MNase that yields >98% mononucleosomes in each cell type, gel-purified the mononucleosomal DNA band (Fig. S1), and sequenced from both ends. In total, we generated 205–226 million pairs of 90 × 90 bp end sequences per cell type (Table S2A). We placed over 94% of these pairs uniquely back onto the human reference genome properly (both ends on the same chromosome, with the right orientation and spanning a reasonable genomic distance) (Table S2A). This results in >11× coverage in both sequence and fragments of mononucleosomes for each cell type (Table S2A).
As a control, we also sequenced randomly sheared genomic DNA fragments of 150–200 bp of WA09-hESC. We achieved the same sequencing and mapping efficiency, reaching a 13X coverage in both sequence and fragments (Table S2A).
Nucleosome occupancy at promoters and other genic sites is influenced by transcript abundance, most strongly in WA09-hESCs
We investigated the relationship of NP with transcript abundance and the sequence G+C content at notable genic sites, including promoters, exon/intron junctions and flanking regions, as well as gene ends. We first sorted the genes into 6 groups based on their transcript abundance, with each group having microarray expression intensity of ≤100, 100–250, 250–500, 500–1000, 1000–3000 or ≥3000 (Fig. 2 and Table S2B). For promoters, which cover regions flanking the transcription start site (TSS), we observed a nucleosome depleted region (NDR) and positioned nucleosomes immediately upstream and downstream of the TSS(Fig. 2A and Fig. S2; Table S2C). The extent of the NDR strongly correlates with the transcript abundance level, with a Pearson correlation coefficient >0.7 for each cell type and of 0.84, the highest, for WA09-hESC (Table S2D). Meanwhile, we also noted a much weaker overall correlation between nucleosome occupancy and the G+C content of promoter sequences (Fig. 2A and Table S2E; see Table S2F for Pearson correlation coefficients at various promoter regions). These correlations apply to exon/intron junctions and flanking regions (Fig. 2B–C). Lastly, a NDR was observed at the transcription termination site (TTS), primarily arising from the very AT-rich sequences there (Fig. 2D and Fig. S2). These findings are consistent with published studies.30,31,38,44 Moreover, our nucleosome occupancy maps closely resemble those of published for hESC (Fig. S3).38
Figure 2.

Nucleosome occupancy at promoters and other genic sites correlates with transcript abundance levels. Nucleosome occupancy and G+C content at promoters (A), exon-intron and intron-exon junctions (B and C), and gene ends (D) are shown. In each panel, genes were divided into 6 groups, represented by the 6 colors as shown, based on their expression intensities from microarray analyses (Table S2B). Genes in each group were then aligned at the indicated positions (e.g., 2 kb upstream and downstream of the TSS). Plots on the left present the average nucleosome occupancy level (Table S2C), represented by the nucleosomal DNA density normalized against the randomly sheared genomic DNA density of WA09-hESC in log2 scale (see Materials and Methods), at each base position shown along the X-axis. Plots on the right denote the average G+C content of the corresponding sequence (Table S2E). See also Fig. S2.
WA09-hESC promoters have the most prominent NDRs among the 3 cell types. For example, the NDRs of the 6 gene expression groups shown in Fig. 2A are significantly larger in WA09-hESCs, when compared to either INM (P = 0.02) or SMC (P = 0.0004) (Table S2D). Meanwhile, no significant difference was observed for the NDRs between INM and SMC (P = 0.2) (Table S2D). Within the NDRs, nucleosome occupancy is actually in negative correlation with the sequence G+C content in WA09-hESCs, with the lowest correlation coefficient reaching −0.57 for WA09-hESC compared to −0.2 for INM and SMC (Table S2F).
Unlike promoters, the gene ends of WA09-hESC have significantly smaller NDRs, compared to INM (P = 0.046) or SMC (P = 0.02) (Fig. 2D and Fig. S2; Table S2D). This indicates the least influence of the AT-rich sequences at TTSs on nucleosome occupancy in WA09-hESC among the 3 cell types.
At exon/intron junctions, WA09-hESC consistently shows the strongest negative correlation between nucleosome occupancy and transcript abundance among the 3 cell types, with Pearson correlation coefficients approximately at −0.8 for WA09-hESC, −0.7 for INM, and −0.6 for SMC (Table S2G). The same conclusion applies to regions flanking the exon/intron junctions (Table S2G). Indeed, the 6 groups of genes shown in Fig. 2B-C, classified based on their transcript abundance, as previously described, are more evenly spaced according to their nucleosome occupancy strength in WA09-hESC. This differs from INM and especially SMC, where nucleosome occupancy divides the 6 gene groups into 3 aggregates, with each having nearly identical nucleosome occupancy level (Fig. 2B-C). The three aggregates roughly correspond to genes being lowly, moderately, or abundantly transcribed, with expression intensity of < 250, 250–500 and > 500, respectively (Fig. 2B–C).
In summary, the findings described above indicate that nucleosome occupancy at promoters and other notable genic regions (Fig. 2) is influenced more by transcript abundance but less by the sequence content in WA09-hESC, when compared to INM and SMC.
Silencing genes, but not activating genes, show substantial changes in promoter NDR
For a total of 118 genes being silenced upon differentiation (genes that are downregulated by >2-fold and have expression intensity of >500 in WA09-hESC but of <500 in both INM and SMC; see Table S3A), we observed a clear change in promoter nucleosome occupancy (Fig. 3A). This is shown by progressively diminishing NDR as WA09-hESC transition to INM and further to SMC (P < 0.001; Table S3B). For a total of 240 genes being activated upon differentiation (genes that are upregulated by ≥2-fold and have expression intensity of <500 in WA09-hESC but of >500 in both INM and SMC; see Table S3A), no such clear change was found (P = 0.2; see Table S3B), although the NDR is slightly more prominent in SMCs (Fig. 3A). No changes were detected for genes of which transcript abundance levels remain largely constant during differentiation (Fig. S4).
Figure 3.
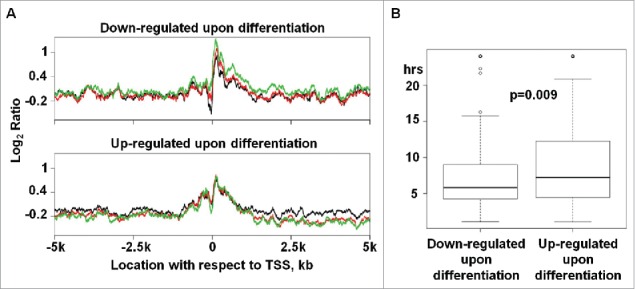
Genes being silenced upon WA09-hESC differentiation display substantial promoter NDR changes and appear to have a shorter mRNA-half life. (A) Top panel presents the average nucleosome occupancy for genes, 118 in total, that are downregulated by >2-fold upon differentiation and have expression intensity of > 500 in WA09-hESC (black) but of < 500 in both INM (red) and SMC (green). Bottom panel presents the average nucleosome occupancy for genes, 240 in total, that are upregulated by ≥ 2-fold upon differentiation and have expression intensity of < 500 in WA09-hESC but of > 500 in both INM and SMC. The average nucleosome occupancy was calculated as described in Fig. 2. See also Table S3A-B and Figs. S4–5. (B) The mRNA half-life distribution of the same downregulated and upregulated genes shown in A (see also Table S3C).
Puzzled by this difference in promoter NP change between gene silencing and activation (Fig. 3A), we investigated the mRNA half-life of these genes. The rationale is that transcript abundance levels, measured here by microarray analysis, are determined by mRNA production, which is regulated by processes including promoter NP, as well as by degradation, which can be assessed with mRNA half-life. Because we did not find global determination of mRNA half-life of hESC in published literature and databases, we instead used values of their mouse counterparts.45 Interestingly, silenced genes have a significantly (P < 0.01) shorter mRNA half-life than activated genes on average (Fig. 3B and Table S3C).
To explore if the promoter NP change shown in Fig. 3A is specific to WA09-hESC differentiation, we investigated genes being silenced or activated during the second stage of the differentiation, INM → SMC. For silenced genes, although the overall nucleosome occupancy increases slightly at the promoter region (Fig. S5), the change at the NDR is not as visible as that of WA09-hESC differentiation (Fig. 3A). For activated genes, no clear NP changes were detected, similar to what is observed during WA09-hESC differentiation (Fig. 3A and S5).
Nucleosome occupancy level at hESC enhancers increases during hESC differentiation
We investigated a total of 5,118 putative active enhancers and 2,287 poised enhancers reported for WA09-hESC by a previous study.46 Both types of enhancers are marked by the presence of chromatin regulators p300 and BRG1, enrichment in histone modification H3K4me1, and low nucleosome occupancy.46 Moreover, while putative active enhancers are enriched in H3K27ac and are near active genes in hESCs, poised enhancers are enriched in H3K27me3 and are associated with genes that are inactive in hESCs but are involved in early embryogenesis.46 Consistent with these features,46 we found that both types of enhancers have the lowest nucleosome occupancy level in WA09-hESCs, but the highest nucleosome occupancy level in SMCs (Fig. 4A). We also examined a total of 7,006 enhancers reported for WA01-hESC.47 We concluded that these enhancers have the lowest nucleosome occupancy level in WA09-hESCs compared to their differentiated derivatives (Fig. 4B), consistent with published findings.38
Figure 4.
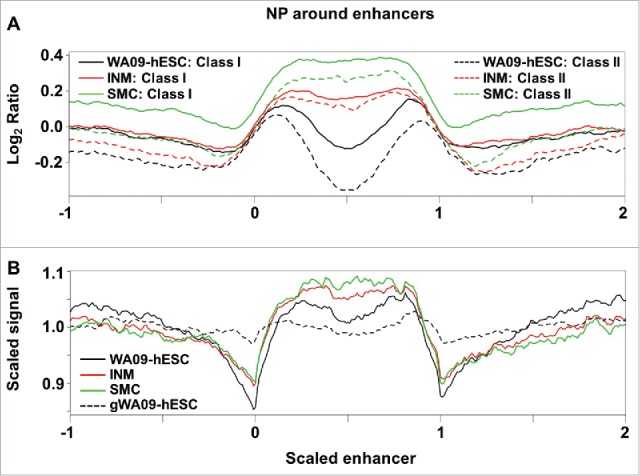
Reported hESC enhancers have the lowest nucleosome occupancy level in WA09-hESC. (A) The nucleosome occupancy level, as defined in Fig. 2, was plotted against scaled enhancer regions of 5,118 putative active enhancers, labeled as “Class I”, and 2,287 poised enhancers, labeled as “Class II”, previously reported46 for WA09-hESC. Scaled enhancer regions are built by treating the entire length of each enhancer as 1 and then expanding to −1 upstream and +1 downstream regions, represented by 0–1, −1–0, and 1–2 respectively in the X-axis. The combined region was then divided into 300 windows, and the average nucleosome occupancy level, represented by the log2 ratio of the Y-axis, was calculated for each window (see Materials and Methods). (B) A total of 7,006 enhancers reported47 for WA01-hESC (H1) were plotted as described.38 Scaled signal, shown in the Y-axis, was obtained by calculating the average fragment coverage in each window as defined in (A) and then performing linear transformation by setting the mean coverage of all windows to 1.
NP at promoters agrees with DNA methylation and histone modification status
We integrated our NP findings with published DNA methylation studies of WA09-hESC.13 As shown in Fig. 5A, only unmethylated promoters display a prominent NDR and, as expected, a large number of methylated genes are silent or lowly expressed. These observations are consistent with the notion that NP precedes DNA methylation in transcription silencing.48 We also incorporated the histone modification data of WA09-hESC from the NIH Roadmap Epigenomics Project. Indeed, promoters enriched with H3K4me3-only exhibit a NP pattern of actively transcribed genes, having a prominent NDR and well positioned nucleosomes respectively upstream and downstream of the TSS (Fig. 5B). Promoters enriched with H3K27me3-only display a NP pattern of silent genes, while promoters with both histone marks have the NP pattern of poised genes (Fig. 5B). These observations support the accuracy of our NP analysis.
Figure 5.

NP at promoters matches published CpG methylation and H3K4me3/H3K27me3 enrichment status in WA09-hESC. (A) Genes were sorted into 5 groups based on their promoter CpG methylation status. Briefly, the methylation data of each CpG within a promoter were obtained from a published study,13 and the promoter status was determined based on the methylation data of all its CpGs. Methylated (M), between methylated and partially methylated (M_P), partially methylated (P), between partially methylated and unmethylated (U_P), or unmethylated (U) correspond to > 80%, 60–80%, 40–60%, 20–40%, or < 20% of the promoter CpGs being methylated respectively. (B) Genes were sorted into 3 groups based on their relative enrichment with H3K4me3 and H3K27me3 as shown, with of ≥ 3, between 3 and −1, and < −1, which largely correspond to open, poised, and closed promoters. For the plot in each panel, the Y-axis represents the average nucleosome occupancy and the X-axis indicates the base pair position, as described in Fig. 2. The table in each panel shows the total number of genes further classified based on their expression intensity (the top row) for each group indicated in the corresponding plot above.
A/T and G/C dinucleotide oscillation was identified in mononucleosomal fragments
Besides transcription, NP is also influenced by sequence composition. In fact, canonical nucleosomal core sequence consists of 147 bp with A/T (AA/AT/TA/TT) and G/C (GG/GC/CG/CC) dinucleotides oscillating at approximately every 10 bp, assisting the winding of the DNA molecule around the histone core.25,26,49 We hence analyzed the sequences of our mononucleosomal fragments of chosen length by studying the genomic regions onto which they were mapped (see Materials and Methods). We indeed observed a strong oscillation in sequence composition for mononucleosomal fragments of 147 bp (Fig. 6A). While not as strong, the oscillation pattern was also visible in mononucleosomal fragments of other lengths. This is especially so for those approximately 10–12 bp increment/decrement away from 147 bp, i.e., 135 bp, 157 bp, 169 bp, and 181 bp (Fig. 6B and Table S4). For mononucleosomal fragments of ≥ 157 bp, the core exhibiting the dinucleotide oscillation is flanked by G+C rich sequences (Fig. 6B and Table S4).
Figure 6.
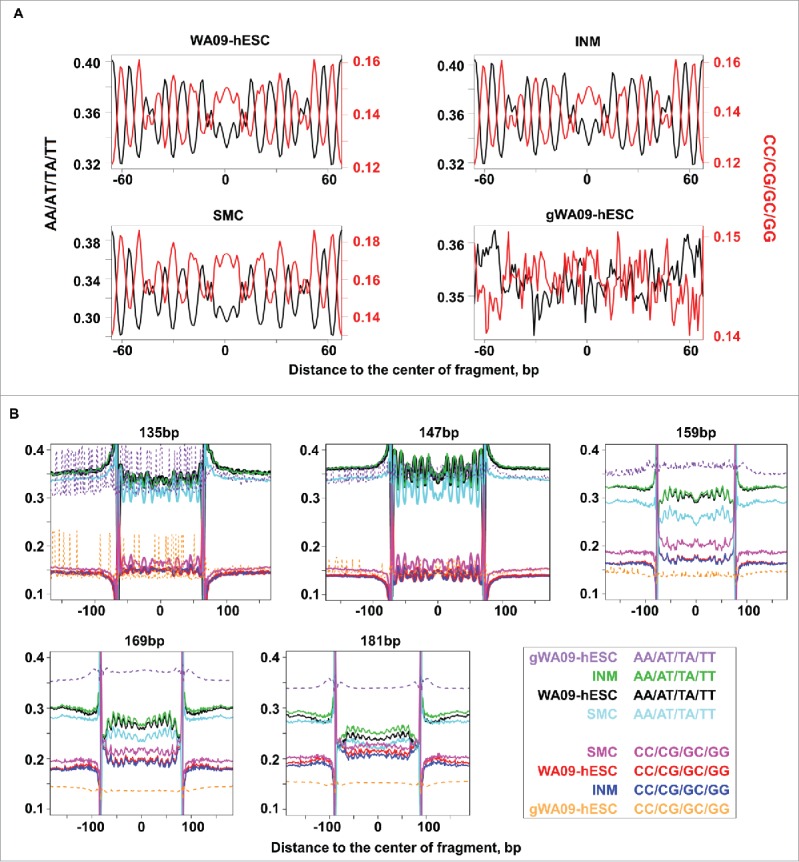
Mononucleosomal fragments display A/T and G/C dinucleotide oscillation. (A) Mononucleosomal (WA09-hESC, INM, and SMC) or randomly sheared genomic DNA and hence non-nucleosomal (gWA09-hESC) fragments of 147 bp were aligned. Then, the average frequencies of AA/AT/TA/TT (black) and GG/GC/CG/CC (red) dinucleotides were computed at each base position and presented at the left and right Y-axis, respectively. The X-axis represents the relative base coordinates from the center of the fragment. Also see Fig. S6 and Table S4. (B) Mononucleosomal or randomly sheared fragments of 135 bp, 147 bp, 159 bp, 169 bp, and 181 bp were plotted. The Y-axis indicates the frequency of either AA/AT/TA/TT or GG/GC/CG/CC of a cell type, with each represented by a color as illustrated. The X-axis extends into neighboring genomic regions. See Table S4 for the complete data of fragments ranging from 135 bp to 209 bp.
Within the same cell type, nearly identical A/T and G/C dinucleotide oscillation patterns were observed between autosomes and the X chromosome (Fig. S6 and Table S4). Among cell types, WA09-hESC resembles INM but slightly differs from SMC (Fig. 6 and Table S4). The oscillation signal is absent in randomly sheared genomic fragments of any length (Fig. 6A and Table S4).
Mononucleosomal sequences have higher G+C content
Consistent with the association of nucleosome occupancy with sequences of higher G+C content,49-51 genomic regions where mononucleosomal fragments were mapped harbor more G and C bases, compared to flanking regions (Fig. 6B and Table S4). Mononucleosomal fragments are also G+C richer than randomly sheared genomic fragments (Fig. 7A). Interestingly, as mononucleosomal fragment length increases, the G+C content of the sequence rises, at each base position (Fig. 6B and Table S4) and in overall percentage (Fig. 7A). These observations were noted in all 3 cell types. Meanwhile, we also found a few differences among the cell types. While mononucleosomal sequences of SMC are about 2% G+C richer (Fig. 6B and 7A and Table S4), WA09-hESC and INM have more mononucleosomal fragments of longer length, with an average length of 165 bp for WA09-hESC and INM vs. 155 bp for SMC (Fig. 7A and Table S5A).
Figure 7.
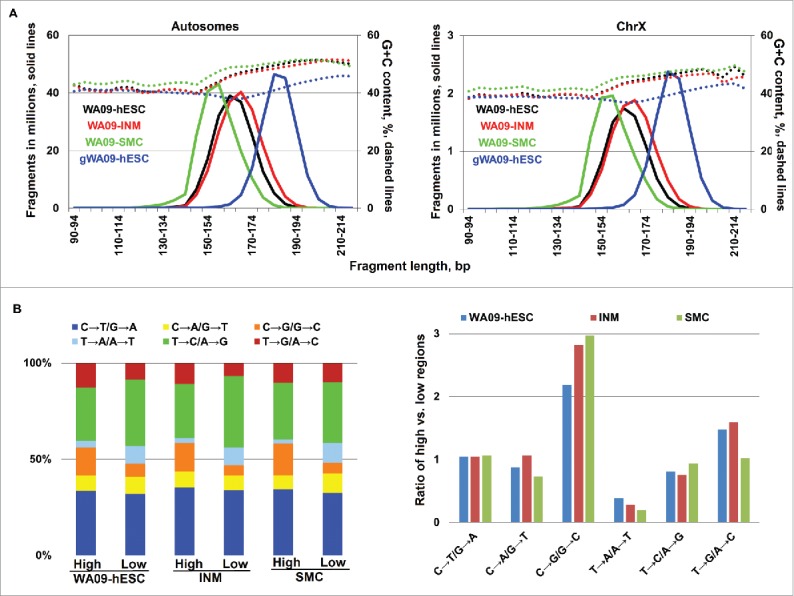
Genomic regions with higher nucleosome occupancy have more C↔G but fewer T↔A substitutions. (A) Mononucleosomal fragments are G+C richer than randomly sheared genomic fragments. The plots also show that the G+C content increases with fragment length, and that hESC and INM have more mononucleosomal fragments of longer length, compared to SMC. See also Table S5A. (B) Genomic regions with higher nucleosome occupancy have more C↔G substitutions and fewer T↔A substitutions, compared to those with lower nucleosome occupancy. The left plot shows the percentage of the 6 base substitution types in genomic regions with higher and lower nucleosome occupancy, represented by “high” and “low,” respectively. The right plot indicates the ratio of each base substitution type in genomic regions with higher vs. lower nucleosome occupancy. See also Table S5B.
Regions with higher nucleosome occupancy have more C↔G but fewer T↔A substitutions
Two factors are currently proposed to explain the higher G+C content of nucleosomal DNA: nucleosome occupancy preference49 or mutation bias.50 In an attempt to test the mutation bias theory,50 we investigated sequence variations in genomic regions with high or low nucleosome occupancy in the 3 cell types. We first utilized sequences from both mononucleosomal fragments and randomly sheared fragments to identify genomic regions with higher or lower nucleosome occupancy in each cell type (see Materials and Methods). We then examined base substitutions (compared to the human reference genome) in both regions. In all 3 cell types, we found that genomic regions with higher nucleosome occupancy have twice as many C↔G mutations but fewer than half T↔A changes (Fig. 7B and Table S5B). This finding is consistent with the notion50 that higher G+C content of nucleosomal DNA arises from mutation bias rather than nucleosome occupancy preference.
WA09-hESC genome appears normal
With the sequences of randomly sheared genomic DNA fragments, we investigated potential structural and sequence variations in the WA09-hESC genome. By examining sequence read pair information, we detected neither translocations nor inversions. We found a comparable amount of copy number variations (CNVs) in the WA09-hESC genome of an individual of Middle East - East European ancestry52 and in a normal (non-diseased) genome of an individual of European ancestry (NA12892) that was sequenced to approximately the same coverage (11.8X). For point/oligo-base changes, we identified about 2.6 million single nucleotide polymorphisms (SNPs) when compared to the human reference genome (Table S5C), with base transitions (C↔T and G↔A) occurring at a higher frequency (∼70%) compared to base transversions (∼30%) (Fig. 8). These observations are comparable to findings from normal human genomes derived from blood samples, including our own analyses with NA12892 and an Asian genome sequenced to a similar coverage53 (Fig. 8 and Table S5C), as well as a published study.54 In summary, the WA09-hESC genome appears normal, with no large genomic changes, and with a sequence mutation rate as low as that identified in normal human genomes, consistent with other studies.3
Figure 8.
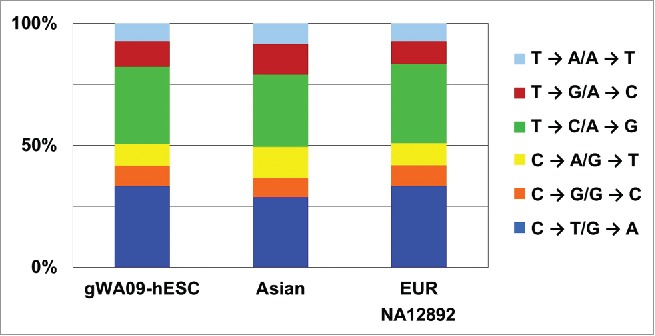
The WA09-hESC genome has a mutation rate as low as normal human genomes. Base substitutions of the WA09-hESC genome are comparable to those of 2 published normal human genomes, EUR (the genome of an individual of an European ancestry) and Asian (the Asian genome53). The base changes were identified by comparing the relevant genomic sequences to the hg18 human genome (also see Table S5C).
Discussion
We performed MNase-seq to investigate NP in a well-defined hESC differentiation system, for which the identity and homogeneity of each cell type was supported by global and individual marker expression analyses. Furthermore, the hESCs under study have maintained their genomic stability and integrity, with no detected genomic rearrangements and with a sequence mutation rate comparable to that of normal human genomes derived from blood samples. Our study contributes to the understanding of the unique chromatin and sequence composition of nucleosomal DNA in hESCs.
Many of our NP findings concur with published studies,13,16,17,23,30,31,38,44 indicating the accuracy of our MNase-seq pipeline. For example, hESC enhancers display lower nucleosome occpancy in hESCs than in their differenciated derivatives,38,46 and NP at promoters agrees with published histone modification and DNA methylation patterns.13 Futhermore, only promoters with unmethylated CpGs exhibit a prominent NDR, consistent with the concept that NP precedes DNA methylation in gene silencing.48 Notably, similar to our histone and DNA methylation findings,16-23 our NP study indicates the uniqueness of hESC chromatin. At promoters and other notable genic sites, nucleosome occupany shows a stronger correlation with transcript abundance but is less influenced by the sequence content in hESC than its differenciated products. We have also detected a shift of NP to G+C richer regions as hESCs differenciate to SMCs. Consistent with the in vivo and in vitro NP comparsion study in C. elegans,55 our observations indicate that chromatin remodeling may be more active in hESC, compared to its differentiated derivatives. Futhermore, our analyses reveal a dynamic NP in hESC, which decreases progressively once differentiation starts. This is consistent with the observation that hESC contains more genes in a poised chromatin state,16,23 to readily resume transcription or to be more irreversibly silenced upon differentiation.
An interesting finding from our study is that genes being silenced, but not genes being activated, show a visible change in nucleosome occupancy at the promoter upon hESC differentiation. This finding was also reported by another group38 and is perhaps related to the possibility that hESCs have the most active chromatin remodeling activity, which decreases as hESCs transition into other cell types, as previously discussed. Interestingly, silenced genes appear to have a shorter mRNA half-life on average, compared to activated genes. Function-wise, while silenced genes encode many known and putative pluripotent markers, activated genes are significantly enriched in ECM-associated groups. A recent study56 reports that during hESC differentiation, genes that have undergone A/B chromatin compartment switching and show correlated gene expression changes are mostly ECM-related and have low G+C content at their promoter. Studies in yeast reveal an association between a gene's mRNA half-life and its promoter sequence.57,58 Based on these publications and our observations, we hypothesize that, for silenced genes, which are enriched in functions maintaining pluripotency, expression decreases by increasing nucleosome occupancy and other repressive epigenetic modifications at the promoter, suppressing transcription, and by rapid decay of existing mRNA molecules. For activated genes, which are enriched in functions of ECM-building, expression increases via higher levels of chromatin change, e.g., B to A compartment switch. Further studies are needed to test this hypothesis.
Our genome-wide analyses identified NP-associated sequence features. These include A/T and G/C dinucleotide oscillation of canonical nucleosomal core sequence25,26 in our mononucleosomal fragments of each cell type. Notably, we found twice as many G↔C substitutions, but fewer than half of A↔T substitutions, in genomic regions with higher nucleosome occupancy than regions with lower nucleosome occupancy. This finding may explain why nucleosomal DNA have higher G+C content.49-51 This result is consistent with a recent study reporting that the higher G+C content of nucleosomal DNA arises from mutation bias rather than nucleosome occupancy preference.50 Frequent G↔C substitutions in cancer have been shown to be associated with over-activity of the APOBEC cytidine deaminases.59 We do not know if any link exists between APOBEC activity and frequent G↔C changes in genomic-regions of higher nucleosome occupancy. One interesting observation is that, compared to WA09-hESC and INM, nucleosomal DNA of SMC is richer in G+C. Meanwhile, 3 APOBEC members, APOBEC3G, APOBEC3C, and APOBEC3F, are expressed 2–3-fold higher in SMC. Whether this is a coincidence or has functional implications remains to be determined. Finally, we caution that the observed high G+C content of nucleosomal DNA may partially arise from the digestion bias of MNase toward AT-rich sequences (although one study has reported that this bias is not substantial in their nucleosome mapping60), besides the strong intrinsic association between nucleosome occupancy and high G+C sequences.49-51 Therefore, our conclusions need to be validated in future studies that correct MNase sequence digestion bias.
Materials and methods
WA09-hESC differentiation and microarray analyses
WA09-hESCs were maintained in StemPro defined media (Invitrogen). Differentiation to INM and SMCs was achieved by supplementation of the defined media with Wnt3a (25 ng/ml) and BMP4 (50 ng/ml) for 4 and 21, days respectively. RNA was purified from approximately 5 million cells per sample using the Qiagen RNeasy Plus Mini kit (Cat. No. 74134). Then, high quality (a 260/280 absorbance ratio of ∼2.0, non-degraded, and free of genomic DNA contamination) samples were analyzed using the Affymetrix Human Gene 1.0 ST array with biological replicates. The moderated t-test implemented in the ‘limma’ package61 was used to identify differentially expressed genes between the cell types, and P-values were adjusted for multiple-hypothesis testing with the Benjamini and Hochberg method.62 PCA analysis was performed using R (www.R-project.org). The packages used are available at the Bioconductor site (www.bioconductor.org).
MNase-seq and mapping sequence reads
Chromatin was processed by following a published protocol31 with MNase (Worthington Biochemical Corp.) to yield >95% mononucleosomal DNA (Fig. S1). DNA samples with a 260/280 absorbance ratio around 1.8 were used for downstream applications. DNA fragments of approximately 150 bp were gel purified as described,63 and sequenced from both ends to yield 90 × 90 bp sequence read pairs using Illumina Genome Analyzer at the BGI. As a control, genomic DNA was extracted following the same protocol but without MNase-digestion, randomly sheared to 100–200 bp fragments, and sequenced from both ends. All read pairs were then mapped to the human genome (hg18) using the Burrows-Wheeler Aligner (BWA) tool64 with the default parameters documented in the bwa-0.5.9 version. Read pairs uniquely placed onto the genome were used for further analyses.
Nucleosome occupancy calculation
The KnownGene annotation (hg18) downloaded from the UCSC genome database (genome.ucsc.edu) was used to match the genes of the Affymetrix Human Gene 1.0 ST array in genomic sequence coordinate. A total of 33,271 transcripts (17,592 genes) with more than 90% overlapping in coordinate were then chosen for nucleosome occupancy analysis. Nucleosome occupancy for base i in the genome was estimated by , where and respectively represent the total count of mononucleosomal fragments (m) and randomly sheared genomic fragments (g) covering base i, with and being the corresponding genome-wide average of and . Data of randomly sheared genomic fragments of WA09-hESC were used in calculations of all 3 cell types, under the assumption that the genome remains the same during the 21-days of cell differentiation. The TSS, exon-intron/intron-exon junction, and gene end data were extracted from the UCSC KnownGene annotation. Average G+C content of each corresponding region was calculated based on the hg18 genome.
Promoter CpG methylation and histone modification analysis
Bisulfite sequencing data of WA09-hESC were obtained from a published study.13 Both H3K4me3 (GSM605316) and H3K27me3 (GSM706066 and GSM667622) ChIP-seq reads of WA09-hESC were downloaded from the NIH Roadmap Epigenomics site (www.roadmapepigenomics.org), and mapped to the hg18 genome with BWA as previously described. Uniquely mapped reads were selected for further analyses.
Gene functional analysis and mRNA half-life
Gene functional annotation and enrichment were analyzed by DAVID.65 Mouse mRNA half-life data were obtained from a published study.45 The mouse-human gene conversion was achieved using the Human and Mouse Ortholog file obtained from Mouse Genome Database (www.informatics.jax.org). As a result, mRNA half-life was assigned to a total of 13,578 human genes.
Mononucleosomal DNA dinucleotide frequency determination
Mononucleosomal DNA sequences were determined as follows. For a mononucleosomal fragment, if its end sequence reads were both mapped perfectly and uniquely onto the human genome, the genomic sequence spanned by the reads would be its sequence. Then, the sequences of all such mapped mononucleosomal fragments of a chosen length were aligned, from which fractions of AA/AT/TA/TT and GG/GC/CG/CC dinucleotides at each base position from dyad were calculated. The same analysis was repeated with randomly sheared genomic fragments for control.
Structural and sequence variation analysis of the WA09-hESC genome
The sequences of randomly sheared genomic DNA of WA09-hESC, along with genomic sequences of an individual of an European ancestry (NA12892) downloaded from www.1000genomes.org and genomic sequences of an Asian,53 were mapped to the hg18 genome with BWA. SNPs, compared to the hg18 genome, were identified using SAMtools66 and GATK.67 CNVs were identified as described,68-70 using . and respectively represent the fragment density of the ith window (200 bp) of the test genome (t: either WA09-hESC or NA12892) and of the normalizing Asian genome (a). and represent the corresponding genome-wide average of and .
To identify genomic regions with higher or lower nucleosome occupancy, we analyzed , where and respectively represent the fragment density of window i of nucleosomal DNA (n) and randomly sheared genomic DNA (g), with and being their corresponding genome-wide average. Furthermore, we set for windows with: 1) and ; or 2) or . Then, we identified genomic regions that are amplified, considered as sites with higher nucleosome occupancy, or deleted, deemed sites with lower nucleosome occupancy, as described.68-70 Next, we examined sequence mutations in these regions as described above.
Accession numbers
MNase-seq and microarray data have been deposited to the GEO database under the accession number GSE46467.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the Emory Biomarker Core for conducting the microarray experiments, as well as the BGI for the sequencing work.
Funding
The study was 545 funded by the National Cancer Institute R01 CA182093 (to SZ) and the National Institute of General Medical Sciences GM085354 (to SD).
References
- 1.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science 1998; 282:1145-7; PMID:9804556; http://dx.doi.org/ 10.1126/science.282.5391.1145 [DOI] [PubMed] [Google Scholar]
- 2.Menendez L, Kulik MJ, Page AT, Park SS, Lauderdale JD, Cunningham ML, Dalton S. Directed differentiation of human pluripotent cells to neural crest stem cells. Nat Protoc 2013; 8:203-12; PMID:23288320; http://dx.doi.org/ 10.1038/nprot.2012.156 [DOI] [PubMed] [Google Scholar]
- 3.Funk WD, Labat I, Sampathkumar J, Gourraud PA, Oksenberg JR, Rosler E, Steiger D, Sheibani N, Caillier S, Stache-Crain B, et al.. Evaluating the genomic and sequence integrity of human ES cell lines; comparison to normal genomes. Stem Cell Res 2012; 8:154-64; PMID:22265736; http://dx.doi.org/ 10.1016/j.scr.2011.10.001 [DOI] [PubMed] [Google Scholar]
- 4.Maitra A, Arking DE, Shivapurkar N, Ikeda M, Stastny V, Kassauei K, Sui G, Cutler DJ, Liu Y, Brimble SN, et al.. Genomic alterations in cultured human embryonic stem cells. Nat Genet 2005; 37:1099-103; PMID:16142235; http://dx.doi.org/ 10.1038/ng1631 [DOI] [PubMed] [Google Scholar]
- 5.Therizols P, Illingworth RS, Courilleau C, Boyle S, Wood AJ, Bickmore WA. Chromatin decondensation is sufficient to alter nuclear organization in embryonic stem cells. Science 2014; 346:1238-42; PMID:25477464; http://dx.doi.org/ 10.1126/science.1259587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee HJ, Hore TA, Reik W. Reprogramming the methylome: erasing memory and creating diversity. Cell Stem Cell 2014; 14:710-9; PMID:24905162; http://dx.doi.org/ 10.1016/j.stem.2014.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gopalakrishnan S, Van Emburgh BO, Robertson KD. DNA methylation in development and human disease. Mutat Res 2008; 647:30-8; PMID:18778722; http://dx.doi.org/ 10.1016/j.mrfmmm.2008.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al.. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009; 462:315-22; PMID:19829295; http://dx.doi.org/ 10.1038/nature08514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiao S, Xie D, Cao X, Yu P, Xing X, Chen CC, Musselman M, Xie M, West FD, Lewin HA, et al.. Comparative epigenomic annotation of regulatory DNA. Cell 2012; 149:1381-92; PMID:22682255; http://dx.doi.org/ 10.1016/j.cell.2012.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hiratani I, Gilbert DM. Replication timing as an epigenetic mark. Epigenetics : official journal of the DNA Methylation Society 2009; 4:93-7; http://dx.doi.org/ 10.4161/epi.4.2.7772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Consortium EP, Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, et al.. An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489:57-74; PMID:22955616; http://dx.doi.org/ 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu G, Cui K, Northrup D, Liu C, Wang C, Tang Q, Ge K, Levens D, Crane-Robinson C, Zhao K. H2A.Z Facilitates Access of Active and Repressive Complexes to Chromatin in Embryonic Stem Cell Self-Renewal and Differentiation. Cell Stem Cell 2013; 12:180-92; PMID:23260488; http://dx.doi.org/ 10.1016/j.stem.2012.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, et al.. Dynamic changes in the human methylome during differentiation. Genome Res 2010; 20:320-31; PMID:20133333; http://dx.doi.org/ 10.1101/gr.101907.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biterge B, Schneider R. Histone variants: key players of chromatin. Cell Tissue Res 2014; 356:457-66; PMID:24781148; http://dx.doi.org/ 10.1007/s00441-014-1862-4 [DOI] [PubMed] [Google Scholar]
- 15.Combes AN, Whitelaw E. Epigenetic reprogramming: enforcer or enabler of developmental fate? Dev Growth Differ 2010; 52:483-91; PMID:20608951; http://dx.doi.org/ 10.1111/j.1440-169X.2010.01185.x [DOI] [PubMed] [Google Scholar]
- 16.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al.. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006; 125:315-26; PMID:16630819; http://dx.doi.org/ 10.1016/j.cell.2006.02.041 [DOI] [PubMed] [Google Scholar]
- 17.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al.. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007; 448:553-60; PMID:17603471; http://dx.doi.org/ 10.1038/nature06008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cui K, Zang C, Roh TY, Schones DE, Childs RW, Peng W, Zhao K. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell 2009; 4:80-93; PMID:19128795; http://dx.doi.org/ 10.1016/j.stem.2008.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruzov A, Tsenkina Y, Serio A, Dudnakova T, Fletcher J, Bai Y, Chebotareva T, Pells S, Hannoun Z, Sullivan G, et al.. Lineage-specific distribution of high levels of genomic 5-hydroxymethylcytosine in mammalian development. Cell Res 2011; 21:1332-42; PMID:21747414; http://dx.doi.org/ 10.1038/cr.2011.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu H, D'Alessio AC, Ito S, Wang Z, Cui K, Zhao K, Sun YE, Zhang Y. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev 2011; 25:679-84; PMID:21460036; http://dx.doi.org/ 10.1101/gad.2036011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA, Marques CJ, Andrews S, Reik W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 2011; 473:398-402; PMID:21460836; http://dx.doi.org/ 10.1038/nature10008 [DOI] [PubMed] [Google Scholar]
- 22.Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, Laiho A, Tahiliani M, Sommer CA, Mostoslavsky G, et al.. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell 2011; 8:200-13; PMID:21295276; http://dx.doi.org/ 10.1016/j.stem.2011.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao XD, Han X, Chew JL, Liu J, Chiu KP, Choo A, Orlov YL, Sung WK, Shahab A, Kuznetsov VA, et al.. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell 2007; 1:286-98; PMID:18371363; http://dx.doi.org/ 10.1016/j.stem.2007.08.004 [DOI] [PubMed] [Google Scholar]
- 24.Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SW, Solovei I, Brugman W, Graf S, Flicek P, Kerkhoven RM, van Lohuizen M, et al.. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol Cell 2010; 38:603-13; PMID:20513434; http://dx.doi.org/ 10.1016/j.molcel.2010.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richmond TJ, Davey CA. The structure of DNA in the nucleosome core. Nature 2003; 423:145-50; PMID:12736678; http://dx.doi.org/ 10.1038/nature01595 [DOI] [PubMed] [Google Scholar]
- 26.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997; 389:251-60; PMID:9305837; http://dx.doi.org/ 10.1038/38444 [DOI] [PubMed] [Google Scholar]
- 27.Venters BJ, Pugh BF. How eukaryotic genes are transcribed. Crit Rev Biochem Mol 2009; 44:117-41; http://dx.doi.org/ 10.1080/10409230902858785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.North JA, Amunugama R, Klajner M, Bruns AN, Poirier MG, Fishel R. ATP-dependent nucleosome unwrapping catalyzed by human RAD51. Nucleic Acids Res 2013; 41:7302-12; PMID:23757189; http://dx.doi.org/ 10.1093/nar/gkt411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chadwick BP, Willard HF. Barring gene expression after XIST: maintaining facultative heterochromatin on the inactive X. Semin Cell Dev Biol 2003; 14:359-67; PMID:15015743; http://dx.doi.org/ 10.1016/j.semcdb.2003.09.016 [DOI] [PubMed] [Google Scholar]
- 30.Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G, Zhao K. Dynamic regulation of nucleosome positioning in the human genome. Cell 2008; 132:887-98; PMID:18329373; http://dx.doi.org/ 10.1016/j.cell.2008.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ozsolak F, Song JS, Liu XS, Fisher DE. High-throughput mapping of the chromatin structure of human promoters. Nat Biotechnol 2007; 25:244-8; PMID:17220878; http://dx.doi.org/ 10.1038/nbt1279 [DOI] [PubMed] [Google Scholar]
- 32.Jiang C, Pugh BF. Nucleosome positioning and gene regulation: advances through genomics. Nature Rev Genet 2009; 10:161-72; PMID:19204718; http://dx.doi.org/ 10.1038/nrg2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teif VB, Vainshtein Y, Caudron-Herger M, Mallm JP, Marth C, Hofer T, Rippe K. Genome-wide nucleosome positioning during embryonic stem cell development. Nat Struct Mol Biol 2012; 19:1185-92; PMID:23085715; http://dx.doi.org/ 10.1038/nsmb.2419 [DOI] [PubMed] [Google Scholar]
- 34.Gaffney DJ, McVicker G, Pai AA, Fondufe-Mittendorf YN, Lewellen N, Michelini K, Widom J, Gilad Y, Pritchard JK. Controls of nucleosome positioning in the human genome. PLoS Genet 2012; 8:e1003036; PMID:23166509; http://dx.doi.org/ 10.1371/journal.pgen.1003036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, Sidow A. Determinants of nucleosome organization in primary human cells. Nature 2011; 474:516-20; PMID:21602827; http://dx.doi.org/ 10.1038/nature10002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prendergast JG, Semple CA. Widespread signatures of recent selection linked to nucleosome positioning in the human lineage. Genome Res 2011; 21:1777-87; PMID:21903742; http://dx.doi.org/ 10.1101/gr.122275.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Struhl K, Segal E. Determinants of nucleosome positioning. Nat Struct Mol Biol 2013; 20:267-73; PMID:23463311; http://dx.doi.org/ 10.1038/nsmb.2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.West JA, Cook A, Alver BH, Stadtfeld M, Deaton AM, Hochedlinger K, Park PJ, Tolstorukov MY, Kingston RE. Nucleosomal occupancy changes locally over key regulatory regions during cell differentiation and reprogramming. Nat Commun 2014; 5:4719; PMID:25158628; http://dx.doi.org/ 10.1038/ncomms5719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hainer SJ, Fazzio TG. Regulation of Nucleosome Architecture and Factor Binding Revealed by Nuclease Footprinting of the ESC Genome. Cell Rep 2015; 13:61-9; PMID:26411677; http://dx.doi.org/ 10.1016/j.celrep.2015.08.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Teif VB, Beshnova DA, Vainshtein Y, Marth C, Mallm JP, Hofer T, Rippe K. Nucleosome repositioning links DNA (de)methylation and differential CTCF binding during stem cell development. Genome Res 2014; 24:1285-95; PMID:24812327; http://dx.doi.org/ 10.1101/gr.164418.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al.. The accessible chromatin landscape of the human genome. Nature 2012; 489:75-82; PMID:22955617; http://dx.doi.org/ 10.1038/nature11232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhong JLK, Winter PS, Crawford GE, Iversen ES, Hartemink AJ. Mapping nucleosome positions using DNase-seq. Genome Res 2016; 26:351-64; PMID:26772197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galan A, Diaz-Gimeno P, Poo ME, Valbuena D, Sanchez E, Ruiz V, Dopazo J, Montaner D, Conesa A, Simon C. Defining the Genomic Signature of Totipotency and Pluripotency during Early Human Development. PloS One 2013; 8:e62135; PMID:23614026; http://dx.doi.org/ 10.1371/journal.pone.0062135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Segal E, Fondufe-Mittendorf Y, Chen L, Thastrom A, Field Y, Moore IK, Wang JP, Widom J. A genomic code for nucleosome positioning. Nature 2006; 442:772-8; PMID:16862119; http://dx.doi.org/ 10.1038/nature04979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharova LV, Sharov AA, Nedorezov T, Piao Y, Shaik N, Ko MS. Database for mRNA half-life of 19 977 genes obtained by DNA microarray analysis of pluripotent and differentiating mouse embryonic stem cells. DNA Res 2009; 16:45-58; PMID:19001483; http://dx.doi.org/ 10.1093/dnares/dsn030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011; 470:279-83; PMID:21160473; http://dx.doi.org/ 10.1038/nature09692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell 2013; 155:934-47; PMID:24119843; http://dx.doi.org/ 10.1016/j.cell.2013.09.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chodavarapu RK, Feng S, Bernatavichute YV, Chen PY, Stroud H, Yu Y, Hetzel JA, Kuo F, Kim J, Cokus SJ, et al.. Relationship between nucleosome positioning and DNA methylation. Nature 2010; 466:388-92; PMID:20512117; http://dx.doi.org/ 10.1038/nature09147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J, et al.. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 2009; 458:362-6; PMID:19092803; http://dx.doi.org/ 10.1038/nature07667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xing K, He XL. Mutation Bias, rather than Binding Preference, Underlies the Nucleosome-Associated G plus C% Variation in Eukaryotes. Genome Biol Evol 2015; 7:1033-8; PMID:25786433; http://dx.doi.org/ 10.1093/gbe/evv053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tillo D, Hughes TR. G+C content dominates intrinsic nucleosome occupancy. BMC Bioinformatics 2009; 10:442; PMID:20028554; http://dx.doi.org/ 10.1186/1471-2105-10-442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.International Stem Cell I, Amps K, Andrews PW, Anyfantis G, Armstrong L, Avery S, Baharvand H, Baker J, Baker D, Munoz MB, et al.. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat Biotechnol 2011; 29:1132-44; PMID:22119741; http://dx.doi.org/ 10.1038/nbt.2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J, Wang W, Li R, Li Y, Tian G, Goodman L, Fan W, Zhang J, Li J, Guo Y, et al.. The diploid genome sequence of an Asian individual. Nature 2008; 456:60-5; PMID:18987735; http://dx.doi.org/ 10.1038/nature07484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, Dooling D, Dunford-Shore BH, McGrath S, Hickenbotham M, et al.. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 2008; 456:66-72; PMID:18987736; http://dx.doi.org/ 10.1038/nature07485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Locke G, Haberman D, Johnson SM, Morozov AV. Global remodeling of nucleosome positions in C. elegans. BMC genomics 2013; 14:284; PMID:23622142; http://dx.doi.org/ 10.1186/1471-2164-14-284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al.. Chromatin architecture reorganization during stem cell differentiation. Nature 2015; 518:331-6; PMID:25693564; http://dx.doi.org/ 10.1038/nature14222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bregman A, Avraham-Kelbert M, Barkai O, Duek L, Guterman A, Choder M. Promoter elements regulate cytoplasmic mRNA decay. Cell 2011; 147:1473-83; PMID:22196725; http://dx.doi.org/ 10.1016/j.cell.2011.12.005 [DOI] [PubMed] [Google Scholar]
- 58.Trcek T, Larson DR, Moldon A, Query CC, Singer RH. Single-molecule mRNA decay measurements reveal promoter- regulated mRNA stability in yeast. Cell 2011; 147:1484-97; PMID:22196726; http://dx.doi.org/ 10.1016/j.cell.2011.11.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al.. Signatures of mutational processes in human cancer. Nature 2013; 500:415-21; PMID:23945592; http://dx.doi.org/ 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Allan J, Fraser RM, Owen-Hughes T, Keszenman-Pereyra D. Micrococcal Nuclease Does Not Substantially Bias Nucleosome Mapping. J Mol Biol 2012; 417:152-64; PMID:22310051; http://dx.doi.org/ 10.1016/j.jmb.2012.01.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 2004; 3:Article3; PMID:16646809; http://dx.doi.org/ 10.2202/1544-6115.1027 [DOI] [PubMed] [Google Scholar]
- 62.Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res 2001; 125:279-84; PMID:11682119; http://dx.doi.org/ 10.1016/S0166-4328(01)00297-2 [DOI] [PubMed] [Google Scholar]
- 63.Spetman B, Lueking S, Roberts B, Dennis JH. Microarray mapping of nucleosome position Epigenetics: A Reference Manual. Craig J and Wong N, eds, Norwich, UK: Horizon Scientific Press, 2011 [Google Scholar]
- 64.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25:1754-60; PMID:19451168; http://dx.doi.org/ 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44-57; PMID:19131956; http://dx.doi.org/ 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 66.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25:2078-9; PMID:19505943; http://dx.doi.org/ 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al.. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20:1297-303; PMID:20644199; http://dx.doi.org/ 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tang J, Le S, Sun L, Yan X, Zhang M, Macleod J, Leroy B, Northrup N, Ellis A, Yeatman TJ, et al.. Copy number abnormalities in sporadic canine colorectal cancers. Genome Res 2010; 20:341-50; PMID:20086242; http://dx.doi.org/ 10.1101/gr.092726.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu D, Xiong H, Ellis AE, Northrup NC, Dobbin KK, Shin DM, Zhao S. Canine spontaneous head and neck squamous cell carcinomas represent their human counterparts at the molecular level. PLoS Genet 2015; 11:e1005277; PMID:26030765; http://dx.doi.org/ 10.1371/journal.pgen.1005277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu D, Xiong H, Ellis AE, Northrup NC, Rodriguez CO Jr., O'Regan RM, Dalton S, Zhao S. Molecular homology and difference between spontaneous canine mammary cancer and human breast cancer. Cancer Res 2014; 74:5045-56; PMID:25082814 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.