

ABSTRACT
Maternal environmental factors can impact on the phenotype of the offspring via the induction of epigenetic adaptive mechanisms. The advanced fetal programming hypothesis proposes that maternal genetic variants may influence the offspring's phenotype indirectly via epigenetic modification, despite the absence of a primary genetic defect. To test this hypothesis, heterozygous female eNOS knockout mice and wild type mice were bred with male wild type mice. We then assessed the impact of maternal eNOS deficiency on the liver phenotype of wild type offspring. Birth weight of male wild type offspring born to female heterozygous eNOS knockout mice was reduced compared to offspring of wild type mice. Moreover, the offspring displayed a sex specific liver phenotype, with an increased liver weight, due to steatosis. This was accompanied by sex specific differences in expression and DNA methylation of distinct genes. Liver global DNA methylation was significantly enhanced in both male and female offspring. Also, hepatic parameters of carbohydrate metabolism were reduced in male and female offspring. In addition, male mice displayed reductions in various amino acids in the liver. Maternal genetic alterations, such as partial deletion of the eNOS gene, can affect liver metabolism of wild type offspring without transmission of the intrinsic defect. This occurs in a sex specific way, with more detrimental effects in females. This finding demonstrates that a maternal genetic defect can epigenetically alter the phenotype of the offspring, without inheritance of the defect itself. Importantly, these acquired epigenetic phenotypic changes can persist into adulthood.
KEYWORDS: Epigenetics, eNOS, Fetal programming, fatty liver, metabolism
Abbreviations
- CML
carboxmethyllysin
- eNOS
endothelial nitric oxide synthase
- iNOS
inducible nitric oxide synthase
- IUGR
intrauterine growth retardation
- LC-MS-MS
liquid chromatography tandem mass spectrometry
- MeDIP
methylated DNA immunoprecipitation
- miRNA
microRNA
- wt
wild type.
Introduction
The ‘fetal origin’ hypothesis proposes that adulthood cardiovascular and metabolic diseases originate through adaptation of the fetus to environmental conditions in early life.1 It was proposed that an event in a critical early period of life leads to sustained alterations of organ structure and function in response to environmental factors. Such events may result in cardiovascular and metabolic diseases in later life. The classical event resulting in fetal programming is maternal undernutrition during pregnancy. This was first recognized in epidemiological studies and later confirmed in animal experiments.1-4 Meanwhile, several other mechanisms caused by environmental conditions in early life leading to lifelong functional and structural alterations have been described, including glucocorticoid exposure of the fetus due to 11β-hydroxysteroid dehydrogenase deficiency of the placenta5,6 or a high protein diet during pregnancy.7 Another mechanism responsible for programming events during intrauterine life might be related to maternal genes affecting the fetal phenotype independently of the fetal DNA-based genome. The first example is described for Drosophila wimp mutation, influencing the offspring's lethal phenotype, even when the mutation is not inherited.8 Our group was the first to translate this to mammals/humans by demonstrating that genetic variation of a maternal gene most likely involved in the control of blood supply to the uterus was associated with a substantial reduction of offspring birth weight without being actually transmitted to the offspring.9,10 Other independent association studies in humans likewise suggest that certain maternal genes may affect the fetal phenotype even without transmission of that particular gene to the fetus.11,12 In other words, a gene of a human individual may influence the physiology of another subject without being present in this particular individual.13 Plausibly, interaction of one organism with the metabolism of another of the same species is seen in mammals mainly during pregnancy, where the placenta serves as interface between both individuals.13
To prove that maternal genes indeed can affect the offspring's phenotype, as suggested by association studies (see above), we bred female heterozygous endothelial nitric oxide synthase (eNOS) knockout mice with male wild type (wt) mice and compared their wild type offspring with offspring from wild type mice. We have chosen eNOS knockout mice to test this hypothesis, because eNOS—like the genetic variations analyzed in our initial association study in humans9,10—plays a pivotal role in the control of vascular and also placental function,14-17 and heterozygous eNOS deficiency has been shown to create an unfavorable intrauterine environment influencing the vascular phenotype in offspring, independently of its genetic transmission.18 We reasoned that the resultant endothelial and vascular dysfunction could also impact central parameters of metabolism as reflected by fatty liver disease. An illustration of the underlying hypothesis of this study is provided in Fig. 1.
Figure 1.
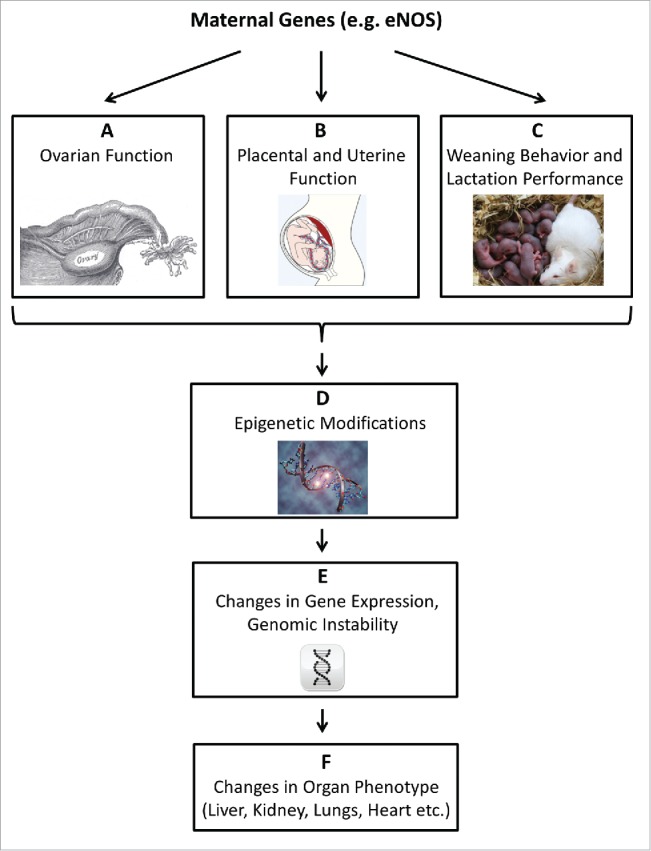
The advanced fetal programming hypothesis. The ‘fetal origin’ hypothesis proposes that adulthood cardiovascular, metabolic, and mental diseases originate through adaptation of the fetus to environmental conditions in early life. We proposed that maternal genetic defects might impact on the offspring phenotype via genomic-epigenomic interactions, without transmittance of the defective gene. Such interactions, here exemplified by the eNOS gene, could be mediated during 3 phases of reproduction (A-C). (A) Maternal gene dysfunction can alter ovary function: eNOS mediates physiological ovarian functions, such as blood flow and angiogenesis and is involved in oocyte meiotic maturation.49,50 (B) Maternal gene dysfunction can alter placental and uterine function: eNOS plays a pivotal role in the control of placental function, and eNOS deficiency is associated with an unfavorable intrauterine environment.16,17 (C) Maternal gene dysfunction may alter weaning behavior and lactation performance: eNOS is involved in behavioral processes and elicits regulatory functions in lactation.51,52 Alterations in maternal eNOS function thus may affect the offspring in the early postnatal phase.51,52 (D) The impact of maternal gene dysfunction on embryonal/fetal/neonatal environmental factors listed in A-C may trigger stable, long lasting epigenetic adaptation in the offspring.53-55 Epigenetic mechanisms encompass DNA methylation, non-coding RNAs, and chromatin modifications.56 (E) Epigenetic modification, specifically DNA methylation and non-coding RNAs, can result in permanently altered gene expression in the offspring.34 On a global scale, both DNA hypermethylation and DNA hypomethylation may cause genomic instability.57,58 (F) Altered gene expression and genomic instability triggered by epigenetic maladaptation can impact on the offspring phenotype by permanently altering organ structure and function.59,60
Results
Phenotype of wt mice born to heterozygous eNOS knockout mothers and wt fathers
First, we verified that offspring born to heterozygous eNOS knockout (eNOS+/−) mothers and wt fathers were wild type with respect to eNOS expression. Gene and protein expression of eNOS and inducible nitric oxide synthase (iNOS) in liver tissue were not altered in male and female wt animals born to eNOS+/− mothers (Fig. 2 A, B; Supplementary Table S1).
Figure 2.
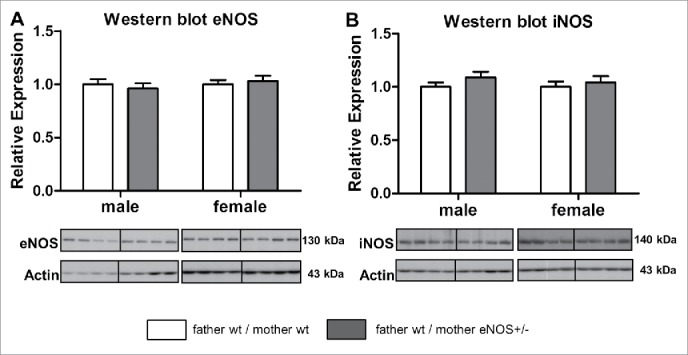
Hepatic eNOS and iNOS protein content in wt offspring. Hepatic eNOS (A) and iNOS (B) protein content analyzed by Western blot (empty bar, wt father / wt mother; gray bar, wt father / eNOS+/− mother ).
Birth Weight, Growth, and Organ Weight
Male wt mice born to eNOS+/− mothers and wt fathers had a significantly lower birth weight when compared to wt male mice born to wt mothers and wt fathers (1.27 ± 0.05 g vs. 1.47 ± 0.05 g, P = 0.004), whereas birth weight of female offspring did not differ significantly [1.26 ± 0.05 g (wt father, eNOS+/− mother) vs. 1.35 ± 0.04 g (wt father, wt mother); Supplementary Fig. S1].
Body weight of male offspring of wt fathers and heterozygous eNOS knockout mothers remained significantly lower during the first days of life (Supplementary Fig. S2). Thereafter, no significant differences in body weight were noticed (Supplementary Fig. S3). At study end at week 24, male mice born to eNOS+/− mothers and wt fathers had a significantly lower body weight when compared to male offspring born to wt mothers and fathers (Supplementary Table S2). Female offspring born to eNOS+/− mothers and wt fathers showed significantly higher body weights than controls starting on day 12 after birth and remaining elevated throughout most of life thereafter (Supplementary Figs. S4 and S5, Supplementary Table S2). Heart weight was significantly higher in female offspring born to eNOS+/− mothers and wt fathers compared to female wt offspring born to wt mothers and fathers (Supplementary Table S2). Relative lung and kidney weights did not differ significantly (Supplementary Table S2). Relative liver weight was not different in female offspring born to eNOS+/− mothers and wt fathers as compared to the control group (Table 1).
Table 1.
Offspring liver weight, morphology, and glycogen content.
| Offspring of both sexes |
Male offspring |
Female offspring |
||||
|---|---|---|---|---|---|---|
| WT (F:WT; M:WT) | WT (F:WT; M:eNOS KO) | WT (F:WT; M:WT) | WT (F:WT; M: eNOS KO) | WT (F:WT; M:WT) | WT (F:WT; M: eNOS KO) | |
| N | 35-48 | 30-35 | 21-22 | 15-18 | 24-27 | 17-18 |
| Absolute Liver Weight (g) | 1.19 ± 0.05 | 1.20 ± 0.04 | 1.56 ± 0.05 | 1.40 ± 0.04 | 0.90 ± 0.02 | 1.00 ± 0.03$ |
| Relative Liver Weight (% of body weight) | 4.31 ± 0.06 | 4.41 ± 0.06 | 4.56 ± 0.07 | 4.59 ± 0.10 | 4.11 ± 0.08 | 4.23 ± 0.06 |
| Lobular Dimension (mm) | 0.07 ± 0.001 | 0.07 ± 0.001 | 0.07 ± 0.001 | 0.07 ± 0.001 | 0.07 ± 0.002 | 0.07 ± 0.001 |
| Connective Tissue Content (% area) | 0.16 ± 0.02 | 0.12 ± 0.02 | 0.12 ± 0.03 | 0.12 ± 0.02 | 0.18 ± 0.03 | 0.12 ± 0.03 |
| Glycogen (mg/g liver) | 14.09 ± 1.02 | 16.44 ± 1.54 | 13.31 ± 1.46 | 17.32 ± 1.83 | 14.93 ± 1.44 | 15.67 ± 2.54 |
| Fat Content (% area) | 2.06 ± 0.26 | 4.33 ± 0.82 | 1.85 ± 0.36 | 1.23 ± 0.29 | 2.25 ± 0.37 | 6.92 ± 1.18 + |
| Lipid Droplet Density (droplets/mm2) | 2701.0 ± 411.2 | 5014.6 ± 732.9 $ | 2537.7 ± 779.2 | 1699.3 ± 416.8 | 2850.8 ± 352.1 | 7777.3 ± 864.1 + |
| N | 13 | 15 | 7 | 7 | 6 | 8 |
| Lobular Inflammation (score) | 0.31 ± 0.13 | 0.53 ± 0.13 | 0.29 ± 0.18 | 0.57 ± 0.20 | 0.33 ± 0.21 | 0.50 ± 0.19 |
| Number of CD68-Positive Immune Cells (score) | 0.85 ± 0.10 | 0.87 ± 0.09 | 0.86 ± 0.14 | 0.86 ± 0.14 | 0.83 ± 0.17 | 0.88 ± 0.13 |
F: Father, M: Mother, WT: wild type, eNOS KO: heterozygous eNOS knockout, $ P < 0.05 vs. WT (F:WT;M:WT), # P < 0.01 vs. WT (F:WT;M:WT), + P < 0.001 vs. WT (F:WT;M:WT)
Blood pressure, heart rate, GFR, and fasting glucose
Blood pressure and heart rate were similar in all groups. Moreover, glomerular filtration rate (GFR) at study end did not differ significantly between groups. Fasting glucose was numerically lower in wt mice born to heterozygous eNOS knockout mothers. This effect was significant, however, only in week 21 (Supplementary Table S2).
Characterization of the liver phenotype
First we assessed the liver morphology. Diameter of liver lobules, liver glycogen concentration, and the connective tissue content were not affected by maternal eNOS genotype (Table 1). However, fat content and density of lipid droplets were significantly higher in female wt mice born to eNOS+/− mothers and wt fathers compared to wt mice born to wt mothers and wt fathers (P < 0.001, see Table 1 and Fig. 3A-C). Examination of lipid droplet size showed a significant increase of droplets in female wt offspring of wt fathers and eNOS+/− mothers, but a non-significant decrease in male animals (Fig. 3D, E). The mean area of lipid droplets in these animals was 5.2 µm2, and 5.6% of droplets were bigger than 25 µm2. These numbers were significantly lower in wt female offspring from wt fathers and wt mothers, with 2.1 µm2 and 0.6%, respectively. None of the groups showed any significant lobular inflammation or an increase in the number of CD68-positive cells (macrophages) (Table 1).
Figure 3.

Lipid deposition in wt offspring liver tissue. (A) Liver sections of wt male and female offspring from wt father and wt mother (wt/wt) and wt father and heterozygous eNOS knockout mother (wt/eNOS+/−), determination of lipid content (B) and lipid droplet density (C) in liver tissue and size distributions of lipid droplets for female (D) and male (E) wt offspring (empty bar, wt father / wt mother; gray bar, wt father / eNOS+/− mother; + P < 0.001 vs. wt father / wt mother).
To extend characterization of the liver phenotype, selected marker proteins related to oxidative stress and hyperglycemia were analyzed by Western blot. Female but not male wt offspring with eNOS+/− mothers and wt fathers had a significantly higher hepatic amount of carboxymethyllysine (CML) (P < 0.01, Fig. 4A). Nitrotyrosine-modified proteins were enhanced in female offspring from eNOS+/− mother, but failed to attain statistical significance (Fig. 4B).
Figure 4.
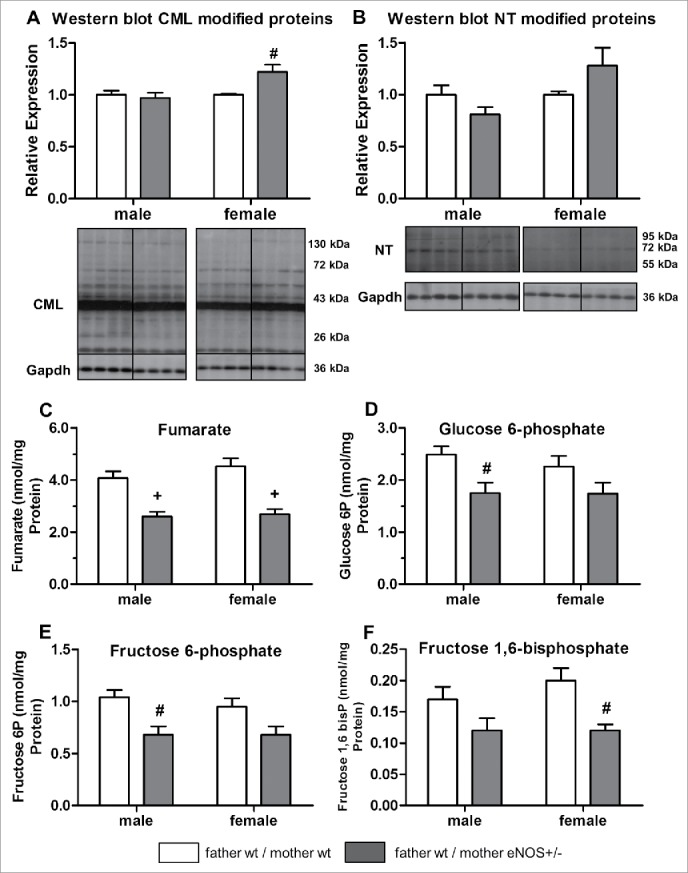
Effects of maternal eNOS deficiency on hepatic protein content and metabolomics in wt offspring. Hepatic content of (A) carboxymethyllysine (CML) and (B) nitrotyrosine (NT) modified proteins (relative expression) as well as (C) fumarate, (D) glucose 6-phosphate, (E) fructose 6-phosphate, and (F) fructose 1,6-bisphosphate concentration (nmol/mg protein) (empty bar, wt father / wt mother; gray bar, wt father / eNOS+/− mother; # P < 0.01 vs. wt father / wt mother; + P < 0.001 vs. wt father / wt mother).
Moreover, selected substrates of glucose metabolism were analyzed in liver tissue by quantitative LC-MS-MS technology. Fumarate concentration was reduced in male and female wt offspring born to eNOS+/− mothers and wt fathers. Male offspring showed significantly lower concentrations of glucose 6-phosphate and fructose 6-phosphate, whereas hepatic fructose 1,6-bisphosphate content was reduced in female mice (Fig. 4 C-F, Supplementary Table S3). Analysis of liver amino acid concentrations revealed that male but not female wt offspring born to eNOS+/− mothers and wt fathers had significantly lower concentration of several, mainly essential amino acids when compared to offspring born to wt mothers and wt fathers (Table 2).
Table 2.
Amino acid concentration in offspring liver tissue.
| Offspring of both sexes |
Male offspring |
Female offspring |
||||
|---|---|---|---|---|---|---|
| WT (F:WT; M:WT) | WT (F:WT; M:eNOS KO) | WT (F:WT; M:WT) | WT (F:WT; M:eNOS KO) | WT (F:WT; M:WT) | WT (F:WT; M:eNOS KO) | |
| N | 48-50 | 30-36 | 21-22 | 15-18 | 27-28 | 15-18 |
| Alanine | 38.26 ± 1.42 | 37.37 ± 1.70 | 35.89 ± 1.63 | 34.57 ± 2.72 | 40.12 ± 2.14 | 40.18 ± 1.88 |
| Glycine | 19.97 ± 0.45 | 18.73 ± 0.63 | 21.02 ± 0.64 | 19.25 ± 0.81 | 19.15 ± 0.59 | 18.12 ± 0.96 |
| Urea | 18.77 ± 0.64 | 18.23 ± 0.69 | 19.28 ± 1.05 | 18.93 ± 0.81 | 18.37 ± 0.79 | 17.53 ± 1.13 |
| Valine | 3.50 ± 0.11 | 3.07 ± 0.12 + | 3.62 ± 0.13 | 2.84 ± 0.15 + | 3.41 ± 0.17 | 3.31 ± 0.18 |
| Leucine | 4.24 ± 0.14 | 3.65 ± 0.19 + | 4.57 ± 0.20 | 3.26 ± 0.20 + | 3.97 ± 0.17 | 4.03 ± 0.29 |
| Isoleucine | 1.90 ± 0.07 | 1.59 ± 0.07 + | 2.02 ± 0.08 | 1.44 ± 0.08 + | 1.80 ± 0.10 | 1.74 ± 0.10 |
| Proline | 2.92 ± 0.16 | 2.70 ± 0.16 | 2.72 ± 0.15 | 2.54 ± 0.24 | 3.07 ± 0.27 | 2.86 ± 0.22 |
| Methionine | 1.31 ± 0.04 | 1.14 ± 0.06 # | 1.36 ± 0.07 | 0.99 ± 0.06 + | 1.28 ± 0.05 | 1.29 ± 0.09 |
| Serine | 4.89 ± 0.19 | 4.42 ± 0.20 $ | 5.00 ± 0.26 | 4.14 ± 0.25 $ | 4.81 ± 0.27 | 4.72 ± 0.30 |
| Threonine | 3.11 ± 0.08 | 2.69 ± 0.13 + | 3.31 ± 0.13 | 2.40 ± 0.14 + | 2.96 ± 0.09 | 2.98 ± 0.19 |
| Phenylalanine | 1.70 ± 0.05 | 1.45 ± 0.08 + | 1.82 ± 0.09 | 1.26 ± 0.08 + | 1.61 ± 0.06 | 1.64 ± 0.13 |
| Aspartic acid | 3.16 ± 0.19 | 2.92 ± 0.11 | 2.97 ± 0.11 | 2.82 ± 0.16 | 3.30 ± 0.33 | 3.04 ± 0.14 |
| Cysteine | 0.26 ± 0.01 | 0.25 ± 0.03 # | 0.26 ± 0.01 | 0.20 ± 0.01 + | 0.25 ± 0.01 | 0.30 ± 0.07 |
| Glutamic acid | 8.63 ± 0.47 | 8.59 ± 0.51 | 8.13 ± 0.38 | 8.06 ± 0.72 | 9.02 ± 0.78 | 9.12 ± 0.71 |
| Lysine | 7.16 ± 0.35 | 6.38 ± 0.38 | 6.96 ± 0.50 | 5.56 ± 0.47 | 7.32 ± 0.49 | 7.19 ± 0.53 |
| Glutamine | 23.46 ± 1.16 | 20.16 ± 1.00 | 25.65 ± 1.40 | 21.68 ± 1.34 | 21.74 ± 1.72 | 19.55 ± 1.48 |
| Arginine | 1.22 ± 0.06 | 1.25 ± 0.11 | 1.07 ± 0.06 | 1.01 ± 0.09 | 1.35 ± 0.10 | 1.48 ± 0.19 |
| Histidine | 4.50 ± 0.13 | 3.78 ± 0.14 + | 4.84 ± 0.17 | 3.71 ± 0.23 + | 4.23 ± 0.18 | 3.84 ± 0.17 |
| Tyrosine | 1.82 ± 0.05 | 1.60 ± 0.07 # | 1.92 ± 0.08 | 1.40 ± 0.08 + | 1.75 ± 0.06 | 1.80 ± 0.10 |
| Tryptophan | 0.50 ± 0.01 | 0.41 ± 0.02 + | 0.49 ± 0.01 | 0.35 ± 0.02 + | 0.51 ± 0.02 | 0.47 ± 0.02 |
Concentration of aminoacids expressed as nmol/mg protein. F: Father, M: Mother, WT: Wild type, eNOS KO: heterozygous eNOS knockout, $ P < 0.05 vs. WT (F:WT;M:WT), # P < 0.01 vs. WT (F:WT;M:WT), + P < 0.001 vs. WT (F:WT;M:WT)
Mechanisms of phenotypic alteration in wt mice born to eNOS+/− mothers
Characterization of the mouse transcriptome
Genome-wide microarray analyses were performed for pooled liver RNA samples of randomly selected mice (n = 30). This approach was complemented by independent RT-PCR based investigation of mRNA transcripts and miRNAs using samples of the total mouse cohort.
Microarray analysis revealed 11,628 and 12,047 genes differentially expressed between male and female offspring of eNOS+/− and wt mothers, respectively (Fig. 5 A, B). Six genes showed P-values ≤ 0.001 in our analyses. Of these, 5 genes (Amy2a5, Clps, Cpa1, Ctrbl, and 2210010C04 Riken cDNA/trypsinogen 7) showed higher expression in female offspring from eNOS+/− mothers and wt fathers than in female offspring from wt mothers and wt fathers, with fold changes (FC) ranging from 2.54 to 4. In addition, Csad showed lower expression (FC = 0.4) in male offspring from eNOS+/− mothers and wt fathers than in male offspring from wt mothers and wt fathers. Next, we performed gene set enrichment analyses (GSEA) based on Gene Ontology (GO; geneontology.org) terms. Similar to the single gene analyses, more significantly enriched gene sets were detected for female compared to male offspring, where terms with permutation P ≤ 0.0001 were only revealed for female mice (Supplementary Files 1 and 2). In particular, considering the biological process domain of GO the top enriched GO terms (permutation P ≤ 0.0001) comprised the term ‘metabolic process’ and its child nodes (Supplementary File 3).
Figure 5.
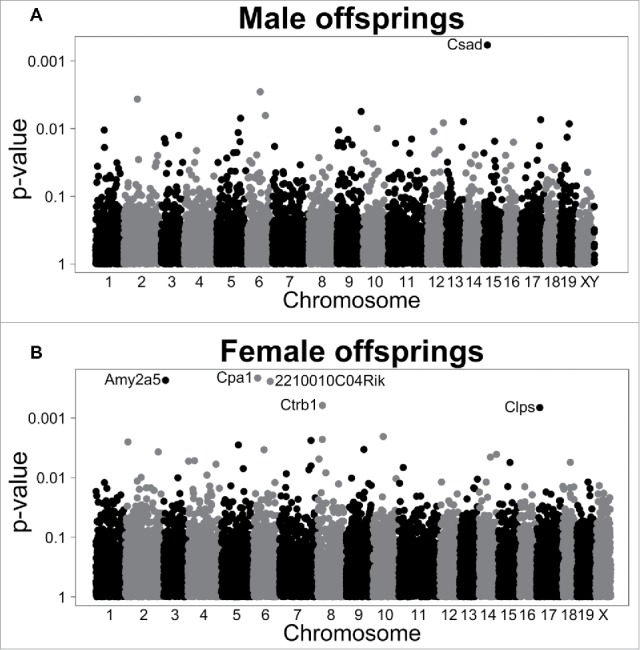
Gene expression in wt offspring. Manhattan plots displaying P-values from differential expression analyses between offspring from wt mothers and wt fathers vs. offspring of eNOS+/− mothers and wt fathers. Analyses were conducted separately in (A) male and (B) female offspring. Genes with P < 0.001 are labeled.
Based on these data, we performed a hypothesis driven approach by analyzing genes potentially involved in liver metabolism and the pathogenesis of fatty liver disease (Supplementary Table S1), since this was the key phenotype in female offspring born to eNOS+/− mothers. Male wt offspring of eNOS+/− mothers and wt fathers expressed significant lower levels of PPARγ and Csad when compared to offspring born to wt mothers and wt fathers. The expression levels of Igf2 and Igf-binding protein 2 (Igf-BP2) were significantly increased. Female wt offspring from eNOS+/− mothers and wt fathers expressed significantly lower levels of Srebf1c and significantly higher levels of Fitm1 and Cdkn1a gene compared to offspring born to wt mothers and wt fathers (Fig. 6 A, B, Supplementary Table S1).
Figure 6.
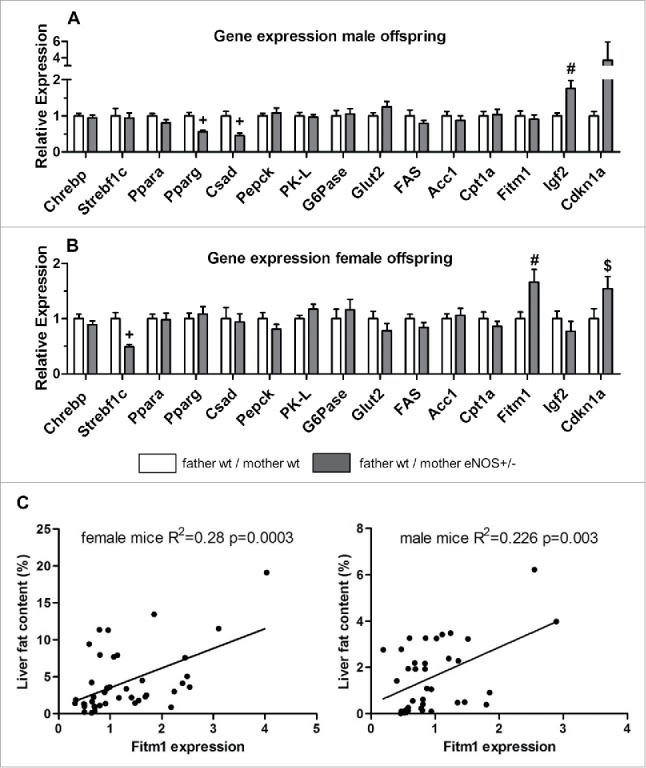
Gene expression in wt offspring. Gene expression (fold expression compared to reference group) was analyzed by real time PCR of (A) male and (B) female wt offspring. (C) Correlation of hepatic Fitm1 gene expression and liver fat content (empty bar, wt father / wt mother; gray bar, wt father / eNOS+/− mother; $ P < 0.05 vs. wt father / wt mother; # P < 0.01 vs. wt father / wt mother; + P < 0.001 vs. wt father / wt mother).
Liver fat ratio correlated significantly with Fitm1 expression (Fig. 6C) in female (correlation coefficient = 0.520) and male (correlation coefficient = 0.476) mice.
Finally, we analyzed the expression levels of selected miRNA implicated in liver metabolism and obesity. For instance, miR-122 and miR-33 regulate hepatic lipid metabolism by influencing the expression of several genes implicated in fatty acid synthesis or oxidation.19 Male, but not female, wt offspring from eNOS+/− mothers and wt fathers expressed significantly more miR-370 (P < 0.05, Fig. 7A,B) when compared to wt offspring born to wt mothers and wt fathers.
Figure 7.
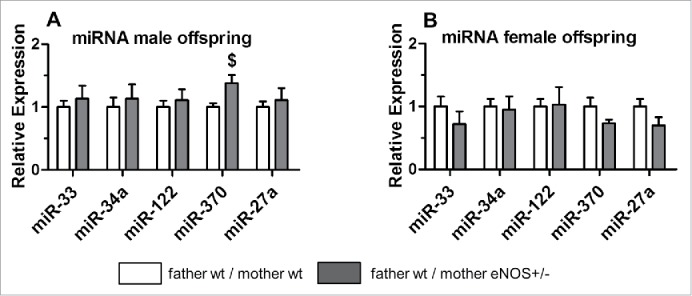
micoRNA expression in wt offspring. MicroRNA (miRNA) expression of selected miRNAs in (A) male and (B) female wt offspring.
However, some of the microarray data could not be confirmed by qRT-PCR [Supplementary Table S4; although the same transcripts were quantified (Supplementary Table S5)] in the total mouse cohort. These discrepancies were most likely caused by gene expression outliers in single animals, distorting the signals in the pooled samples used for microarrays.
Characterization of epigenetic factors
To investigate if epigenetic mechanisms contributed to phenotypic differences, global DNA methylation levels were determined in liver tissue by LC-MS-MS analysis. Significantly higher content of 5-methylcytosine was found in female and male wt offspring born to eNOS+/− mothers and wt fathers (Fig. 8A). Correlation of liver fat content with DNA methylation in female and male (Fig. 8B) wt offspring revealed a significant correlation in female mice (correlation coefficient = 0.533), whereas in male mice there was no correlation (correlation coefficient = −0.158). The expression of the Fitm1 gene correlated significantly with total liver DNA methylation in female (correlation coefficient = 0.369) but not in male offspring (correlation coefficient = −0.047) (Fig. 8C). Next, we performed specific DNA methylation of the candidate gene Fitm1 by MeDIP analysis. DNA methylation revealed significant lower methylation of Fitm1 gene exon 1 in female wt offspring of eNOS+/− mother and wt father (P < 0.05, Fig. 9) compared to wt offspring born to wt mothers. Moreover, DNA methylation of a CpG island of another candidate gene (Cdkn1a) was significantly increased in female wt offspring from eNOS+/− mothers (P < 0.01, Fig. 10).
Figure 8.
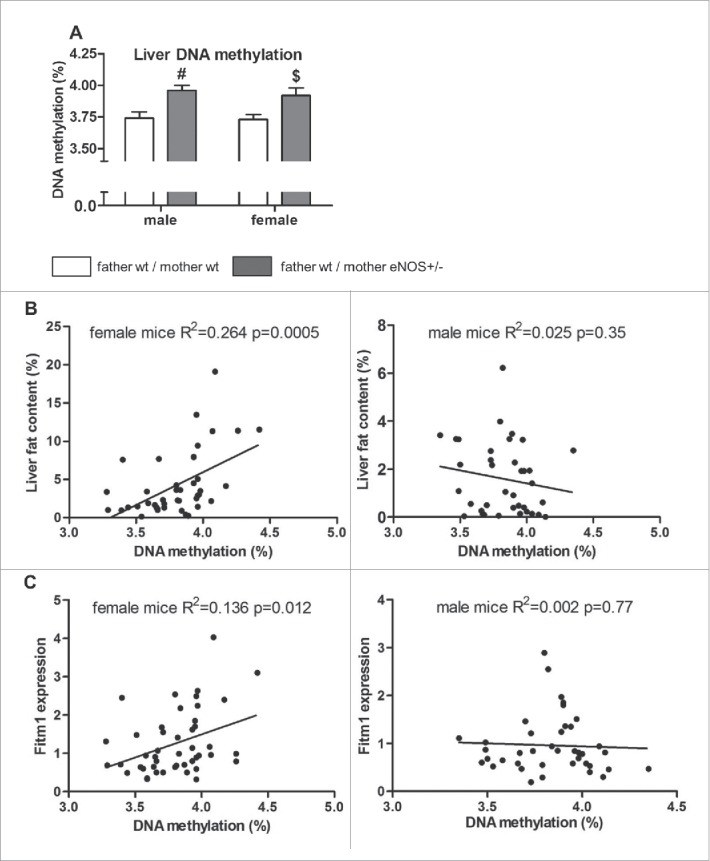
Global DNA methylation in wt offspring. Liver DNA methylation (in % of methylated to total cytosine residues) in (A) male and female wt offspring as well as correlation of liver DNA methylation with (B) liver fat content (in % of red oil positive area in liver tissue) and (C) hepatic Fitm1 gene expression in female and male wt offspring (empty bar, wt father / wt mother; gray bar, wt father / eNOS+/− mother; $ P < 0.05 vs. wt father / wt mother; #: P < 0.01 vs. wt father / wt mother).
Figure 9.
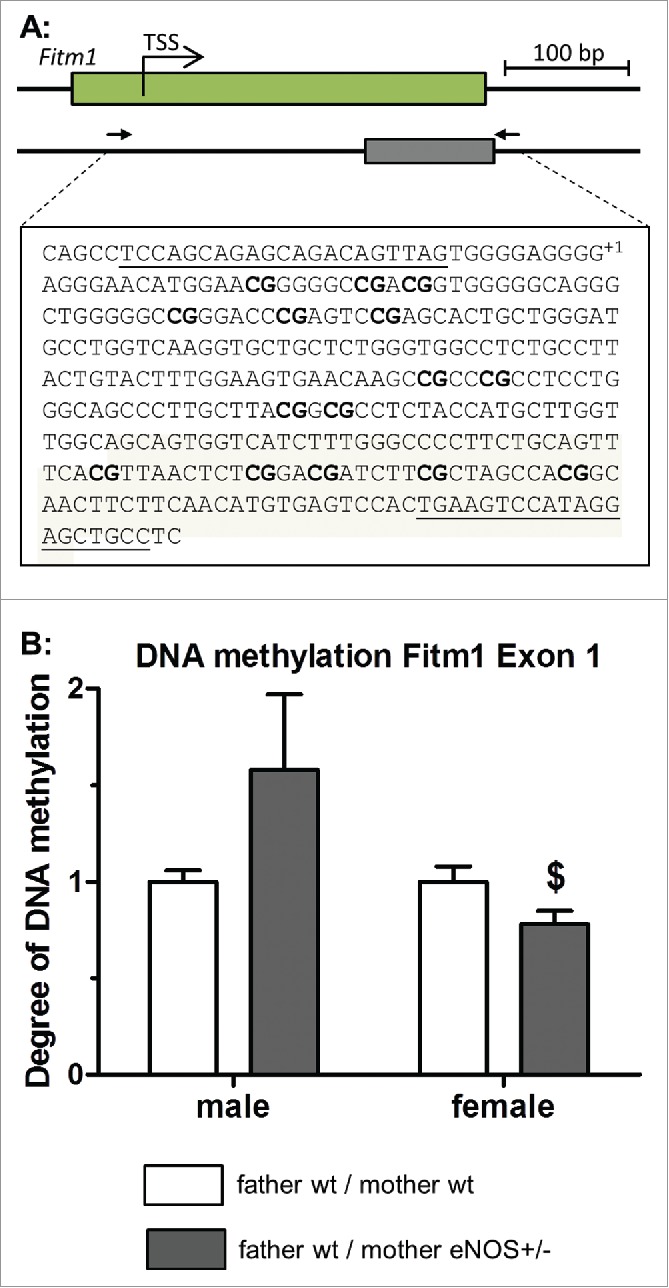
MeDIP DNA methylation analysis of Fitm1 gene. (A) Genomic organization of Fitm1 exon 1, putative transcription start site (TSS), and position of identified CpG island. Amplified sequence is shown in detail (primer binding sites in bold letters and analyzed CpG dinucleotides bold / underlined, TSS indicated with +1), (B) degree of DNA methylation of Fitm1 exon 1 in liver tissue of wt offspring (empty bar, wt father / wt mother; gray bar, wt father / eNOS+/− mother; $ P < 0.05 vs. wt father / wt mother).
Figure 10.
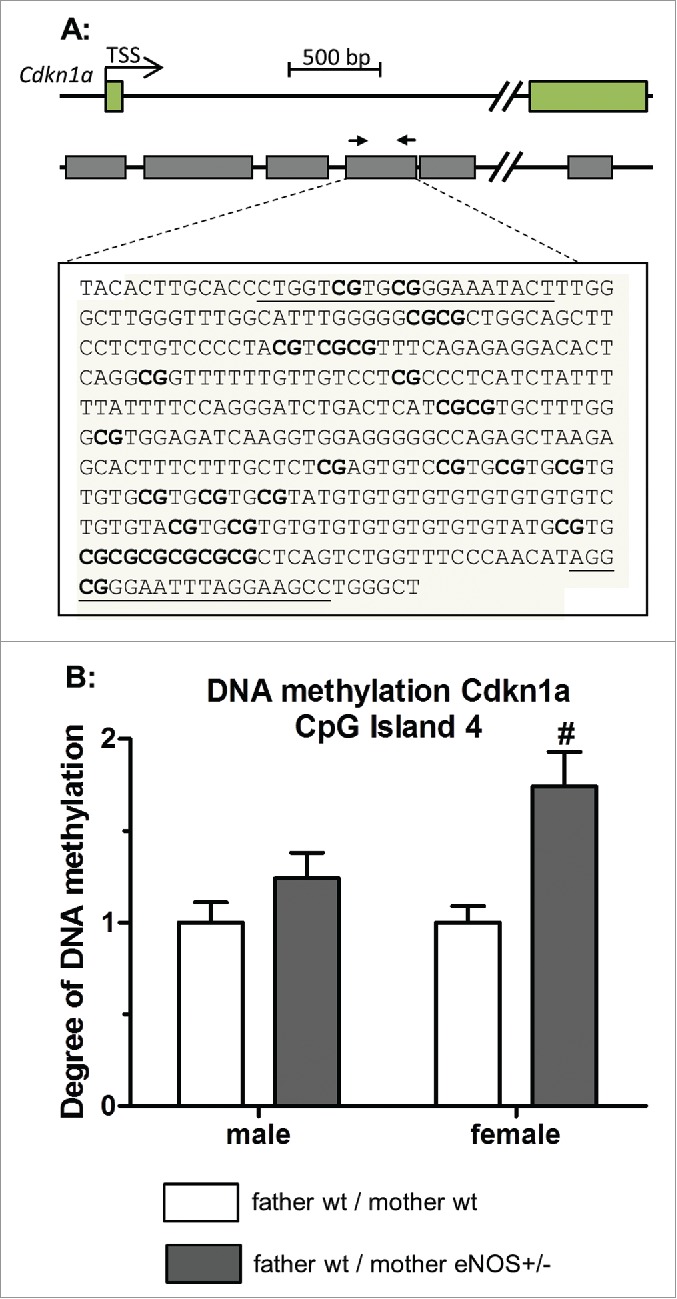
MeDIP DNA methylation analysis of Cdkn1a gene. (A) Genomic organization of the Cdkn1a gene showing first exons, transcription start site (TSS), and the position of identified CpG islands; amplified sequence of CpG island 4 is shown in detail (primer binding sites in bold letters and analyzed CpG dinucleotides bold / underlined). (B) Degree of DNA methylation of CpG island 4 in liver tissue of wt offspring (empty bar, wt father / wt mother; gray bar, wt father / eNOS+/− mother;# P < 0.01 vs. wt father / wt mother).
Discussion
This study was designed to test the advanced fetal programming hypothesis (Fig. 1) stating that maternal genes may affect the fetal phenotype independently of transmission of the gene to the fetus.9-12 To test this hypothesis, we bred female heterozygous eNOS knockout mice with male wild type mice and compared the phenotype of their wild type offspring to the phenotype of offspring with wild type parents. We have chosen eNOS knockout mice to test this hypothesis because eNOS plays a pivotal role in placental function.14-17 The unfavorable intrauterine environment due to reduced maternal eNOS expression has been shown to induce endothelial dysfunction in the offspring, independently of genetic transmission.18 Therefore, we performed an initial screening to identify organs that are affected by the advanced fetal programming process. This analysis revealed that the liver is, in particular, affected by maternal heterozygous eNOS deficiency in female wild type offspring, whereas other organs, such as kidney and heart, seem to be less affected. In the next step, we performed a detailed analysis using morphological and biochemical techniques as well as liver metabolomics to get a thorough understanding of the resulting liver phenotype and underlying molecular mechanisms.
Our study revealed that female offspring born to eNOS+/− deficient female mice developed an increased absolute liver weight, liver fat accumulation, and reduced liver concentrations of fructose 1,6-bisphosphate and fumarate (Figs. 3 and 4). Male offspring born to eNOS+/− female mice had a reduced birth weight (Table 1). Liver concentrations of glucose 6-phosphate, fructose 6-phosphate, and fumarate were reduced (Fig. 4). The concentration of various amino acids (for example, tryptophan, valine, leucine, isoleucine, threonine, phenylalanine) was likewise reduced in the liver of male mice born to female eNOS+/− mice (Table 2). In contrast to female offspring, liver fat accumulation was not affected in male offspring (Fig. 3).
The amount of methylated cytosine as percentage of total cytosine was significantly increased in wild type offspring born to female heterozygous eNOS knockout mice (Fig. 8A). Total liver DNA methylation was significantly related to liver fat accumulation in female offspring—the main phenotypic finding in our study (Fig. 8B).
Nitric oxide (NO) is involved in the local regulation of vascular resistance, promotes angiogenesis, and is a potential regulator of placental steroid biosynthesis and glucose uptake.14 Therefore, deficiency of eNOS is related to the development of intrauterine growth retardation (IUGR).20 It may promote the development of hypoxia due to reduced uterine blood flow and placental oxygenation in pregnant mice, thereby restricting nutrient transport capacity and fetal growth.15-17 This hypothesis is supported by earlier studies showing that the unfavorable intrauterine environment due to diminished maternal eNOS expression can promote endothelial dysfunction in the offspring, independently of genetic transmission.18 Embryonic development is very sensitive to even moderate hypoxia. Hence, maternal heterozygous eNOS deficiency may be causal, because of the crucial role of eNOS in placental function,14-17 which may contribute to fetal tissue hypoxia similar to offspring of mice with complete lack of eNOS (eNOS-/- mice),21 which develop hypertension, and renal and cardiac dysfunction in adult life.
In view of the support with relatively hypoxygenated blood via the portal vein, the liver is particularly vulnerable during hypoxia, and fatty liver is a common complication, for instance, in obstructive sleep apnea.22 Decreased oxygen availability triggers the switch from mitochondrial oxidative phosphorylation to anaerobic glycolysis and affects lipid metabolism and accumulation, generally reducing oxygen consumption while maintaining ATP production.23 Continuous (even mild) placental hypoxia in heterozygous eNOS knockout mothers together with the alterations of placental hormone synthesis due to also mild NO deficiency may lead to epigenetic alterations in growing offspring and increases the sensibility of offspring to metabolic dysfunction in later life, such as the development of a phenotype similar to non-alcoholic fatty liver disease.24 These alterations of placental microcirculation together with the alteration of placental hormone synthesis might cause alterations in the activity of key enzymes involved in the regulation of DNA methylation. DNA methylation together with other epigenetic mechanisms is a key factor of organ development and differentiation in early life. Thus, any NO-related alterations of this tightly regulated differentiation process during early life might affect organ function in later life. However, it is unclear so far why the offspring liver seems to be particularly vulnerable to these NO effects in the placenta. Moreover, early postnatal mechanisms might play a role as well, since the offspring was not adopted by foster mothers; it is also possible that the observed effects could be due to the postnatal environment (amount or quality of the milk in the heterozygous eNOS knockout mice or way of nursing of heterozygous eNOS knockout mice). Moreover, maternal heterozygous eNOS deficiency might also affect the maturation of the genetically normal oocytes in the maternal ovary (Fig. 1).
We demonstrated differences in total and gene specific DNA methylation, hepatic fat accumulation and expression of distinct candidate genes of wild type offspring born to heterozygous eNOS knockout mothers and wild type fathers and a clear correlation of DNA methylation and hepatic fat accumulation in female offspring (Fig. 8B). Genes involved in hepatic lipid metabolism or mitochondrial activity do not contribute to the observed lipid accumulation in female offspring (Fig. 6B, Supplementary Table S1). Changes of lipid content may also be related to alteration in hepatic lipid accumulation, influx or export.25 Fat storage inducing transmembrane protein (FITM) 1 belongs to a recently identified family of proteins of the endoplasmic reticulum that induce lipid droplet accumulation, while it is not involved in triglyceride biosynthesis. FITM proteins are regulated by PPAR α and γ and their upregulation leads to accumulation of lipid droplets.26 Fitm1 gene showed altered DNA methylation in the intragenic region of exon 1 in female wild type offspring from eNOS+/− mother, which is in line with the higher level of Fitm1 gene expression in these animals (Figs. 6B and 9). Brenet et al.27 showed that not only promoter methylation status, but also DNA methylation of intragenic regions, especially of the first exon, influences gene expression. Otherwise, there is no direct evidence that methylation of this genomic region influences Fitm1 gene expression. The positive correlation between Fitm1 gene expression and DNA methylation in female offspring suggests further effects of DNA methylation, with a possible indirect effect on repressor regions of the Fitm1 gene (Fig. 8B).
Liver Cdkn1a expression was also increased in the wild type offspring from female heterozygous eNOS knockout mice (Fig. 6B), a factor previously described to be upregulated in fatty liver.28,29 Bioinformatic analysis predicted Cdkn1a to be a future epigenetic biomarker for obesity, susceptible for early life changes in promoter methylation, through its implication in cell cycle progression and adipogenesis and the high abundance of CpG islands.30 We found differences in Cdkn1a DNA methylation, but further studies are need to elucidate how CpG island methylation influences Cdkn1a gene expression.
The observed decrease in Srebf1c expression could be due to compensatory regulation to avoid further hepatic fat accumulation. Similarly, Anavi et al.31 found that hepatic Srebf1c expression in rats is reduced after infusion of a lipid emulsion.
The observed differences in gene expression do not account for all phenotypic alterations conditioned by maternal eNOS+/− deficiency. Microarray analysis of gene expression failed to identify further promising candidates since subsequent RT-PCR analyses revealed that most of these genes were not equally altered in all animals (Supplementary Table S4). In line with this, other studies of the transcriptome in animal models of fetal programming showed that the most differently regulated genes seem not to be related to the observed phenotype,32,33 indicating that our current understanding of the cascade of biochemical alterations induced by early life stimuli, such as maternal mild NO deficiency during pregnancy, is far from being complete. In particular, we do not understand why the resulting phenotype is different in male and female offspring, although the stimulus—mild NO deficiency—is the same for female and male offspring. Sex differences in fetal programming to the same stimuli, however, are broadly described.5 A general answer to this question might be the observation that the timing of DNA methylation is different in males and females during early life development and, moreover, male and female sex steroid hormone synthesized by the fetus and the placenta might also modulate the effects of mild NO deficiency during pregnancy on epigenetic and phenotypic alterations in the offspring in a gender-dependent manner.
In male wt offspring born to eNOS+/− mothers and wt fathers, the liver tissue concentration of most amino acids was slightly reduced, by about 30%. Exceptions were those amino acids that are either directly involved in ammonia metabolism (glutamic acid, glutamine, aspartic acid, arginine) or gluconeogenesis (alanine, glycine, serine). The reduction of the amino acid concentration closely parallels the percent increase in liver glycogen content of these animals, indicating that the observed decrease might be due to the way of calculating this parameter, since the liver amino acid concentration was given as amount of amino acids per gram wet weight.
We also analyzed the expression of miRNA related to lipid homeostasis, but the observed differences did not explain the hepatic phenotype in the offspring. In addition, other epigenetic mechanisms such as histone modification or further alterations in miRNA expression could be involved.34 Thus, epigenetic modifications of yet unknown genes might have caused the observed differences in gene expression.
Fetal programming is a multifactorial process, and minor differences in promoter methylation and expression of a large number of genes may significantly contribute to the observed outcome. This complex scenario and the limitations of current biomedical technologies explain—together with the above-described reasons—why it has been impossible to describe all epigenetic alterations induced by parental genetic or early life parental nutritional stimuli in detail.
Our observations support the advanced fetal programming hypothesis10,35 and propose a non-environmental mechanism of fetal programming driven by altered parental gene function. Maternal, and possibly also paternal, genes may influence the epigenome of maturing sperm, oocyte, and later embryo/fetus and alter the phenotype of the fetus in later life, independently of the fetal genome.35 This hypothesis has major implications: i) It breaks with the classical laws of inheritance. According to classical genetics the phenotype of wild type offspring born to wild type mothers or heterozygous eNOS knockout mice should be identical, but this was not the case. By contrast, our study showed that a parental gene affects the phenotype in the offspring without genetic transmission. ii) It suggests reassessing one of the most important tools currently used to understand gene function: murine transgenic or knockout animal models. Results of this study indicate that genetically manipulated animal models may not only reflect causality between a certain genetic alteration and a resulting phenotype. Altered gene function may additionally induce permanent epigenetic changes, thus impacting the offspring phenotype even without transmittance of the modified maternal or paternal gene.
Material and methods
Breeding protocol
The entire study protocol was approved by the animal welfare comity of the state of Berlin, Germany. We used the eNOS knockout mice36 of the C57BL/6J strain and their wild type (wt) littermate. The breeding procedure is described in Supplementary Fig. S6. Female wt mice were crossbred with homozygous male eNOS knockout mice. The resulting female heterozygous eNOS knockout (eNOS+/−) mice were then again crossed with wt male mice. Only wt offspring of this breeding procedure (F2 generation, see Supplementary Fig. S6) entered the study. These mice were compared to wt mice resulting from crossing wt male and wt female mice. Study design and experimental protocols were conducted according to the local institutional guidelines for the care and use of laboratory animals.
Study protocol
Male and female offspring were kept for 24 weeks and analyzed separately. Body weight, length, and abdominal diameter of the F2 generation were measured daily until day 13; thereafter, body weight was measured daily until day 40 and weekly thereafter until week 20 of the experiment. Blood pressure was measured using the tail cuff method at week 24, as previously described.37 GFR was calculated from urine and plasma creatinine at week 23. Fasting glucose level was determined at study week 13 and 21.
Histology
Liver tissue was stained with hematoxylin eosin to determine lobular dimensions and the extent of lobular inflammation as described in supplementary methods. Red oil staining was done as described elsewhere.38 For further details see Supplementary methods.
Immunohistochemical staining
Liver sections were immunostained for CD68 protein as described in Supplementary material. The number of CD68-positive macrophages in the liver was quantified as described previously.39
Liver glycogen content
Hepatic glycogen content was determined using the amyloglucosidase method, as described previously.40 See Supplementary material for further details.
Western blotting
Immunoblotting of liver proteins was examined as previously described.37,41 For further details see Supplementary methods.
Determination of central carbon metabolites in liver tissue
Metabolite concentrations in liver tissue homogenate were determined by gas chromatography-mass spectrometry (GC-MS) analysis and liquid chromatography-tandem mass spectrometry (LC-MS-MS) as described previously42,43 with minor modifications. Detailed method was described in Supplementary material.
Quantitative real time PCR
Determination of gene expression level with quantitative real time PCR was performed as previously described.44 See Supplementary methods for further details.
MeDIP assay
Immunoprecipitation of methylated genomic DNA (MeDIP assay) was performed as described by Weber et al.45 with minor modifications. Briefly, liver DNA was sonicated and precipitated with antibody against 5-methylcytosine. The amount of methylated DNA was quantified with quantitative real time PCR, comparing the appearance of specific DNA sequence in the precipitated and input DNA. See Supplementary methods for further details.
Microarray gene expression analysis and GSEA
High quality RNA samples from the liver of 9 male and female offspring of wt fathers and wt mothers, as well as 6 male and female wt offspring from wt fathers and eNOS+/− knockout mothers were selected for gene expression profiling. Pooled samples consisting of 3 samples each were analyzed using Affymetrix GeneChip Mouse Gene ST 1.0 arrays (Affymetrix, Santa Clara, CA). Microarray data was pre-processed using the RMA (Robust Multichip average)46 algorithm in Expression Console 1.1 software (Affymetrix). Only genes with signal intensities > 27 = 128 on minimum 2 out of the 5 arrays with male/female mouse pools were further investigated. Differences in gene expression levels between male and female mice with wt or eNOS+/− mother were analyzed by package limma-3.8.247 of statistical software R-2.13.0 (www.r-project.org). A P-value < 0.05 was considered to indicate a significant difference. Manhattan plots were used to display P-values from limma analyses.
Based on the fold changes from limma analyses mentioned above GSEA of GO terms was performed by Bioconductor package clusterProfiler_2.4.248 (www.bioconductor.org) for female as well for male offspring. To be more precise, GSEA was conducted separately for each of the 3 GO domains (biological process, cellular component, and molecular function) using a minimal gene set size of 30 and 9999 permutations. To assign probe set IDs of the Affymetrix array to GO terms for GSEA, packages clusterProfiler and mogene10sttranscriptcluster.db _8.4.0 were first applied to assign probe set IDs to Entrez IDs (www.ncbi.nlm.nih.gov/gene) and, thereby, to GO terms. If several probe set IDs were present for one Entrez ID, the result (fold change) of the probe set showing the smallest P-value in limma analysis was used as input for GSEA.
Quantification of total DNA methylation
Five-methyl-2'-deoxycytidine content was determined by LC-MS-MS analysis (details see Supplementary Material). Briefly, genomic DNA samples were hydrolyzed by treatment with nuclease P1 and alkaline phosphatase. After hydrolysis, 2'-deoxycytidine and 5-methyl-2'-deoxycytidine were measured by LC-MS-MS analysis using the respective stable isotope-labeled analogs [15N3]deoxycytidine and 5-[2H3]methyl-deoxycytidine as internal standards. DNA methylation status is given as percentage of 5-methyl-2'-deoxycytidine content relative to total cytosine residues.
Statistics
Statistical analysis was performed using IBM SPSS statistics, version 19. All values are presented as mean ± SEM unless noticed differently. For all data Mann-Whitney-U test was performed and a P-value < 0.05 was considered to indicate a significant difference.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgements
We gratefully acknowledge Monika Elbl, Andrea Jarmuth and Monika Seiler for excellent technical assistance.
Funding
This study was supported by the Deutsche Forschungsgemeinschaft to BH, the chinese National Natural Science Foundation of China (no. 81300557) to JL, and a grant of the Robert Bosch Foundation to MS, SW, UH and ES.
References
- 1.Barker DJP. The developmental origins of adult disease. J Am Coll Nutr 2004; 23:588S-95S; PMID:15640511; http://dx.doi.org/ 10.1080/07315724.2004.10719428 [DOI] [PubMed] [Google Scholar]
- 2.Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med 2008; 359:61-73; PMID:18596274; http://dx.doi.org/ 10.1056/NEJMra0708473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ravelli AC, van der Meulen JH, Michels RP, Osmond C, Barker DJ, Hales CN, Bleker OP. Glucose tolerance in adults after prenatal exposure to famine. Lancet 1998; 351:173-7; PMID:9449872; http://dx.doi.org/ 10.1016/S0140-6736(97)07244-9 [DOI] [PubMed] [Google Scholar]
- 4.Vehaskari VM, Aviles DH, Manning J. Prenatal programming of adult hypertension in the rat. Kidney Int 2001; 59:238-45; PMID:11135076; http://dx.doi.org/ 10.1046/j.1523-1755.2001.00484.x [DOI] [PubMed] [Google Scholar]
- 5.Seckl JR, Cleasby M, Nyirenda MJ. Glucocorticoids, 11beta-hydroxysteroid dehydrogenase, and fetal programming. Kidney Int 2000; 57:1412-7; PMID:10760076; http://dx.doi.org/ 10.1046/j.1523-1755.2000.00984.x [DOI] [PubMed] [Google Scholar]
- 6.Aufdenblatten M, Baumann M, Raio L, Dick B, Frey BM, Schneider H, Surbek D, Hocher B, Mohaupt MG. Prematurity is related to high placental cortisol in preeclampsia. Pediatr Res 2009; 65:198-202; PMID:19047954; http://dx.doi.org/ 10.1203/PDR.0b013e31818d6c24 [DOI] [PubMed] [Google Scholar]
- 7.Thone-Reineke C, Kalk P, Dorn M, Klaus S, Simon K, Pfab T, Godes M, Persson P, Unger T, Hocher B. High-protein nutrition during pregnancy and lactation programs blood pressure, food efficiency, and body weight of the offspring in a sex-dependent manner. Am J Physiol Regul Integr Comp Physiol 2006; 291:R1025-30; PMID:16675628; http://dx.doi.org/ 10.1152/ajpregu.00898.2005 [DOI] [PubMed] [Google Scholar]
- 8.Parkhurst SM, Ish-Horowicz D. wimp, a dominant maternal-effect mutation, reduces transcription of a specific subset of segmentation genes in Drosophila. Genes Dev 1991; 5:341-57; PMID:2001838; http://dx.doi.org/ 10.1101/gad.5.3.341 [DOI] [PubMed] [Google Scholar]
- 9.Hocher B, Slowinski T, Stolze T, Pleschka A, Neumayer HH, Halle H. Association of maternal G protein beta3 subunit 825T allele with low birthweight. Lancet 2000; 355:1241-2; PMID:10770310; http://dx.doi.org/ 10.1016/S0140-6736(00)02094-8 [DOI] [PubMed] [Google Scholar]
- 10.Hocher B, Slowinski T, Bauer C, Halle H. The advanced fetal programming hypothesis. Nephrol Dial Transplant 2001; 16:1298-9; PMID:11390742; http://dx.doi.org/ 10.1093/ndt/16.6.1298 [DOI] [PubMed] [Google Scholar]
- 11.Masuda K, Osada H, Iitsuka Y, Seki K, Sekiya S. Positive association of maternal G protein beta3 subunit 825T allele with reduced head circumference at birth. Pediatr Res 2002; 52:687-91; PMID:12409514 [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Zuckerman B, Pearson C, Kaufman G, Chen C, Wang G, Niu T, Wise PH, Bauchner H, Xu X. Maternal cigarette smoking, metabolic gene polymorphism, and infant birth weight. JAMA 2002; 287:195-202; PMID:11779261; http://dx.doi.org/ 10.1001/jama.287.2.195 [DOI] [PubMed] [Google Scholar]
- 13.Petry CJ, Ong KK, Dunger DB. Does the fetal genotype affect maternal physiology during pregnancy? Trends Mol Med 2007; 13:414-21; PMID:17900986; http://dx.doi.org/ 10.1016/j.molmed.2007.07.007 [DOI] [PubMed] [Google Scholar]
- 14.Vatish M, Randeva HS, Grammatopoulos DK. Hormonal regulation of placental nitric oxide and pathogenesis of pre-eclampsia. Trends Mol Med 2006; 12:223-33; PMID:16616640; http://dx.doi.org/ 10.1016/j.molmed.2006.03.003 [DOI] [PubMed] [Google Scholar]
- 15.Skarzinski G, Khamaisi M, Bursztyn M, Mekler J, Lan D, Evdokimov P, Ariel I. Intrauterine growth restriction and shallower implantation site in rats with maternal hyperinsulinemia are associated with altered NOS expression. Placenta 2009; 30:898-906; PMID:19709742; http://dx.doi.org/ 10.1016/j.placenta.2009.07.014 [DOI] [PubMed] [Google Scholar]
- 16.Kusinski LC, Stanley JL, Dilworth MR, Hirt CJ, Andersson IJ, Renshall LJ, Baker BC, Baker PN, Sibley CP, Wareing M, et al.. eNOS knockout mouse as a model of fetal growth restriction with an impaired uterine artery function and placental transport phenotype. Am J Physiol Regul Integr Comp Physiol 2012; 303:R86-93; PMID:22552791; http://dx.doi.org/ 10.1152/ajpregu.00600.2011 [DOI] [PubMed] [Google Scholar]
- 17.Kulandavelu S, Whiteley KJ, Qu D, Mu J, Bainbridge SA, Adamson SL. Endothelial nitric oxide synthase deficiency reduces uterine blood flow, spiral artery elongation, and placental oxygenation in pregnant mice. Hypertension 2012; 60:231-8; PMID:22615111; http://dx.doi.org/ 10.1161/HYPERTENSIONAHA.111.187559 [DOI] [PubMed] [Google Scholar]
- 18.Costantine MM, Ghulmiyyah LM, Tamayo E, Hankins GDV, Saade GR, Longo M. Transgenerational effect of fetal programming on vascular phenotype and reactivity in endothelial nitric oxide synthase knockout mouse model. Am J Obstet Gynecol 2008; 199:250.e1-7; PMID:18771972; http://dx.doi.org/ 10.1016/j.ajog.2008.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore KJ, Rayner KJ, Suárez Y, Fernández-Hernando C. The role of microRNAs in cholesterol efflux and hepatic lipid metabolism. Annu Rev Nutr 2011; 31:49-63; PMID:21548778; http://dx.doi.org/ 10.1146/annurev-nutr-081810-160756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schiessl B, Mylonas I, Hantschmann P, Kuhn C, Schulze S, Kunze S, Friese K, Jeschke U. Expression of endothelial NO synthase, inducible NO synthase, and estrogen receptors alpha and beta in placental tissue of normal, preeclamptic, and intrauterine growth-restricted pregnancies. J Histochem Cytochem 2005; 53:1441-9; PMID:15983116; http://dx.doi.org/ 10.1369/jhc.4A6480.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kulandavelu S, Whiteley KJ, Bainbridge SA, Qu D, Adamson SL. Endothelial NO synthase augments fetoplacental blood flow, placental vascularization, and fetal growth in mice. Hypertension 2013; 61:259-66; PMID:23150513; http://dx.doi.org/ 10.1161/HYPERTENSIONAHA.112.201996 [DOI] [PubMed] [Google Scholar]
- 22.Musso G, Cassader M, Olivetti C, Rosina F, Carbone G, Gambino R. Association of obstructive sleep apnoea with the presence and severity of non-alcoholic fatty liver disease. A systematic review and meta-analysis. Obes Rev 2013; 14:417-31; PMID:23387384; http://dx.doi.org/ 10.1111/obr.12020 [DOI] [PubMed] [Google Scholar]
- 23.Goda N, Kanai M. Hypoxia-inducible factors and their roles in energy metabolism. Int J Hematol 2012; 95:457-63; PMID:22535382; http://dx.doi.org/ 10.1007/s12185-012-1069-y [DOI] [PubMed] [Google Scholar]
- 24.Thompson LP, Al-Hasan Y. Impact of oxidative stress in fetal programming. J Pregnancy 2012; 2012:582748; PMID:22848830; http://dx.doi.org/ 10.1155/2012/582748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reddy JK, Rao MS. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am J Physiol Gastrointest Liver Physiol 2006; 290:G852-8; PMID:16603729; http://dx.doi.org/ 10.1152/ajpgi.00521.2005 [DOI] [PubMed] [Google Scholar]
- 26.Kadereit B, Kumar P, Wang W-J, Miranda D, Snapp EL, Severina N, Torregroza I, Evans T, Silver DL. Evolutionarily conserved gene family important for fat storage. Proc Natl Acad Sci USA 2008; 105:94-9; PMID:18160536; http://dx.doi.org/ 10.1073/pnas.0708579105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brenet F, Moh M, Funk P, Feierstein E, Viale AJ, Socci ND, Scandura JM. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS One 2011; 6:e14524; PMID:21267076; http://dx.doi.org/ 10.1371/journal.pone.0014524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takasaki M, Honma T, Yanaka M, Sato K, Shinohara N, Ito J, Tanaka Y, Tsuduki T, Ikeda I. Continuous intake of a high-fat diet beyond one generation promotes lipid accumulation in liver and white adipose tissue of female mice. J Nutr Biochem 2012; 23:640-5; PMID:21775120; http://dx.doi.org/ 10.1016/j.jnutbio.2011.03.008 [DOI] [PubMed] [Google Scholar]
- 29.Dudley KJ, Sloboda DM, Connor KL, Beltrand J, Vickers MH. Offspring of mothers fed a high fat diet display hepatic cell cycle inhibition and associated changes in gene expression and DNA methylation. PLoS ONE 2011; 6:e21662; PMID:21779332; http://dx.doi.org/ 10.1371/journal.pone.0021662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campión J, Milagro FI, Martínez JA. Individuality and epigenetics in obesity. Obes Rev 2009; 10:383-92; PMID:Can't; http://dx.doi.org/ 10.1111/j.1467-789X.2009.00595.x [DOI] [PubMed] [Google Scholar]
- 31.Anavi S, Ilan E, Tirosh O, Madar Z. Infusion of a lipid emulsion modulates AMPK and related proteins in rat liver, muscle, and adipose tissues. Obesity (Silver Spring) 2010; 18:1108-15; PMID:20057367; http://dx.doi.org/ 10.1038/oby.2009.489 [DOI] [PubMed] [Google Scholar]
- 32.Mischke M, Pruis MGM, Boekschoten MV, Groen AK, Fitri AR, van de Heijning BJM, Verkade HJ, Müller M, Plösch T, Steegenga WT. Maternal Western-style high fat diet induces sex-specific physiological and molecular changes in two-week-old mouse offspring. PLoS One 2013; 8:e78623; PMID:24223833; http://dx.doi.org/ 10.1371/journal.pone.0078623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lillycrop KA, Rodford J, Garratt ES, Slater-Jefferies JL, Godfrey KM, Gluckman PD, Hanson MA, Burdge GC. Maternal protein restriction with or without folic acid supplementation during pregnancy alters the hepatic transcriptome in adult male rats. Br J Nutr 2010; 103:1711-9; PMID:20211039; http://dx.doi.org/ 10.1017/S0007114509993795 [DOI] [PubMed] [Google Scholar]
- 34.Laker RC, Wlodek ME, Connelly JJ, Yan Z. Epigenetic origins of metabolic disease: The impact of the maternal condition to the offspring epigenome and later health consequences. Food Science and Human Wellness 2013; 2:1-11; PMID:NOT_FOUND; http://dx.doi.org/ 10.1016/j.fshw.2013.03.002 [DOI] [Google Scholar]
- 35.Hocher B. More than genes: the advanced fetal programming hypothesis. J Reprod Immunol 2014; 104-105:8-11; PMID:24721253; http://dx.doi.org/ 10.1016/j.jri.2014.03.001 [DOI] [PubMed] [Google Scholar]
- 36.Gödecke A, Decking UK, Ding Z, Hirchenhain J, Bidmon HJ, Gödecke S, Schrader J. Coronary hemodynamics in endothelial NO synthase knockout mice. Circ Res 1998; 82:186-94; PMID:9468189; http://dx.doi.org/ 10.1161/01.RES.82.2.186 [DOI] [PubMed] [Google Scholar]
- 37.Quaschning T, Voss F, Relle K, Kalk P, Vignon-Zellweger N, Pfab T, Bauer C, Theilig F, Bachmann S, Kraemer-Guth A, et al.. Lack of endothelial nitric oxide synthase promotes endothelin-induced hypertension: lessons from endothelin-1 transgenic/endothelial nitric oxide synthase knockout mice. J Am Soc Nephrol 2007; 18:730-40; PMID:17287431; http://dx.doi.org/ 10.1681/ASN.2006050541 [DOI] [PubMed] [Google Scholar]
- 38.Lillie R, Ashburn L. Supersaturated solutions of fat stains in dilute isopropanol for demonstration of acute fatty degeneration not shown by Herxheimers's technique. Archs Path 1943; :432(36). [Google Scholar]
- 39.van den Broek MAJ, Shiri-Sverdlov R, Schreurs JJW, Bloemen JG, Bieghs V, Rensen SS, Dejong CHC, Olde Damink SWM. Liver manipulation during liver surgery in humans is associated with hepatocellular damage and hepatic inflammation. Liver Int 2013; 33:633-41; PMID:23356550; http://dx.doi.org/ 10.1111/liv.12051 [DOI] [PubMed] [Google Scholar]
- 40.Bergmeyer (Hrsg) Methoden der enzymatischen Analyse. Weinheim/Bergstr: Verlag Chemie; 1962. [Google Scholar]
- 41.Vignon-Zellweger N, Rahnenführer J, Theuring F, Hocher B. Analysis of cardiac and renal endothelin receptors by in situ hybridization in mice. Clin Lab 2012; 58:939-49; PMID:23163110 [PubMed] [Google Scholar]
- 42.Hofmann U, Maier K, Niebel A, Vacun G, Reuss M, Mauch K. Identification of metabolic fluxes in hepatic cells from transient 13C-labeling experiments: Part I. Experimental observations. Biotechnol Bioeng 2008; 100:344-54; PMID:18095337; http://dx.doi.org/ 10.1002/bit.21747 [DOI] [PubMed] [Google Scholar]
- 43.Maier K, Hofmann U, Reuss M, Mauch K. Dynamics and control of the central carbon metabolism in hepatoma cells. BMC Syst Biol 2010; 4:54; PMID:20426867; http://dx.doi.org/ 10.1186/1752-0509-4-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chaykovska L, von Websky K, Rahnenführer J, Alter M, Heiden S, Fuchs H, Runge F, Klein T, Hocher B. Effects of DPP-4 inhibitors on the heart in a rat model of uremic cardiomyopathy. PLoS One 2011; 6:e27861; PMID:22125632; http://dx.doi.org/ 10.1371/journal.pone.0027861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, Schübeler D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 2005; 37:853-62; PMID:16007088; http://dx.doi.org/ 10.1038/ng1598 [DOI] [PubMed] [Google Scholar]
- 46.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 2003; 31:e15; PMID:12582260; http://dx.doi.org/ 10.1093/nar/gng015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smyth G. Limma: linear models for microarray data. In: Bioinformatics and Computational Biology Solutions using R and Bioconductor. New York: Springer; 2005. [Google Scholar]
- 48.Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012; 16:284-7; PMID:22455463; http://dx.doi.org/ 10.1089/omi.2011.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chmelíková E, Jeseta M, Sedmíková M, Petr J, Tůmová L, Kott T, Lipovová P, Jílek F. Nitric oxide synthase isoforms and the effect of their inhibition on meiotic maturation of porcine oocytes. Zygote 2010; 18:235-44; PMID:Can't; http://dx.doi.org/ 10.1017/S0967199409990268 [DOI] [PubMed] [Google Scholar]
- 50.Rosselli M, Keller PJ, Dubey RK. Role of nitric oxide in the biology, physiology and pathophysiology of reproduction. Hum Reprod Update 1998; 4:3-24; PMID:9622410; http://dx.doi.org/ 10.1093/humupd/4.1.3 [DOI] [PubMed] [Google Scholar]
- 51.Gammie SC, Huang PL, Nelson RJ. Maternal aggression in endothelial nitric oxide synthase-deficient mice. Horm Behav 2000; 38:13-20; PMID:10924282; http://dx.doi.org/ 10.1006/hbeh.2000.1595 [DOI] [PubMed] [Google Scholar]
- 52.Cieslar SRL, Madsen TG, Purdie NG, Trout DR, Osborne VR, Cant JP. Mammary blood flow and metabolic activity are linked by a feedback mechanism involving nitric oxide synthesis. J Dairy Sci 2014; 97:2090-100; PMID:24508428; http://dx.doi.org/ 10.3168/jds.2013-6961 [DOI] [PubMed] [Google Scholar]
- 53.Gu L, Liu H, Gu X, Boots C, Moley KH, Wang Q. Metabolic control of oocyte development: linking maternal nutrition and reproductive outcomes. Cell Mol Life Sci 2015; 72:251-71; PMID:25280482; http://dx.doi.org/ 10.1007/s00018-014-1739-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Szyf M. The early life environment and the epigenome. Biochim Biophys Acta 2009; 1790:878-85; PMID:19364482; http://dx.doi.org/ 10.1016/j.bbagen.2009.01.009 [DOI] [PubMed] [Google Scholar]
- 55.Kuzawa CW, Thayer ZM. Timescales of human adaptation: the role of epigenetic processes. Epigenomics 2011; 3:221-34; PMID:22122283; http://dx.doi.org/ 10.2217/epi.11.11 [DOI] [PubMed] [Google Scholar]
- 56.Januar V, Desoye G, Novakovic B, Cvitic S, Saffery R. Epigenetic regulation of human placental function and pregnancy outcome: considerations for causal inference. Am J Obstet Gynecol 2015; 213:S182-96; PMID:26428498; http://dx.doi.org/ 10.1016/j.ajog.2015.07.011 [DOI] [PubMed] [Google Scholar]
- 57.Toyota M, Suzuki H. Epigenetic drivers of genetic alterations. Adv Genet 2010; 70:309-23; PMID:20920753; http://dx.doi.org/ 10.1016/B978-0-12-380866-0.60011-3 [DOI] [PubMed] [Google Scholar]
- 58.Duncan BK, Miller JH. Mutagenic deamination of cytosine residues in DNA. Nature 1980; 287:560-1; PMID:6999365; http://dx.doi.org/ 10.1038/287560a0 [DOI] [PubMed] [Google Scholar]
- 59.Yu H-L, Dong S, Gao L-F, Li L, Xi Y-D, Ma W-W, Yuan L-H, Xiao R. Global DNA methylation was changed by a maternal high-lipid, high-energy diet during gestation and lactation in male adult mice liver. Br J Nutr 2015; 113:1032-9; PMID:25778733; http://dx.doi.org/ 10.1017/S0007114515000252 [DOI] [PubMed] [Google Scholar]
- 60.Gabory A, Attig L, Junien C. Developmental programming and epigenetics. Am J Clin Nutr 2011; 94:1943S-1952S; PMID:2204916; http://dx.doi.org/ 10.3945/ajcn.110.000927 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.