

Abstract
Implementing a center-wide precision medicine strategy at a major cancer center is a true multidisciplinary effort and requires comprehensive alignment of a broad screening strategy with a clinical research enterprise that can use these data to accelerate development of new treatments. Here, we describe the genomic screening approach at Memorial Sloan Kettering Cancer Center, a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology designated MSK-IMPACT, and how it enables and supports a large clinical trial portfolio enriched for multi-histology, biomarker-selected, ‘basket’ studies of targeted therapies.
Introduction
The therapeutic value of targeting oncogenic alterations responsible for the growth and metastasis of cancer has now been well established in a variety of clinical settings including human epidermal growth factor (HER2[s1])-positive breast cancer [1,2], BRAF mutant melanoma [3], lung cancers with alterations of epidermal growth factor receptor (EGFR)/anaplastic lymphoma kinase (ALK)/ROS1 [4-6] and KIT/platelet-derived growth factor receptor alpha (PDGFRA) mutant gastrointestinal stromal tumors (GISTs) [7]. Technical improvements in our ability to profile tumors molecularly as well as the rapidly expanding portfolio of therapeutic agents targeting a variety of cell-signaling networks have converged to create an unparalleled opportunity to pursue precision medicine initiatives in the clinic. Acknowledging this changing medical landscape, the US National Institutes of Health recently announced a US$215 million Precision Medicine Initiative, which will initially focus on cancer therapeutics [8].
Recognizing these important trends, Memorial Sloan Kettering Cancer Center has been engaged in a multiyear strategic plan to integrate precision medicine comprehensively into our entire clinical research enterprise. This multidisciplinary effort has required the close collaboration of hospital leadership, oncology, pathology, molecular diagnostics, computational biology, clinical and translational researchers and pharmaceutical partners to meet the needs of our patients in a rapidly changing environment. Herein, we present our experience as a potential template for institutions contemplating similar initiatives.
Molecular screening strategy
From the outset, our precision medicine strategy has been based on the concept of offering molecular screening to a broad base of patients with advanced cancer with the long-term goal of screening all new patients. The primary hurdle to achieving this goal is that the majority of patients cared for at our institution (and elsewhere) do not have tumor types for which molecular testing is required to guide standard-of-care therapy. Whereas, for example, tumor testing for EGFR mutations and ALK fusions is now broadly accepted as a component of standard care in patients with advanced lung adenocarcinomas, for many common solid tumors including cancers of the prostate, ovary, uterus and others the choice of standard therapy is not dictated by somatic mutational status and thus molecular profiling remains investigational. In other cancers, including those of the breast and esophagus, limited molecular profiling is performed, but the standard methodologies employed (fluorescent in situ hybridization and immunohistochemistry for ERBB2/HER2) do not enable a broader assessment of somatic mutational events. Thus, to ensure patients would have access to molecular screening regardless of cancer type, we initiated a clinical trial (NCT01775072) to consent patients to broad mutational profiling and paired this clinical study with institutional resources to support the in-depth genetic testing of tumor types where it is not currently reimbursable or where reimbursement is insufficient to cover the cost. This research consent was modeled after consents developed for the The Cancer Genome Atlas that cover the potential privacy risks associated with next-generation sequencing as well as the sharing of genomic data with access-controlled databases such as dbGaP (http://www.ncbi.nlm.nih.gov/gap).
We also recognized that the technologic characteristics of our screening platform would be crucial to the success of this initiative. To be clinically useful, we concluded that a tumor-profiling platform must be compatible with the types of archival tumor material typically available. Specifically, the assay must work with formalin-fixed paraffin-embedded tumor tissue, small core biopsies and fine needle aspirates, and needed to be sufficiently sensitive to enable the detection of actionable alterations on samples with low tumor purity. Moreover, to maximize utility across a wide variety of cancer types, the profiling methodology needed to be able to evaluate a large number of genes simultaneously and detect all major classes of actionable genomic alterations including base substitutions (point mutations), small insertions or deletions, copy number alterations and structural rearrangements. Designing a single test capable of detecting the vast majority of known clinically actionable alterations, thereby forgoing the need for disease-specific panels and multiple separate testing platforms, was also essential to simplify laboratory workflow and to facilitate scaling of this initiative over time. Finally, the throughput of the tumor-profiling platform needed to be such that final results would be available within a clinically useful timeframe, which we determined to be one month or less.
Selecting a profiling platform
We evaluated multiple technology platforms for our screening program. Enrichment by amplification, or amplicon capture, relies on a highly multiplexed PCR[s2] reaction involving locus-specific primer pairs simultaneously amplifying target regions in the genome. Although amplicon capture can produce deep sequence coverage with very little DNA, it is generally suitable only for a more limited number of genes. Assays that employ this approach therefore typically target ‘hotspots’ of recurrent somatic mutations rather than the full coding sequence of target genes. Moreover, amplicon capture methods are not well suited for detection of copy number gains and losses or translocations. By comparison, enrichment by hybridization, or hybridization capture, utilizes biotinylated synthetic DNA probes or ‘baits’ that are complementary for the targeted genomic DNA regions, allowing their subsequent enrichment. Hybridization capture typically requires more input DNA than amplicon capture, but can be scaled to a larger number of genes (up to the whole exome). Panels based on hybridization capture can be designed to target all coding sequences of all genes. Non-exonic regions that harbor recurrent somatic alterations such as the telomerase reverse transcriptase (TERT) promoter can also be included in the assay design. Importantly, hybridization capture methods enable the detection and quantitative assessment of copy number alterations and selected structural rearrangements [9,10]. Structural rearrangements are detected by including baits for select intronic regions (a technique sometimes called intron tiling) for commonly rearranged genes to sequence across the fusions points because these typically involve introns. This technique can miss rearrangements that occur in highly repetitive intronic sequences or in genes where breakpoints can occur across a wide range of introns. Although detection of fusion genes can be more comprehensive utilizing RNA sequencing, this increased efficiency comes with significant additional logistic complexities [11,12] and the major targetable fusions in solid tumors (involving ALK, RET, ROS1) are amenable to the intron tiling approach. Based on these considerations, we ultimately settled on a hybridization capture panel for genomic DNA sequencing to drive our precision medicine initiative. Creating a custom hybrid capture panel also provided control over the content of our assay and provided room for growth as new targets of interest were identified in parallel discovery efforts such as The Cancer Genome Atlas and the International Cancer Genome Consortium.
Somatic variant calling strategy
Appropriate selection of a targeted therapeutic agent based on molecular profiling is grounded on the principle of detecting somatically acquired genetic aberrations that are associated with malignant transformation. Definitively distinguishing somatic alterations from inherited germline variants was therefore of paramount importance to our testing strategy. To this end, we decided to profile each tumor in conjunction with the patient’s blood as a matched normal (germline) control. In the absence of germline comparison, variants identified from tumor sequencing must be filtered according to databases of common single nucleotide polymorphisms (SNPs). Using matching germline DNA allows somatic mutations to be unambiguously called. By comparison, bioinformatic filtering of probable germline polymorphisms based on SNP databases can lead to false-positive mutation calls at sites of rare inherited SNPs (so-called ‘private’ SNPs not present in databases), including cancer-susceptibility alleles.
Although the decision to sequence matched germline DNA offers significant advantages in the bioinformatic efficiency of making somatic variant sequence calls in the tumor sample, it also enables the detection of pathogenic variants in the genes that are sequenced in the blood. Incidental findings can emerge as a result of tumor sequencing that relate to a patient’s inherited susceptibility to cancer or other diseases, with unanticipated yet significant consequences for family members who share these variants [13,14]. To address this possibility, all patients undergoing molecular profiling at our center are consented and given the ability to ‘opt-out’ of return of incidental germline findings that can be tangential to the clinical utility of the test itself. The automated data analysis pipeline used to make somatic calls filters germline variants such that they are not called or seen by the molecular pathologist during the review and signout process of somatic MSK-IMPACT testing. Very rarely, large germline deletions or duplications have been incidentally identified as the result of routine quality control procedures employed by the bioinformatic pipeline. In these circumstances, these incidental findings are submitted for review by an Institutional Review Board (IRB) Genomics Advisory Panel that determines their suitability for return to the patient. Of note, incidental somatic findings can also be uncovered as a result of sequencing blood as a source of normal control DNA. For instance, somatic mutations in myeloid-leukemia-associated genes have been associated with advanced age and could identify patients with a pre-leukemic state [15-17].
Aside from the incidental (or accidental) detection of germline variants in the matched normal DNA, patients who desire systematic detection and annotation of their germline DNA for pathogenic or possible-pathogenic variants sign an additional consent and receive pre-test genetic counseling in the form of an IRB-approved video. During this consent process, patients are counseled on the potential implications for the patient and their family of this type of analysis. For these patients, somatic results are released as soon as they are available because germline annotation typically takes longer than somatic mutation calling. This germline annotation effort has already resulted in reporting of pathologic variants in genes such as BRCA1 and BRCA2 among others included in the assay design and these could predict for clinical benefit from novel therapeutics such as poly-ADP ribose polymerase (PARP) inhibitors.
Integrated mutation profiling of actionable cancer targets (MSK-IMPACT)
The final design of our profiling platform, MSK-IMPACT, is shown in Figure 1. Technical aspects of this assay have been previously published [10]. Briefly, to call somatic mutations, we require a minimum coverage depth at that position of 20 reads. To call mutations at known hotspots we require at least eight mutant reads and at least 2% mutant allele frequency. To call mutations outside of known hotspots we require at least ten mutant reads and at least 5% mutant allele frequency. Importantly, we are only rarely at these read number cutoffs because our mean coverage is about 600x. To call copy number alterations at least a twofold gain or loss relative to matched normalized baseline for that sample is used. Narrow amplicons are considered more likely to be significant than broad gains. Of note, the clinical reports include the fold change so that clinicians can differentiate between high and low copy number gains and losses.
Figure 1.
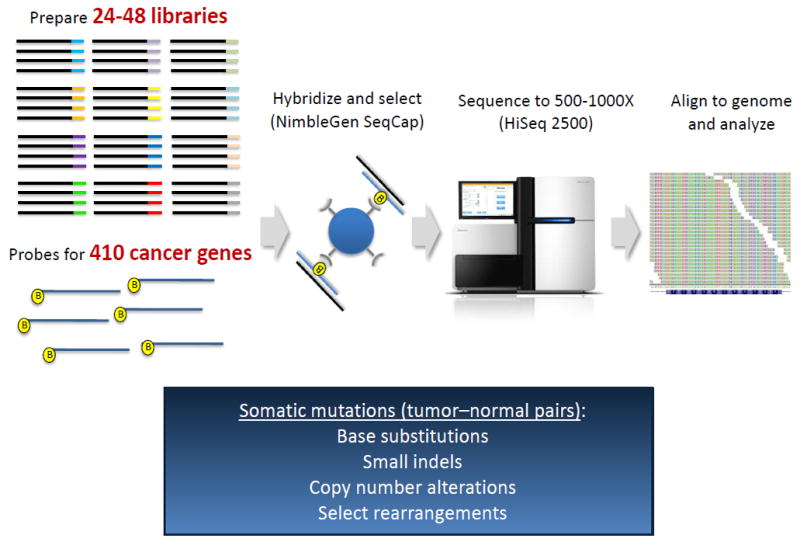
[s4]. Schematic Overview of MSK-IMPACT. MSK-IMPACT is a custom designed hybridization capture-based next-generation sequencing assay that utilizes biotinylated synthetic DNA probes or baits that are complementary for the targeted genomic DNA regions, allowing their subsequent enrichment. Tumor and matched normal are sequenced in parallel using an Illumina® HiSeq 2500. The assay includes full exon coverage of 410 genes, in addition to select intronic regions. MSK-IMPACT can detect all classes of genomic alterations including base substitutions, small indels, copy number alterations and select rearrangements. Up to 48 libraries, representing paired tumor–normal samples from 24 unique patients, can be sequenced simultaneously. Testing is performed in a Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory and results entered into the medical record.
To ensure results could be returned into the patient’s medical record within a clinically useful timeframe (two-to-three weeks) we undertook an extensive validation effort so that MSK-IMPACT could be performed within a CLIA-certified laboratory. For tumor types where genomic testing is required to guide selection of routine first-line therapy (for example lung cancer and melanoma) faster tests are also available, for instance immunohistochemical tests for mutated EGFR and BRAF and ALK fusion. In its first iteration, initiated in January 2014, MSK-IMPACT included full exon coverage of 341 genes, in addition to select intronic regions to enable detection of actionable fusions. Since February 2015, a second iteration of MSK-IMPACT was implemented expanding coverage to a total of 410 genes (see Table S1 in the supplementary material online for complete gene list). We expect to update the content of this panel every 6–12 months in response to emerging scientific data regarding important alterations found in cancer as well as with changes to our clinical trial portfolio. Of particular interest has been expanding the coverage of recurrent structural rearrangements because this typically entails additional tiling of introns in the affected genes. Notably, as of June 2015, over 5000 patient tumor samples have been studied, the vast majority with matched normal DNA.
Whole exome sequencing in the clinic
Some institutions have chosen to pursue whole exome sequencing (WES) as the basis of their screening program. In evaluating this approach for our own needs, we identified several potential drawbacks. First, although the costs of sequencing continue to fall, WES remains considerably more expensive than hybrid capture sequencing. Moreover, the depth of coverage obtained with WES sequencing is typically significantly lower than with target capture technologies meaning that material with low tumor content, or mutations of lower allelic frequency, might not be detected with this approach. WES could thus miss important clinically actionable alterations such as EGFR and BRAF mutations in low-purity tumors. Additionally, a hybrid capture-based approach might be better suited to identify actionable fusions through more-extensive tiling of intronic regions in genes such as ALK and ROS1 (see Table S2 in supplementary material online for list of such introns tiled in MSK-IMPACT). Finally, although WES can facilitate the discovery of new drug targets or development of richer mutational signatures, it is unlikely that analysis of the additional genes covered by a WES approach would alter patient care, because the vast majority of available targeted therapies are directed toward the proteins encoded by genes already present in our 410 gene panel.
In general, institutions that have adopted clinical WES have done so by limiting the scope of their screening efforts to a small subset of the total number of patients they manage. Because a major objective of our precision medicine strategy was to identify and enroll patients with rare but potentially actionable mutations to clinical trials, a broader screening strategy that covered most if not all known clinically actionable alterations and was feasible in thousands of patients per year was better suited to our needs.
Using genomic data to enhance and accelerate clinical research
Instituting a universal tumor genetic profiling program is only the necessary foundation for a center-wide precision medicine strategy. We believe that using the genomic data generated by a universal screening initiative requires a comprehensive alignment of the clinical research enterprise. The following sections describe our view of the capabilities needed to implement a precision medicine approach to drug development.
Mining genomic data
As our universal tumor genetic profiling program expands to include tens of thousands of patients, mining these data offers a unique opportunity to make novel observations and, in doing so, better align our clinical trial portfolio with the patients under active treatment at our center. To promote these uses, de-identified genomic data generated by our screening program are clinically annotated and made available internally, on an institution-wide basis, to scientific and clinical investigators through the MSKCC cBioPortal (http://cbioportal.mskcc.org/). The cBioPortal is a web-based resource for exploring, visualizing and analyzing multidimensional cancer genomics data [18,19]. cBioPortal is free open-source software available for download (https://github.com/cBioPortal/cbioportal) by groups or institutions that want to create their own portals. An intuitive interface makes it easy for researchers without bioinformatics expertise to explore genetic alterations interactively across multiple patient samples, genes and pathways and to associate these factors to clinical outcomes. By linking the MSK-IMPACT data in the cBioPortal to the MSKCC institutional database, we have the capability to merge the genomic results automatically with clinical data such as patient diagnosis (stage, histology, primary tumor site), vital status (alive or dead), date of last follow-up, date of last scan, tumor markers and treatment administration records. Broad access to this unique dataset within our institution has already been used to make previously unrecognized associations between clinical phenotypes and genomic alterations and has prompted the development of new therapeutic studies. Memorial Sloan Kettering Cancer Center also intends to participate in data sharing with the wider scientific community through the American Association for Cancer Research (AACR) GENIE consortium and other similar initiatives.
Clinical trial portfolio
Accelerating the development of genomically selected treatments requires the availability of clinical studies to treat the large number of patients with potentially targetable alterations identified through our screening efforts. To accomplish this, we have increased access to targeted therapies at our center in a number of ways. First, as an institution, we have greatly expanded our participation in early-phase studies. These studies, which now frequently include large expansion cohorts to evaluate efficacy in molecularly enriched populations, have become an invaluable resource in providing patients with access to novel therapies matched to their specific genomic profile. In addition, individual disease specialty teams have opened studies for recurrent actionable genomic alterations observed in their respective patient populations. The presence of a universal screening program such as MSK-IMPACT onsite has made such studies increasingly feasible because they do not need to rely on slower and tissue-intensive single gene screening assays run at a central reference laboratory to identify rare genomic subpopulations within a particular histology, for example see [20].
Multi-histology, biomarker-selected, ‘basket’ studies are a particularly efficient way to expand access to ‘matched’ targeted therapies and therefore the development of such studies has been a major focus at our center. The term basket study has been used to describe a wide variety of study designs, and it is therefore worthwhile to define our use of this term with greater precision. Some investigators have referred to studies that profile patients with a single or limited number of related cancer types and assign them to one of several matched treatment arms as basket studies. We would instead refer to these as ‘molecular allocation’ studies. Three common features of molecular allocation studies are: (i) patients are enrolled and screened before an actionable alteration has been identified; (ii) tumors are profiled using a central assay; (iii) the number of eligible cancer types are limited because investigational agents must be selected based on the previously defined prevalence of targetable mutations within the tumor types of interest. Therefore, molecular allocation studies greatly expand access to matched treatment within a single disease type, but do not fully address the needs of an institution implementing universal highly multiplexed screening across all tumor types. A major weakness of this study design is that the agents included are often chosen based upon availability and do not often represent the best-in-class inhibitor for a particular mutant target. Additionally, such studies are often insufficiently powered to validate genotype–response associations for rare mutations, in particular those with a prevalence of less than 5% of the study population.
By our definition, a basket study is a clinical trial in which eligibility is defined based upon the presence of a particular genetic alteration rather than a particular cancer type. In our experience, basket studies are most successful when they share a number of common features. First, only patients with tumors already known to harbor a qualifying genomic alteration are enrolled. This design uncouples molecular screening from the individual therapeutic study and necessitates availability of a separate screening program, such as the center-wide strategy we have implemented, to identify potentially eligible patients. Second, the qualifying molecular alteration (sometimes referred to as an integral biomarker) does not need to be confirmed by a central assay before treatment. We do advocate that tumor (either archival or through fresh biopsy) and plasma be banked to serve as bridging samples for central companion diagnostic testing and validation. Third, patients can enroll regardless of tumor type; however, the tumor types with the highest anticipated rate of positivity for the integral biomarker are enrolled to their own cohort. This flexible statistical design enables independent efficacy assessment in tumor types that enroll in sufficient quantities, most often using a Simon two-stage design. Fourth, the qualifying genomic alteration is typically, but not always, of low overall incidence but present across a wide variety of cancer types making individual tumor-type-specific studies impractical. A sample schema of a basket study is shown in Figure 2.
Figure 2.

Sample basket study schema. The typical study design and biostatistical plan for a multi-histology, biomarker-selected, basket study is shown. Several disease cohorts are pre-specified and another all-comers ‘other’ cohort allows enrollment of remaining disease types. Pertains[s5] indicate the expected frequency of the biomarker of interest within each disease type. Basket studies typically utilize a two-stage design targeting a response rate of at least 30% enabling relatively small sample sizes.
Patient and protocol matching
The ability to identify patients with rare genomic alterations of interest and ‘match’ them to the most appropriate study is also crucial. Treating physicians cannot realistically be aware of every precision medicine study being conducted at a large center. This difficulty has been exacerbated by the proliferation of multi-histology basket studies because the protocol’s Principal Investigator might not be a member of the disease-specialty team where the patient is being treated. Furthermore, because early phase precision medicine studies frequently open and close cohorts and amend eligibility, it is increasingly difficult for any one physician to be aware of all the protocol opportunities available to their patients. Therefore, the ability of a protocol’s Principal Investigator to identify patients who potentially qualify for participation in their precision medicine study, regardless of whether they have an existing treatment relationship, becomes a crucial programmatic capability.
To address this need, we created a protocol–patient matching system. Upon obtaining an IRB waiver, the Principal Investigator of a precision medicine study works with a data analyst to build a virtual cohort of patients that can be tracked dynamically during their time at the center. In its most basic form, this system can search all molecular reports on a daily basis to provide a Principal Investigator with a list of all living patients with a tumor harboring a specific genomic alteration (for example a BRAF V600E mutation). However, the search criteria can be further refined to include additional information available in our institutional database including diagnosis, stage, disease status, chemotherapy administration records, laboratory values and upcoming appointments. Figure 3 provides a high level schema of this system and how a cohort can be created and tracked. Principal investigators use a simple web interface to track eligible patients and can even set email alerts that are automatically sent to the treating oncologist when pre-specified events occur (such as a scan showing progression in a patient with a qualifying genomic alteration). This capability has proved crucial for accruing to our multi-histology basket studies and has scaled up well as the number of patients sequenced and number of precision medicine studies have increased dramatically.
Figure 3.

Schematic overview of protocol matching system. The protocol matching system at Memorial Sloan Kettering Cancer Center is based on multiple canonical data stores including a scheduling system, molecular and surgical pathology reports, and electronic medical records feeding into a centralized institutional database. This database is in turn used to create automated queries that can use genomic and clinical data to identify patients potentially eligible for a precision medicine study and to notify the study Principal Investigator and the treating physician. Patient genomic and clinical data are also fed in a de-identified fashion into the cBioPortal for investigational data mining and visualization.
Concluding remarks
Implementing a center[s3]-wide precision medicine strategy at a major cancer center is a true multidisciplinary effort and requires comprehensive alignment of broad screening strategy with a clinical research enterprise that can use these data to accelerate development of new treatments. The full realization of this vision makes clinical approaches that were previously unfeasible become a possibility and holds the promise of a new era of stratified medicine.
Supplementary Material
Highlights.
Comprehensive precision medicine strategies require a comprehensive multidisciplinary approach
Screening assays must be sufficiently broad to identify the full landscape of actionable genomic alterations
Profiling must be applied broadly, irrespective of whether molecular testing is required to guide standard-of-care therapy
Multi-histology biomarker-selected ‘basket’ studies support treatment based on identified actionable alterations
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Slamon DJ, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 2.Swain SM, et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med. 2015;372:724–734. doi: 10.1056/NEJMoa1413513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chapman PB, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735–742. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 5.Shaw AT, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 6.Shaw AT, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371:1963–1971. doi: 10.1056/NEJMoa1406766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Demetri GD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 8.Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372:793–795. doi: 10.1056/NEJMp1500523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Won HH, et al. Detecting somatic genetic alterations in tumor specimens by exon capture and massively parallel sequencing. J Vis Exp. 2013;80:e50710. doi: 10.3791/50710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng DT, et al. MSK-IMPACT: a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015 doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng Z, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20:1479–1484. doi: 10.1038/nm.3729. [DOI] [PubMed] [Google Scholar]
- 12.Wu YM, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013;3:636–647. doi: 10.1158/2159-8290.CD-13-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green RC, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–574. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Catenacci DV, et al. Tumor genome analysis includes germline genome: are we ready for surprises? Int J Cancer. 2015;136:1559–1567. doi: 10.1002/ijc.29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Busque L, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44:1179–1181. doi: 10.1038/ng.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaiswal S, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie M, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cerami E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao J, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paik PK, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015;5:842–849. doi: 10.1158/2159-8290.CD-14-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.