

Abstract
Objective:
This was a longitudinal single-center cohort study to comprehensively explore multimodal progression markers for Parkinson disease (PD) in patients with recently diagnosed PD (n = 123) and age-matched, neurologically healthy controls (HC; n = 106).
Methods:
Thirty tests at baseline and after 24 months covered nonmotor symptoms (NMS), cognitive function, and REM sleep behavior disorder (RBD) by polysomnography (PSG), voxel-based morphometry (VBM) of the brain by MRI, and CSF markers. Linear mixed-effect models were used to estimate differences of rates of change and to provide standardized effect sizes (d) with 95% confidence intervals (CI).
Results:
A composite panel of 10 informative markers was identified. Significant relative worsening (PD vs HC) was seen with the following markers: the Unified Parkinson's Disease Rating Scale I (d 0.39; CI 0.09–0.70), the Autonomic Scale for Outcomes in Parkinson's Disease (d 0.25; CI 0.06–0.46), the Epworth Sleepiness Scale (d 0.47; CI 0.24–0.71), the RBD Screening Questionnaire (d 0.44; CI 0.25–0.64), and RBD by PSG (d 0.37; CI 0.19–0.55) as well as VBM units of cortical gray matter (d −0.2; CI −0.3 to −0.09) and hippocampus (d −0.15; CI −0.27 to −0.03). Markers with a relative improvement included the Nonmotor Symptom (Severity) Scale (d −0.19; CI −0.36 to −0.02) and 2 depression scales (Beck Depression Inventory d −0.18; CI −0.36 to 0; Montgomery-Åsberg Depression Rating Scale d −0.26; CI −0.47 to −0.04). Unexpectedly, cognitive measures and select laboratory markers were not significantly changed in PD vs HC participants.
Conclusions:
Current CSF biomarkers and cognitive scales do not represent useful progression markers. However, sleep and imaging measures, and to some extent NMS, assessed using adequate scales, may be more informative markers to quantify progression.
One of the great challenges in Parkinson disease (PD) therapeutics is the definition of clinically meaningful endpoints that measure progression, or lack thereof.1 To date, numerous clinical trials with putative neuroprotective agents have yielded negative results.2–4 It remains unclear whether trial design or the agent tested, or both, are the reasons for past failures.
Currently, several parameters are typically selected as endpoints for use in symptomatic and neuroprotective treatment trials, e.g., the Unified Parkinson's Disease Rating Scale (UPDRS), quality of life measurements,5 or the “need for levodopa therapy.”2
Reports of selective, longitudinal investigations of nonmotor signs (NMS),6–8 neuroimaging, and laboratory analyses9 have been previously published as potential markers of progression.10 These investigations were largely conducted in smaller cohorts and focused on single symptoms/measures and frequently relied on more advanced PD cases, and lacked age-matched healthy controls.
The de novo Parkinson (DeNoPa) cohort represents a prospective, longitudinal, single-center, observational study of participants with de novo PD and matched neurologically healthy controls (HC). The study's aim is to improve early diagnosis and to assess proper progression markers, scored at baseline with biannual follow-up.11
We hypothesized that a composite panel of multimodal progression markers covering overall NMS, cognitive measures, sleep, imaging, and CSF indices would be instructive in understanding the natural history of early PD, and to thereby delineate specific and sensitive outcome measures for future intervention trials. To this end, we applied the same panel of tests to a group of HC to separate PD changes from the process of normal aging.
METHODS
Study participants.
Longitudinal data were collected at baseline (BL) and at 24 months follow-up (24FU). Detailed inclusion criteria have been described previously11 and are summarized in appendix e-1 on the Neurology® Web site at Neurology.org.
After BL assessments, all patients started dopaminergic therapy according to accepted guidelines12 with stable medication until the 24FU visit. The levodopa equivalent dosage (LED) at 24FU was determined as described.13 The clinical diagnosis was reassessed in the on state for all patients at 24FU by consensus of 2 teams of independent neurologists (C.T./F.S.-D. and B.M./J.E.; for details, see appendix e-1).
Standard protocol approvals, registrations, and patient consents.
We received approval from the ethical standards committee on human experimentation (Landesärztekammer Frankfurt) for all experiments using human participants. Written informed consent was obtained from all study participants (consent for research).
Procedures.
Protocols, data collection, and data/sample storage have been described previously.11 Briefly, our approach is of an exploratory nature. We examined longitudinal assessments using validated and published tools: NMS were the most relevant diagnostic tests in DeNoPa11 and were also investigated longitudinally by means of the Movement Disorder Society (MDS) UPDRS part I,14 the NMS Questionnaire, the Nonmotor Symptom Scale (NMSS), and the Autonomic Scale for Outcomes in Parkinson's Disease (Scopa-AUT). The PD Questionnaire, which has been the primary endpoint in randomized studies,5 was used together with assessment of depression by the self-reported Beck Depression Inventory (BDI) and the Montgomery-Åsberg Depression Rating Scale (MADRS). Cognition was tested with the Mini-Mental State Examination (MMSE), the Montreal Cognitive Assessment Test (MoCA), and the Clock Drawing Test. Further tests for different cognitive domains are given in appendix e-1. Sleep was assessed using the Parkinson's Disease Sleep Scale, the Epworth Sleepiness Scale (ESS), and the REM Sleep Behavior Disorder (RBD) Screening Questionnaire (RBD-SQ). In addition, we performed video-supported polysomnography (PSG) to classify RBD. Voxel-based morphometry (VBM) of gray matter and hippocampus by 1.5T volumetric MRI was included because significant atrophy has been described in correlation with PD progression and cognitive decline.15 We also investigated serum urate.16 We tested known CSF markers that have been previously discussed as potential diagnostic markers of PD state or its progression, including total α-synuclein (α-syn), β-amyloid 1-42, total and phosphorylated tau protein (t-tau and p-tau), as well as neurofilament light chain proteins.17 See appendix e-1 for experimental details.
Statistical analysis.
We used linear mixed-effect models to identify differences in the rates of change in selected markers between PD and controls. For each marker, we estimated a random intercept model including group (PD vs HC), time (BL vs 24FU), and their interaction as predictors. Models were estimated using restricted maximum likelihood estimation, taking into account all available data points at BL and 24FU. Differential change in PD is apparent in a significant group multiplied by the time interaction. We present model-based effect sizes for differential change, standardized by the observed SD in the PD group at baseline.18 Significance testing was based on 95% confidence intervals using parametric bootstrap with 2,000 resamples.
In addition, we explored whether changes in individual markers were influenced by selected BL characteristics. To this end, we conducted robust multiple regression analyses in the PD sample predicting 24FU scores of individual markers (while controlling for respective BL scores) from LED and dopamine agonists at 24FU and selected sociodemographic and clinical factors at BL. Robust regression was used to deal with non-normality (e.g., outliers) in the distribution of markers.19 All analyses were carried out with the statistical platform R,20 using the package lme4 for linear mixed models and robustbase for robust regressions.
We did not correct for multiple testing since our approach was of an exploratory nature (see also the Discussion and appendix e-1).21 However, marker candidates were chosen according to their clinical relevance and following a review of the literature.
RESULTS
Of the 269 recruited participants, 254 (94%) underwent follow-up assessments (147 PD and 107 HC). Four patients and 2 controls died before 24FU. Reassessment of the clinical working diagnosis led to the identification of a distinct neurologic disorder in 24 (16%) patients shown in figure e-1. The reassessment of our previously described panel of diagnostic markers11 in the remaining 123 patients with PD and 106 HC at BL remained significant.
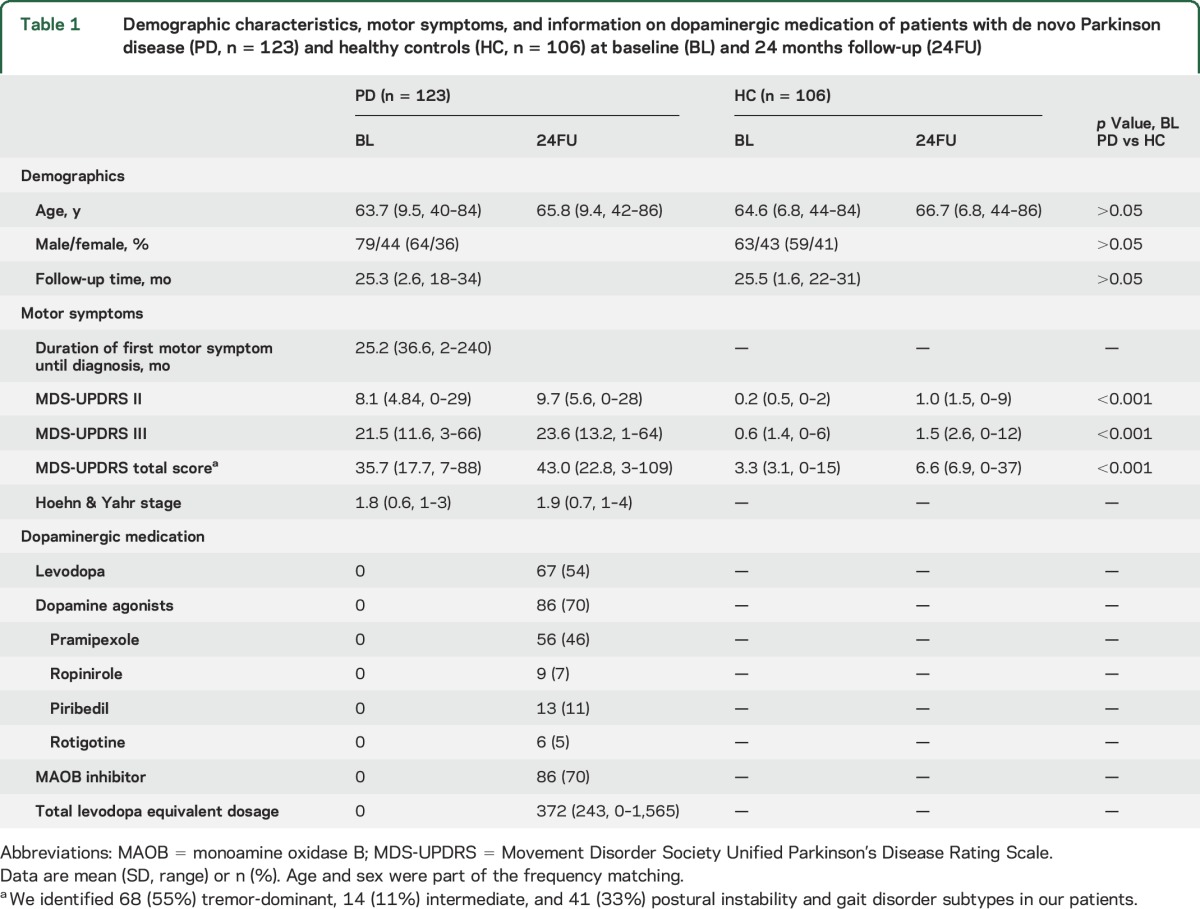
The demographics, motor characteristics, and details of dopaminergic treatment at 24FU are shown in table 1.
Table 1.
Demographic characteristics, motor symptoms, and information on dopaminergic medication of patients with de novo Parkinson disease (PD, n = 123) and healthy controls (HC, n = 106) at baseline (BL) and 24 months follow-up (24FU)
Panel of progression markers.
Table 2 list the results of 30 tests performed in the entire DeNoPa cohort, which we used as surrogate markers for PD and its progression. Ten of the 30 markers showed statistically significant rates of change relative to HC over the 2-year observational period, mainly in the categories of NMS, depression, sleep, and imaging (table 2 and figures 1–3).
Table 2.
Parameters of linear mixed effect models

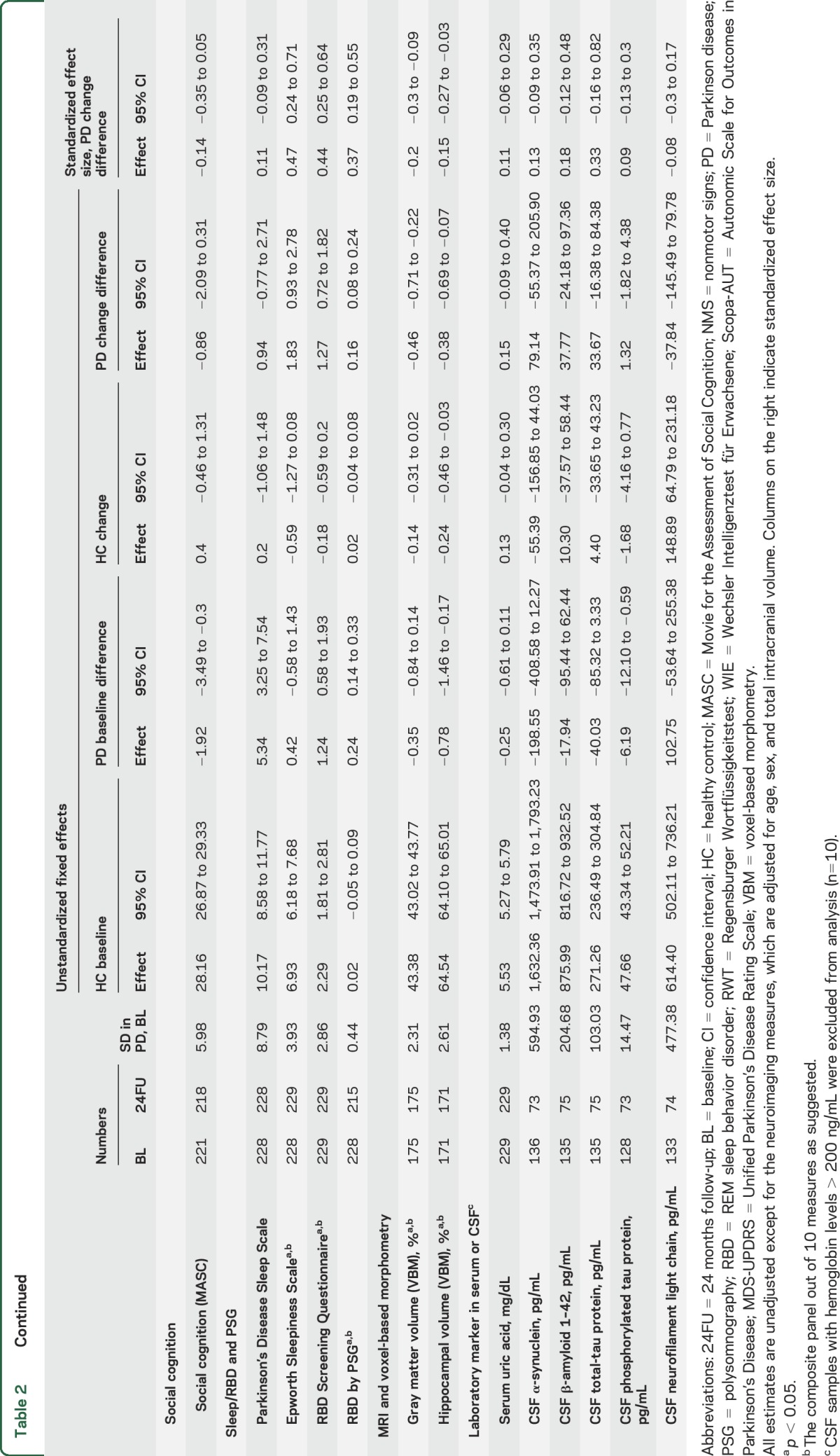
Figure 1. Longitudinal changes in nonmotor symptoms (NMS), impaired quality of life (QoL), and depression.

Estimated means and 95% confidence intervals of Movement Disorder Society–Unified Parkinson's Disease Rating Scale I (MDS-UPDRS I) (A), NMS Questionnaire (NMSQuest) (B), NMS Severity Scale (NMSS) (C), Scopa-AUT (D), QoL measure (PDQ-39) (E), and the depression scales: Beck Depression Inventory (BDI) (F) and Montgomery-Åsberg Depression Rating Scale (MADRS) (G) at baseline (BL) and 2-year follow-up (24FU). Blue line represents healthy controls (HC); red line represents participants with Parkinson disease (PD). *Significant change in PD vs HC (p < 0.05).
Figure 2. Longitudinal changes in sleep parameters.

Estimated means and 95% confidence intervals of the Parkinson's Disease Sleepiness Scale (PDSS-2) (A), Epworth Sleepiness Scale (ESS) (B), REM Sleep Behavior Disorder by Questionnaire (RBD-SQ) (C), and by video-supported polysomnography (PSG) (D), at baseline (BL) and 2-year follow-up (24FU). Blue line represents healthy controls (HC); red line represents participants with Parkinson disease (PD). *Significant change in PD vs HC (p < 0.05).
Figure 3. Longitudinal changes in 1.5T volumetric MRI.

Estimated means and 95% confidence intervals of voxel-based morphometry (VBM) in total gray matter volume (GMV) (A) and hippocampal volume (HCV) (B) at baseline (BL) and 2-year follow-up (24FU). Blue line represents healthy controls (HC); red line represents participants with Parkinson disease (PD). *Significant change in PD vs HC (p < 0.05). (C) Example of gray matter segmentation in 3 orthogonal cutplanes. For the analysis, native space maps were summed to generate an absolute volume per class and subject. (D) Example of smoothed gray matter maps (in Montreal Neurological Institute space) shown with the a priori defined hippocampal analysis region overlaid in red. Within this region, the mean average gray matter probability was calculated yielding one average value of hippocampal gray matter per subject.
Clinical measures.
Nonmotor symptoms significantly worsened in PD vs HC as assessed by the MDS-UPDRS I score (table 2 and figure 1A). The worsening of autonomic features relative to controls was reflected by a statistically significant increase in the Scopa-AUT scores (table 2 and figure 1D). This increase was mainly driven by an increase in cardiovascular items of the Scopa-AUT (data not shown). In contrast, the severity score for NMS (by NMSS) showed improvement in relation to controls (table 2 and figure 1C). Assessment of depression showed a significant improvement in the self-reported BDI and investigator-rated MADRS (table 2 and figure 1, F and G).
Most of our preselected cognitive measures were worse in PD than in controls during follow-up. Interestingly, slopes were parallel for PD and HC, revealing no significant incremental changes in PD over controls in single tests (table 2). Also, the tests for executive function showed a parallel (and nonsignificant) decrease for both groups.
Excessive daytime sleepiness, as assessed by the ESS, significantly worsened with PD progression compared to controls. RBD significantly increased for the PD group by questionnaire at follow-up, while it decreased in HC (table 2 and figure 2, A–C).
Technical investigations.
PSG also showed a significant increase of the prevalence of RBD in patients with PD while in HC the prevalence increased only slightly, as expected, with increasing age (table 2 and figure 2D). The severity of RBD by RBD severity score22 in PD did not differ between baseline and 24FU (data not shown).
Volumetric imaging measures of cortical and hippocampal structures by MRI decreased significantly at 24FU in patients with PD (figure 3, A and B), which was not associated with age, PD phenotype (table e-1), or changes in cognitive measures (data not shown). Statistical parametric maps and change plots of VBM measures of cortical and hippocampal volume for both groups indicate regions where atrophy rates differed significantly in patients with PD vs controls (figure 3, C and D).
Serum uric acid levels did not significantly differ between PD and HC at baseline and at 24FU. Total CSF α-syn levels were lower in patients with PD compared to HC at BL and 24FU, but the rates of changes were not significant. Furthermore, a nonsignificant increase in CSF for t-tau was observed (figure e-2). Results for CSF ratios were not significantly altered (data not shown).
Predictors of change during 24-month observation.
Multiple regression analysis in the PD group, which tested the joint statistical influence of 8 potentially confounding factors, showed only 24 (10%) significant effects regarding changes within the panel of 30 investigated markers (table e-1). For example, the presence of RBD at BL indicated worse cognitive decline on MoCA. The LED, as another surrogate marker for clinical severity, was significantly associated with decreased cognitive function (by the memory screening test MMSE and the attention/working memory test Digit Span Backward) and increasing RBD as well as serum uric acid. The intake of dopamine agonists was associated with improvement of depression (BDI), worse performance on the memory task Verbal Learning and Memory Test, and increase on CSF p-tau but no other marker.
DISCUSSION
We performed a 2-year follow-up investigation on clinical measures and biomarkers through clinical and technical assessment, including PSG, MRI, and serum/CSF measures in the largest single-center cohort of 123 clinically diagnosed early PD participants and 106 matched healthy controls. Overall, we found that 20 of 30 tests showed no statistically significant and no specific progression in our DeNoPa patients. The remaining 10 markers showed statistically significant changes reflecting either progression in PD or change due to treatment or both when compared to controls: these encompassed increasing sleep disturbances and worsening of NMS, whereas NMS severity (NMSS) and depression (by MADRS and BDI) improved. Regarding imaging markers, general gray matter volume and hippocampal volume specifically and significantly decreased over the 2-year period.
Clinical measures.
Several smaller studies6,7,10 and one longitudinal study8 have previously suggested the use of NMS as candidate progression markers. NMS worsened in PD as measured by the MDS-UPDRS I in DeNoPa, while the severity of NMS measured by NMSS improved in our cohort. Autonomic dysfunction, as assessed by Scopa-AUT, also showed statistically significant worsening in PD compared to HC. The only systematic longitudinal investigation on NMS showed an increase in NMS domains after 24FU, but NMS severity and more specific autonomic features had not been addressed. Also, these patients were at different stages of disease and the majority had received symptomatic treatment already at BL.8 While the NMS tools cover various NMS, the MDS-UPDRS I captures a few key symptoms. Both the NMSS and MDS-UPDRS I show strong convergent validity.23 The discrepancy of performance of self-reported vs investigator-reported NMS in DeNoPa could also be the result of different sensitivity of scales and questionnaires, and points towards the need for an improved and widely standardized assessment of NMS for longitudinal investigations.
We recorded an improvement of depression in our patients with PD on 2 independent scales and individual differences in improvement were not significantly associated with dopaminergic dosage. The improvement of depression and quality of life in the early honeymoon phase of PD (generally considered to represent the first 5–10 years after diagnosis) is in accordance with other studies,8 and is also related to dopamine agonists.24
Following analysis of various domains of cognition, we identified clear differences in cognitive performance in PD compared to controls (see table 2; PD baseline differences), but only cross-sectionally and not with any significant change longitudinally. Most previous longitudinal studies did not include control groups.25 Therefore previous studies in PD may have overinterpreted normal cognitive aging and we have thus corrected this deficit in our study design.
RBD and reduced subjective quality of sleep are recognized inherent symptoms during prodromal and fully developed motor PD.26 Daytime sleepiness progressively worsens in PD, confirming that it may be dependent on dopaminergic therapy27 or an intrinsic factor of disease progression. RBD may be a useful marker to monitor progression of PD either with PSG or with a similar effect size by a 10-item self-rating questionnaire (RBD-SQ). The presence of RBD indicates worse cognitive decline by MoCA, as has been shown previously.28
Technical investigations.
To date, DeNoPa comprises the largest collection and interrogation of longitudinal data of PSG, MRI volumetry, and CSF markers.
Our MRI structural measures showed significant overall and hippocampal gray matter volume loss in PD. The hippocampal region was selected, since, according to the Braak staging scheme,29 α-syn pathology is expected to emerge there before affecting the neocortex, and hippocampal involvement has been suggested in PD dementia.30 However, effect sizes were similar in both measures and correlations with changes in cognitive markers were nonsignificant in this early PD cohort (data not shown).
A longitudinal CSF study found increasing tau and tau/Aβ1-42 levels,9 but significantly decreasing CSF α-syn levels in PD.31 In our study, none of the CSF markers (or their ratios) that were tested reached statistical significance for progression. Different demographics and sample preparation could account for this discrepancy. It could also be explained by the small number (n = 85; 37%) of DeNoPa participants who consented to serial lumbar puncture (37%) at 24FU. A similarly small annual rate of change in CSF markers has also been observed in patients with established Alzheimer disease (AD). The stability of our markers in this early stage of motor PD could result from a slowing of the neurodegeneration process in the symptomatic phase similar to that seen in AD.32 This underlines the need for more specific progression marker candidates for the early phase of the disease, which will emerge from this and other ongoing studies.33
We explored many obvious candidates in a large cohort of PD participants and looked for progression markers for future use in clinical trial cohorts and tried to exclude noninformative ones. The fact that we identified no more than 33% of the markers analyzed having significantly changed during our 24-month observational period highlights the mandate of finding reliable progression markers. These results have immediate consequences for clinical research in PD by restricting the potential usage of some markers, and will take more time to eventually change the clinical care delivered to patients with PD.
There are limitations of clinical practice–led exploratory studies, such as ours, which merit discussion. First, DeNoPa is a real-life observational survey study. Therefore, we symptomatically treated patients' motor symptoms. The effects of dopaminergic treatment on NMS vary depending up on the underlying mechanisms; i.e., a symptom influenced by dopaminergic deficit may respond positively to dopaminergic treatment (such as depression and NMS),24 whereas a non-dopamine-responsive symptom may worsen with such treatment (such as daytime sleepiness, especially in the early titration phase; further discussion can be found in appendix e-1).27 Therefore, we cannot remove any influence conferred by therapy on our investigational markers, but purposefully included 8 markers that are likely independent of any introduced symptomatic therapy,34 such as MRI volumetry and blood and CSF markers.
Second, most of our proposed marker candidates showed promising results as early diagnostic markers, which does not necessarily qualify them as progression markers. Further marker candidates specific for progression of the disease need to be explored. Third, we did not control for multiple testing. Although this is an appropriate exploratory strategy for detecting true changes in patients with PD (i.e., reducing type II errors), it may lead to false-positive findings. However, the detection of 10 significant changes among 30 tested is clearly more than would be expected by chance alone, thus corroborating the significance of our results as a whole.35
The use of our proposed panel of outcome measures needs further validation before being used to rate progression across centers. One limitation is the responsiveness of depression and some NMS to therapy,36 requiring new elaborated NMS scales. This should be undertaken independently by multicenter cohorts enrolling larger numbers of participants; this is currently underway.33
Future studies need to investigate the validity of these proposed markers, standardize the assessment of nonmotor features, and identify more sensitive and disease-specific marker candidates that reflect underlying biological processes (such as propagation of α-syn pathology, inflammation, and neuronal death). In this way, validated and improved markers will be used as clinically meaningful endpoints of progression that are essential in the development of treatments for PD.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the participants in the study and their families as well as referring neurologists; Olivia Steuer and Carolin Sengstock for technical assistance with sample collection; Maria Rudolph, Miriam Schanze, Valentina Krenz, and Friederike Panzer for support with data entry; Anne-Marie Williams for revision of the manuscript; and Michael Schlossmacher and Gabrielle Strobel for advice and review of the manuscript.
GLOSSARY
- 24FU
24 months follow-up
- α-syn
α-synuclein
- AD
Alzheimer disease
- BDI
Beck Depression Inventory
- BL
baseline
- DeNoPa
de novo Parkinson
- ESS
Epworth Sleepiness Scale
- HC
healthy control
- LED
levodopa equivalent dosage
- MADRS
Montgomery-Åsberg Depression Rating Scale
- MDS
Movement Disorder Society
- MMSE
Mini-Mental State Examination
- MoCA
Montreal Cognitive Assessment Test
- NMS
nonmotor signs
- NMSS
Nonmotor Symptom Scale
- p-tau
tau protein phosphorylated at threonine 181
- PD
Parkinson disease
- PSG
polysomnography
- RBD
REM sleep behavior disorder
- RBD-SQ
REM Sleep Behavior Disorder Screening Questionnaire
- Scopa-AUT
Autonomic Scale for Outcomes in Parkinson's Disease
- t-tau
total tau protein
- UPDRS
Unified Parkinson's Disease Rating Scale
- VBM
voxel-based morphometry
Footnotes
Supplemental data at Neurology.org
Editorial, page 128
Contributor Information
Collaborators: DeNoPa Study Group, Manfred Altenhain, Hans Baiker, Mechthild Bauer, Rudolf Brodhuhn, Monica Canelo, Oliver Erdmann, Gudrun Falkenstein, Werner Feldeisen, Andreas Fetzer, Michael Fischer, Robert Flapper, Bernd-Joachim Forsting, Peter Gensicke, Erdmann Haacke, Herbert Hansal, Frank Hellmann, Violetta Herrberg, Wolfgang Herschmann, Odo Hildenhagen, Torsten Jörn, Zoltan Jakubovich, Stefan Kalok, Jürgen Kleebach, Thomas Klitsch, Günter Knaak, Horst Kretzschmar, Jürgen Köhler, Jörg Kühne, Christoph Lassek, Jens Leferink, Sybille Leferink, Christoph Lehmenkühler, Martina Lorenz, Hans-Joachim Müller, Heiko Müller, Andreas Nachtmann, Sabine Niehaus, Inna Paseka, Walter Paulus, Holger Pausch, Eckhardt Petter, Regine Pfeil, Markus Cornelius Quosigk, Klaus Radau-Pfeil, Martin Ruppenthal, Reiner Schimmelpfenning, Burkhard Schirmer, Michael Schorr, Michael Spillner, Roland Sporleder, Herma Steinberg, Klaus-Dieter Toepfer, Gerald van Nasse, Carsten Weber-Isele, Frank Weinhold, Frank-Lothar Welter, Birgit Willeke, and Monika Wüstenhagen
AUTHOR CONTRIBUTIONS
B.M., F.S.-D., and C.T. designed the study and were responsible for recruitment and data processing. J.Z., E.T., and T.F. oversaw all statistical analyses. T.W., J.E., E.T., M.S., E.L., and N.K.F. were responsible for performing investigations and collecting and entering data. H.Z. and P.T. were involved in sample analyses and data interpretation. C.T. oversaw patient recruitment, reassessed independently the clinical diagnosis and characterization, and assisted in the interpretation of data. B.M., C.T., and J.Z. wrote the manuscript. F.S.-D., T.F., N.K.F., P.T., and H.Z. coedited the manuscript. B.M., C.T., and F.S.-D. had full access to the clinical primary data. All authors had access to the data generated in the study including the statistical analysis and decided to submit the paper for publication.
STUDY FUNDING
The study was supported by unrestricted research grants from the Paracelsus-Elena-Klinik, Kassel, Germany, Teva Pharma/Lundbeck, GE Healthcare, and the Hermann und Lilly Schilling Foundation; and unrestricted grants from the University Medical Centre Göttingen, the Paracelsus-Elena-Klinik, Kassel, Germany, the Michael J Fox Foundation for Parkinson's Research (MJFF), and Teva Pharma. The study sponsors provided support through an unrestricted grant and had no influence on the study design, collection and analysis of data, the writing of the paper, or the decision to submit the paper. The sponsors have been informed about the final manuscript and the submission for publication.
DISCLOSURE
B. Mollenhauer has received speaker honoraria from Orion Corporation and GlaxoSmithKline; holds or has pending patents re: Method of differentially diagnosing dementias, Novel ELISA-based quantification of α-synuclein proteins in cerebrospinal fluid and peripheral blood products using 384-well plates, and MicroRNA expression profiling of cerebrospinal fluid; serves as a consultant for Bayer Schering Pharma AG, Roche, Biogen, and GE Healthcare; and receives research support from Teva Pharmaceutical Industries Ltd., Desitin Pharmaceuticals, GmbH, Boehringer Ingelheim, GE Healthcare and the Michael J. Fox Foundation for Parkinson's Research as well as from the EU (within the Joint Programming for Neurodegenerative disorders). J. Zimmerman reports no disclosures relevant to the manuscript. F. Sixel-Döring serves/has served on scientific advisory boards for Orion Corporation and Medtronic Inc. and UCB; has received funding for travel from Boehringer Ingelheim, Cephalon, TEVA and MundiPharm; and has received speaker honoraria from Abbvie, Bayer Healthycare, Boehringer Ingelheim, Desitin, GSK, Meda Pharma, Medtronic Inc., Novartis, Orion Corporation, and UCB. N. Focke received funding from the Stifterverband für die Deutsche Wissenschaft (Dr. Werner Jackstädt-Stipend), served on an advisory board for GE Healthcare, and received honoraria from GlaxoSmithKline. T. Wicke reports no disclosures relevant to the manuscript. J. Ebentheuer has received speaker honoraria from Desitin and UCB. M. Schaumburg is supported by the Deutsche Parkinson Vereinigung (DPV). E. Lang, E. Trautmann, H. Zetterberg, and P. Taylor report no disclosures relevant to the manuscript. T. Friede serves as a consultant to Novartis, Biogen Idec, Grünenthal, AstraZeneca, Almirall, SGS, and Pharmalog. C. Trenkwalder serves on scientific advisory boards for Teva Pharma, Boehringer Ingelheim, UCB, Novartis, Mundipharma International Limited, and Britannia; has received speaker honoraria from Boehringer Ingelheim, Cephalon, Inc., UCB, Novartis, and GlaxoSmithKline; and receives research support from Teva Pharmaceutical Industries Ltd. and Mundipharm Intern. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Biglan KM, Holloway RG. Surrogate endpoints in Parkinson's disease research. Curr Neurol Neurosci Rep 2003;3:314–320. [DOI] [PubMed] [Google Scholar]
- 2.Parkinson Study Group. Effects of tocopherol and deprenyl on the progression of disability in early Parkinson's disease. N Engl J Med 1993;328:176–183. [DOI] [PubMed] [Google Scholar]
- 3.Investigators NN-P. A randomized clinical trial of coenzyme Q10 and GPI-1485 in early Parkinson disease. Neurology 2007;68:20–28. [DOI] [PubMed] [Google Scholar]
- 4.Parkinson Study Group PI. Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology 2007;69:1480–1490. [DOI] [PubMed] [Google Scholar]
- 5.Schuepbach WM, Rau J, Knudsen K, et al. . Neurostimulation for Parkinson's disease with early motor complications. N Engl J Med 2013;368:610–622. [DOI] [PubMed] [Google Scholar]
- 6.Erro R, Picillo M, Vitale C, et al. . Non-motor symptoms in early Parkinson's disease: a 2-year follow-up study on previously untreated patients. J Neurol Neurosurg Psychiatry 2013;84:14–17. [DOI] [PubMed] [Google Scholar]
- 7.Hely MA, Morris JG, Reid WG, Trafficante R. Sydney Multicenter Study of Parkinson's disease: non-L-dopa-responsive problems dominate at 15 years. Mov Disord 2005;20:190–199. [DOI] [PubMed] [Google Scholar]
- 8.Antonini A, Barone P, Marconi R, et al. . The progression of non-motor symptoms in Parkinson's disease and their contribution to motor disability and quality of life. J Neurol 2012;259:2621–2631. [DOI] [PubMed] [Google Scholar]
- 9.Zhang J, Mattison HA, Liu C, et al. . Longitudinal assessment of tau and amyloid beta in cerebrospinal fluid of Parkinson disease. Acta Neuropathol 2013;126:671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maetzler W, Liepelt I, Berg D. Progression of Parkinson's disease in the clinical phase: potential markers. Lancet Neurol 2009;8:1158–1171. [DOI] [PubMed] [Google Scholar]
- 11.Mollenhauer B, Trautmann E, Sixel-Doring F, et al. . Nonmotor and diagnostic findings in subjects with de novo Parkinson disease of the DeNoPa cohort. Neurology 2013;81:1226–1234. [DOI] [PubMed] [Google Scholar]
- 12.Parkinson-Syndrome: Diagnostik und Therapie. Available at: http://www.dgn.org/leitlinien-online-2012/inhalte-nach-kapitel/2346-ll-09-2012-parkinson-syndrome-diagnostik-und-therapie.html. Accessed October 6, 2015. [Google Scholar]
- 13.Tomlinson CL, Stowe R, Patel S, Rick C, Gray R, Clarke CE. Systematic review of levodopa dose equivalency reporting in Parkinson's disease. Mov Disord 2010;25:2649–2653. [DOI] [PubMed] [Google Scholar]
- 14.Goetz CG, Tilley BC, Shaftman SR, et al. . Movement Disorder Society-sponsored revision of the Unified Parkinson's Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord 2008;23:2129–2170. [DOI] [PubMed] [Google Scholar]
- 15.Hanganu A, Bedetti C, Degroot C, et al. . Mild cognitive impairment is linked with faster rate of cortical thinning in patients with Parkinson's disease longitudinally. Brain 2014;137:1120–1129. [DOI] [PubMed] [Google Scholar]
- 16.Ascherio A, LeWitt PA, Xu K, et al. . Urate as a predictor of the rate of clinical decline in Parkinson disease. Arch Neurol 2009;66:1460–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall S, Ohrfelt A, Constantinescu R, et al. . Accuracy of a panel of 5 cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or Parkinsonian disorders. Arch Neurol 2012:1–8. [DOI] [PubMed] [Google Scholar]
- 18.Feingold A. Effect sizes for growth-modeling analysis for controlled clinical trials in the same metric as for classical analysis. Psychol Methods 2009;14:43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maronna R, Martin D, Yohai V. Robust Statistics: Theory and Methods. New York: Wiley; 2006. [Google Scholar]
- 20.R Core Team. R: a Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. Available at: http://www.R-project.org. Accessed June 1, 2013. [Google Scholar]
- 21.Rothman KJ. No adjustments are needed for multiple comparisons. Epidemiology 1990;1:43–46. [PubMed] [Google Scholar]
- 22.Sixel-Doring F, Schweitzer M, Mollenhauer B, Trenkwalder C. Intraindividual variability of REM sleep behavior disorder in Parkinson's disease: a comparative assessment using a new REM sleep behavior disorder severity scale (RBDSS) for clinical routine. J Clin Sleep Med 2011;7:75–80. [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez-Martin P, Chaudhuri KR, Rojo-Abuin JM, et al. . Assessing the non-motor symptoms of Parkinson's disease: MDS-UPDRS and NMS Scale. Eur J Neurol 2015;22:37–43. [DOI] [PubMed] [Google Scholar]
- 24.Thobois S, Lhommee E, Klinger H, et al. . Parkinsonian apathy responds to dopaminergic stimulation of D2/D3 receptors with piribedil. Brain 2013;136:1568–1577. [DOI] [PubMed] [Google Scholar]
- 25.Muslimovic D, Schmand B, Speelman JD, de Haan RJ. Course of cognitive decline in Parkinson's disease: a meta-analysis. J Int Neuropsychol Soc 2007;13:920–932. [DOI] [PubMed] [Google Scholar]
- 26.Sixel-Doring F, Trautmann E, Mollenhauer B, Trenkwalder C. Associated factors for REM sleep behavior disorder in Parkinson disease. Neurology 2011;77:1048–1054. [DOI] [PubMed] [Google Scholar]
- 27.Ataide M, Franco CM, Lins OG. Daytime sleepiness in Parkinson's disease: perception, influence of drugs, and mood disorder. Sleep Disord 2014;2014:939713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anang JB, Gagnon JF, Bertrand JA, et al. . Predictors of dementia in Parkinson disease: a prospective cohort study. Neurology 2014;83:1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003;24:197–211. [DOI] [PubMed] [Google Scholar]
- 30.Tam CW, Burton EJ, McKeith IG, Burn DJ, O'Brien JT. Temporal lobe atrophy on MRI in Parkinson disease with dementia: a comparison with Alzheimer disease and dementia with Lewy bodies. Neurology 2005;64:861–865. [DOI] [PubMed] [Google Scholar]
- 31.Stewart T, Liu C, Ginghina C, et al. . Cerebrospinal fluid alpha-synuclein predicts cognitive decline in Parkinson disease progression in the DATATOP cohort. Am J Pathol 2014;184:966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fagan AM, Xiong C, Jasielec MS, et al. . Longitudinal change in CSF biomarkers in autosomal-dominant Alzheimer's disease. Sci Transl Med 2014;6:226ra230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The Parkinson Progression Marker Initiative (PPMI). Prog Neurobiol 2011;95:629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mollenhauer B, Locascio JJ, Schulz-Schaeffer W, Sixel-Doring F, Trenkwalder C, Schlossmacher MG. Alpha-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: a cohort study. Lancet Neurol 2011;10:230–240. [DOI] [PubMed] [Google Scholar]
- 35.Sherman RA, Funder DC. Evaluating correlations in studies of personality and behavior: beyond the number of significant findings to be expected by chance. J Res Personality 2009;43:1053–1063. [Google Scholar]
- 36.Smith KM, Eyal E, Weintraub D; ADAGIO Investigators. Combined rasagiline and antidepressant use in Parkinson disease in the ADAGIO study: effects on nonmotor symptoms and tolerability. JAMA Neurol 2015;72:88–95. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.