

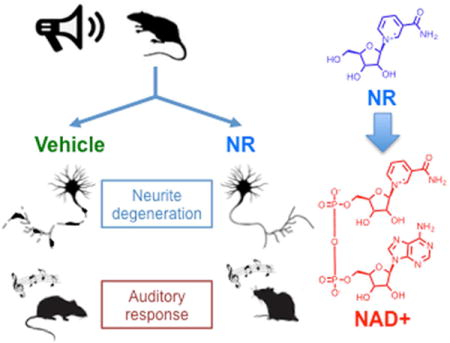
SUMMARY
Intense noise exposure causes hearing loss by inducing degeneration of spiral ganglia neurites that innervate cochlear hair cells. Nicotinamide adenine dinucleotide (NAD+) exhibits axon-protective effects in cultured neurons, however, its ability to block degeneration in vivo has been difficult to establish due to its poor cell permeability and serum instability. Here, we describe a strategy to increase cochlear NAD+ levels in mice by administering nicotinamide riboside (NR), a recently described NAD+ precursor. We find that administration of NR, even after noise exposure, prevents noise-induced hearing loss (NIHL) and spiral ganglia neurite degeneration. These effects are mediated by the NAD+-dependent mitochondrial sirtuin, SIRT3, since SIRT3-overexpressing mice are resistant to NIHL and SIRT3 deletion abrogates the protective effects of NR and expression of NAD+ biosynthetic enzymes. These findings reveal that administration of NR activates a NAD+-SIRT3 pathway that reduces neurite degeneration caused by noise exposure.
Graphical abstract
INTRODUCTION
Noise exposure is a major cause of hearing loss worldwide (Hammer et al., 2013). Noise exposure causes damage to diverse cochlear structures, including the spiral ganglia nerve fibers that normally form synaptic contacts with cochlear hair cells (Spoendlin, 1975). These synapses enable the spiral ganglia to convey acoustic information from the cochlea to higher order brain stem structures. Following intense noise exposure, hair cells release neurotransmitters that lead to excitotoxic damage in neurites, resulting in synaptic disruption and neurite degeneration that is evident after 24 h (Kujawa and Liberman, 2009; Lin et al., 2011; Spoendlin, 1975). If noise exposure is moderate, neurite regeneration can occur, which can restore synaptic connectivity and auditory capacity (Puel et al., 1998). However, persistent noise exposure or intense acoustic trauma can result in permanent neurite degeneration (Spoendlin, 1975).
Spiral ganglia neurite degeneration is linked to mitochondrial dysfunction. Following noise exposure, glutamate release induces the formation of mitochondria-derived reactive oxygen species (Jager et al., 2000; Ohlemiller et al., 1999; Puel et al., 1998; Puel et al., 1995; Ruel et al., 2005). Thus, impaired mitochondrial function may be an early step in NIHL.
Studies over the past decade have suggested that NAD+ may be useful for blocking axonal degeneration; however, the idea that NAD+ exerts axon-protective effects is controversial. Milbrandt and colleagues first showed that application of NAD+ to sensory neurons prevents axonal degeneration elicited by transection (Araki et al., 2004). Although this study suggested that the effects of NAD+ are transcription-dependent and occur at micromolar concentrations, another study showed that the effects of NAD+ are transcription-independent and require application of millimolar concentrations to axons (Wang et al., 2005). Other studies cast doubt on the idea that NAD+-biosynthetic enzymes exert their axon-protective effects through NAD+ since their protective effects do not correlate with their effects on NAD+ levels (Sasaki et al., 2009). Additionally, the intracellular target of NAD+ has been controversial. Initial studies suggested a role for the sirtuin SIRT1 in cultured neurons (Araki et al., 2004). However, this could not be replicated in SIRT1 knockout animals (Wang et al., 2005). The diverse inconsistencies seen in these and other studies make it unclear whether NAD+ influences a physiologically relevant axon-degeneration pathway.
The inconsistencies seen in studies of NAD+ may relate to the use of cultured neurons. Removal of neurons from their native environment and culturing them results in altered gene expression relative to neurons in vivo (Diaz et al., 2002). Additionally, Schwann cells and oligodendrocytes can be lost during culturing. These cells have a major role in regulating axonal integrity and influence axonal metabolism by transferring metabolites to axons (Saab et al., 2013). Since these cells are often lost during culturing, it is difficult to extrapolate studies on axon degeneration performed in vitro to axons in vivo that retain their interactions with diverse supporting cells. Thus, it remains unclear if NAD+ exerts an axon protective effect, and if this effect is seen in animals.
It is difficult to determine if NAD+ prevents axon degeneration in vivo. NAD+ is readily degraded by serum hydrolases (Chi and Sauve, 2013), making it difficult to test its effects in animals. Additionally, NAD+ is highly polar and, like other nucleotides and dinucleotides, is not readily taken up by cells (Bortell et al., 2001; Yang et al., 2007). As a result, millimolar extracellular concentrations are needed to induce micromolar changes in intracellular NAD+ concentrations (Bortell et al., 2001; Yang et al., 2007). Nicotinamide, an NAD+ precursor has been tested for effects on axon degeneration in an encephalomyelitis model (Kaneko et al., 2006). However, because nicotinamide inhibits sirtuins, NAD+-dependent deacetylating/deacylating enzymes (Guarente, 2013). it is unclear if the effects of nicotinamide reflect its ability to increase NAD+ or its pleiotropic inhibitory effects. Thus, it remains unclear if increasing cellular NAD+ levels is an effective approach for blocking axonal degeneration in vivo.
Here we examine whether augmentation of intracochlear NAD+ levels protects mice from NIHL. Using WldS mice, which overexpress an NAD+ biosynthetic enzyme (Conforti et al., 2000), we show that genetic stabilization of NAD+ levels markedly protects mice from NIHL. In order to test the effect of increasing intracellular NAD+ levels, we used the NAD+ precursor, nicotinamide riboside (NR). NR has been shown to inhibit axon degeneration elicited by axon transection in cultured neurons (Sasaki et al., 2006). We find that NR administration provides an efficient route to increase NAD+ levels in the cochlea in mice, and also protects mice from NIHL and noise-induced spiral ganglia neurite retraction. Importantly, we also show that the protective effects of NR can be achieved even when NR is administered after noise exposure, suggesting that NR might be clinically useful to prevent hearing loss after unexpected noise exposure in humans. We show that the effects of NR and the noise resistance of WldS mice are both promoted by the NAD+-dependent mitochondrial sirtuin, SIRT3. These data demonstrate the translational value of NR for pharmacologic augmentation of NAD+ and activation of a SIRT3-dependent pathway that inhibits NIHL.
RESULTS
WldS mice are markedly resistant to NIHL
To address whether NIHL might be influenced by NAD+, we first examined mice that exhibit enhanced endogenous NAD+ biosynthesis. The WldS mouse expresses a gene fusion comprising the NAD+ biosynthetic enzyme Nicotinamide mononucleotide adenylyl transferase 1 and Ube4a (Conforti et al., 2000). WldS is highly expressed in neurons, and its expression markedly delays degeneration of the distal sciatic axonal segment following axonal transection (Lunn et al., 1989). WldS expression prevents the drop in axonal NAD+ levels that normally occurs after axonal injuries (Wang et al., 2005). Thus, the WldS mouse provides a genetic approach to stabilize NAD+ levels.
Unless indicated, the C57BL/6 mouse strain was used as the background for all transgenic and knockout animals, as well as pharmacologic studies, due to its highly susceptibility to NIHL (Coling et al., 2003; Mizutari et al., 2013; Yan et al., 2013). This sensitivity has made C57BL/6 a widely utilized model to test approaches for blocking NIHL (Coling et al., 2003; Mizutari et al., 2013; Someya et al., 2010; Yan et al., 2013). Eight to ten week-old mice were used for all experiments, as mice of this age have no evidence of age-associated hearing loss (Someya et al., 2010). The WldS mice were backcrossed onto the C57BL/6 background (see Methods).
To test whether WldS-mediated NAD+ biosynthesis protects from NIHL, we measured auditory brainstem responses (ABR) elicited by tone burst stimuli after acoustic trauma. An ABR occurs when a mouse hears the tone burst stimulus. Acoustic trauma was elicited by a 90 dB octave band noise exposure for 120 min. To quantify the degree of hearing loss, we measured “threshold shifts.” These shifts refer to the increased level of sound intensity that is required to elicit an ABR. The threshold for detecting sound stimuli is determined by exposing mice to 5 msec tone bursts at a specific frequency and volume. The minimum sound intensity that evokes an ABR, and also shows an increased magnitude with increasing sound intensity, is designated the sound threshold for the tested frequency (Willott, 2005). If the threshold shift is only seen 24 hr after noise exposure, it is designated a “transient threshold shift,” while threshold shifts that remain 14 d after noise exposure are designated “permanent threshold shifts.”
We measured transient and permanent threshold shifts in noise-exposed WldS mice using 8,000 Hz, 16,000 Hz and 32,000 Hz tone bursts. In wild-type mice, 24 hr after noise exposure the transient threshold shift at 32,000 Hz was 38 dB (Fig. 1). At 16,000 Hz, a threshold shift of 21 dB was observed. A smaller shift of 10 dB was noted at 8,000 Hz (Fig. 1). These threshold shifts persisted at 14 d. The persistence of these threshold shifts indicates permanent hearing loss. The more prominent hearing loss at higher frequencies following noise exposure is typical of NIHL (Wang et al., 2002).
Figure 1. Genetic augmentation of NAD+ protects against NIHL.
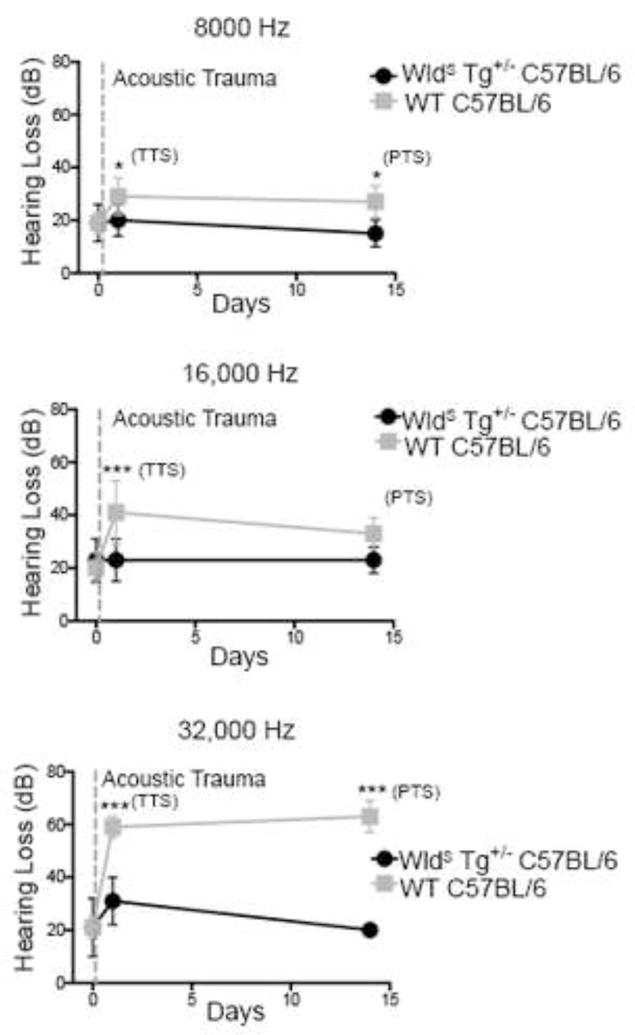
WldS mice have reduced NIHL. WldS C57BL/6 transgenic animals (black) and littermate control C57BL/6 animals (grey) were exposed to 2 hr of 90 dB noise and then evaluated for changes in hearing 24 hr and 2 wks later by measuring auditory brainstem responses. WldS C57BL/6 animals showed marked protection as measured by both the levels of transient hearing loss at 24 hr and permanent hearing loss at 14 d in comparison to wild-type controls, most notably in the higher frequencies (16,000 and 32,000 Hz) typically affected by noise exposure. This demonstrates that genetically enhanced NAD+ biosynthesis can prevent hearing loss associated with noise exposure. Each point is the average threshold shift measured on a minimum of 7 mice per group. Statistical analysis was performed using a two-way ANOVA followed by a Bonferroni post-test (*P < 0.05, ***P < 0.001). Error bars represent SD.
In contrast, the WldS mice exhibited clear protection from NIHL. At 24 hr following acoustic trauma, mice exhibited no threshold shift at 8,000 and 16,000 Hz, and a 10 dB threshold shift at 32,000 Hz (Fig. 1). By 14 d, no threshold shift was seen at any frequency. These data show that WldS animals exhibit marked resistance to both transient and permanent hearing loss following acoustic trauma.
Administration of NR prevents decreases in cochlear NAD+ after noise exposure and protects mice from NIHL
We next sought to achieve the beneficial effects of the WldS mutation using a pharmacologic approach. Because NAD+ is highly cell impermeable, it is not suitable as a therapeutic agent. We recently described a synthetic route to prepare the NAD+ precursor, NR, in amounts suitable for administration to animals (Yang et al., 2007). This compound is markedly less polar than NAD+, and treatment with NR increases NAD+ levels in various mammalian cell lines by as much as 50–170% (Yang et al., 2007; Canto et al., 2012). NR is a salvageable precursor of NAD+, that is phosphorylated by NR kinases as the first step in its conversion to NAD+ (Bogan and Brenner, 2008). NR may be particularly bioavailable in neurons due to the expression of genes in the NR kinase pathway (Sasaki et al., 2006). Notably, NR kinase 2, as well as other enzymes involved in NAD+ biosynthesis are induced in sensory neurons following axon injury (Sasaki et al., 2006).
We therefore asked if administration of NR to mice could prevent a decrease in cochlear NAD+ levels following noise exposure. Prior to noise exposure, cochlear NAD+ levels were essentially identical in control and WldS mice (Supplementary Fig. 1a). Following noise exposure, NAD+ levels were decreased by almost 60% (Supplementary Fig. 1b). This noise-induced decrease was absent in mice harboring the WldS allele (Supplementary Fig. 1b). Similarly, in mice treated with NR (1000 mg/kg) twice daily for 5 days prior to noise exposure and continuing for 48 hours after noise exposure, there was no significant noise-induced reduction in cochlear NAD+ levels (Supplementary Fig. 1b). Thus, NR and WldS prevent the drop in NAD+ levels seen following noise exposure.
We then asked if administration of NR could prevent NIHL. In these experiments, NR was administered by intraperitoneal injection at 1000 mg/kg twice daily, beginning 5 d prior to acoustic trauma, until 14 d following the trauma.
Compared to vehicle-treated mice, NR-treated mice exhibited negligible transient threshold shifts at 24 hr at 8000 Hz and 16,000 Hz (6 and 8 dB respectively), and a reduced threshold shift at 32,000 Hz (16 dB) (Fig. 2). NR-treated mice were also protected from permanent hearing loss at all 3 frequencies (Fig. 2).
Figure 2. NR prevents NIHL.
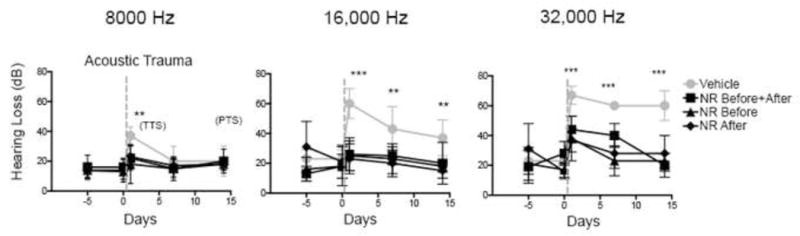
Administration of NR prevents NIHL. C57BL/6 mice were tested for hearing loss at 24 hr, 7 d and 14 d after exposure to 2 hr of 90 dB noise. NR was administered at 1,000 mg/kg by intraperitoneal injection twice daily either for 5 d prior to noise exposure and 14 d after (NR All), 5 d prior to noise exposure (NR Before) or 14 d after exposure (NR After). NR treatment in all groups showed protection against transient hearing loss at 24 hr and permanent hearing loss at 14 d in comparison to vehicle controls, most notably at higher frequencies affected by noise exposure (16,000 and 32,000 Hz). Each point is the average threshold shift measured in 7 mice per group. Statistical analysis was performed using a two-way ANOVA followed by a Bonferroni post-test (**P < 0.01, ***P < 0.001). Error bars represent SD.
We next asked if the effect of NR is seen in other mouse strains. We subjected BalbC or CBA mice to NR (1000 mg/kg) twice daily as above. As with C57BL/6 mice, NR-treated BalbC and CBA mice exhibited negligible transient threshold shifts at 24 hr at 8000 Hz and 16,000 Hz, and a reduced threshold shift at 32,000 Hz (Supplemental Fig. 2). Both of these strains of NR-treated mice were also protected from permanent hearing loss at all 3 frequencies (Supplemental Fig. 2). Thus, NR markedly reduces NIHL in diverse genetic backgrounds.
Administration of NR after noise exposure prevents NIHL
We next asked if NR exhibits protective effects when administered following acoustic trauma. In these experiments, NR was administered for the 5 d prior to acoustic trauma, or for the 14 d following acoustic trauma. In both cases, NR administration prevented both transient threshold shifts at 24 hr and permanent threshold shifts at 14 d (Fig. 2). These data indicate that NR administered after exposure to acoustic trauma is sufficient to prevent transient and permanent hearing loss.
SIRT3-overexpressing mice are highly resistant to NIHL
If NR mediates its effects by conversion to NAD+, then its effects would require a NAD+-regulated enzyme. NAD+-regulated mitochondrial enzymes are possible candidates for several reasons. First, treatment of cells with NR preferentially increases NAD+ levels in mitochondria (Canto et al., 2012). Thus, NR may have a more pronounced effect on mitochondrial NAD+-dependent enzymes compared to cytosolic enzymes. Second, WldS has been detected in various subcellular compartments, including mitochondria (Avery et al., 2012), which raises the possibility that it may exert its protective effects by influencing mitochondrial NAD+ levels. Third, NIHL is associated with increased mitochondria-derived reactive oxygen species, a hallmark of mitochondrial dysfunction (Ohlemiller et al., 1999). Lastly, overexpression of SIRT3, a mitochondrial NAD+-dependent deacetylase, was recently shown to potentiate the axon-protective effects of NAD+ in a KCl deprivation model of axon degeneration (Magnifico et al., 2013). SIRT3 deacetylates a large subset of the mitochondrial proteome (Hebert et al., 2013; Rardin et al., 2013). The other mitochondrial sirtuins, SIRT4 and SIRT5, have much more limited roles in mitochondria as deacetylases and appear to function to remove other acyl modifications (Herskovits and Guarente, 2013). We therefore tested whether SIRT3 mediates the effects of NAD+.
To test this idea, we generated SIRT3-overexpressing mice. Cre-inducible SIRT3-overexpressing mice were generated and crossed with a β-actin/Cre mouse line to create mice that exhibit global SIRT3-overexpression, with an increase in SIRT3 levels of ~3.5 fold (Fig. 3a–c, Supplementary Fig. 3).
Figure 3. SIRT3 transgene overexpression prevents NIHL.
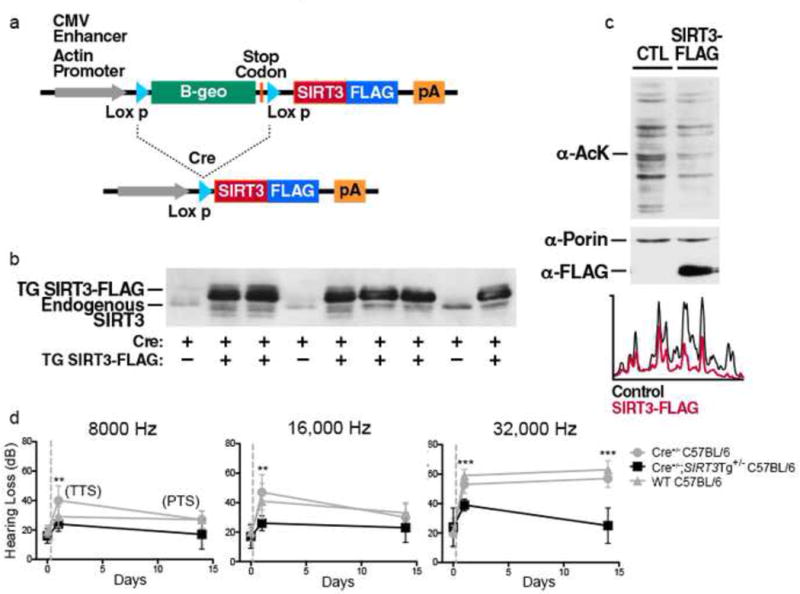
(a) Schematic representation of the transgene and its modification in the presence of Cre. Cre expression results in a genomic deletion that leads to SIRT3 expression under the control of the actin promoter. The mice used in this study were crossed with a ß-actin/Cre mouse to generate offspring overexpressing SIRT3 in all cells.
(b) Confirmation of SIRT3 expression in SIRT3-overexpressing mice. Expression of SIRT3-FLAG is detected by western blotting analysis in mouse liver using specific antiserum for mouse SIRT3. The endogenous mouse SIRT3 protein is seen as a lower abundance and lower molecular weight protein (minus FLAG).
(c) Overexpression of SIRT3 is associated with a global decrease in mitochondrial protein acetylation. We wanted to determine if the FLAG-tagged SIRT3 transgene is functional. Since SIRT3 mediates the deacetylation of mitochondrial proteins, we asked if there was reduced acetylation of mitochondrial proteins in the SIRT3-overexpressing mice. Mitochondria isolated from liver of SIRT3-overexpressing mice and a littermate control were analyzed by western blotting using an anti-acetyllysine antibody. Immunostaining for the mitochondrial protein porin was used to confirm equal loading (middle panel). The autoradiogram was analyzed by a line scan (below) and shows a global decrease in mitochondrial protein acetylation (Wild type, black; SIRT3-overexpressing mice, red). These data indicate that the FLAG epitope does not inhibit the function of the SIRT3 protein and that the SIRT3-oveerexpressing mice have increased SIRT3 activity in vivo.
(d) WT C57BL/6 animals, Cre+/− C57BL/6 mice and Cre+/−; SIRT3 Tg+/− C57BL/6 mice were tested for hearing loss just prior to a 2 hr 90dB noise exposure, 24 hr after noise exposure, and 14 days after noise exposure. C57BL/6 Mice with SIRT3 overexpression were protected against both transient and permanent NIHL most markedly at higher frequencies. Statistical analysis was performed using a two-way ANOVA followed by a Bonferroni post-test (**P < 0.01, ***P < 0.001). Error bars represent SD.
We then asked if 8–10 week old SIRT3-overexpressing mice exhibit protection from NIHL. At any given NAD+ concentration, Michaelis-Menten kinetics predicts that increased SIRT3 expression will give a corresponding increase in SIRT3-mediated deacetylation. As expected, wild-type mice and mice only expressing the Cre allele were highly susceptible to noise exposure (Fig. 3d). However, noise-exposure in SIRT3-overexpressing mice results in no significant 24 hr threshold shift at 8,000 and 16,000 Hz, and a mild 10 dB threshold shift at 32,000 Hz (Fig. 3d). By 14 d, no threshold shift was seen at any frequency. Taken together, these data show that SIRT3 overexpression prevents both transient and permanent hearing loss.
SIRT3 is necessary for the protective effects of the WldS gene and NR administration
We next asked if SIRT3 mediates the protective effects of WldS. We generated WldS mice containing a deletion of the SIRT3 gene (SIRT3−/−; WldS). WldS mice exhibited marked resistance to NIHL at all frequencies, at both 24 hr and 14 d (Fig. 4). However, the SIRT3−/−; WldS mice were highly susceptible to NIHL at both time points, resembling the hearing loss seen in wild-type animals (Fig. 4). Thus, SIRT3 is required for the protective effects of WldS on NIHL.
Figure 4. WldS mice are protected against NIHL in a SIRT3-dependent fashion.
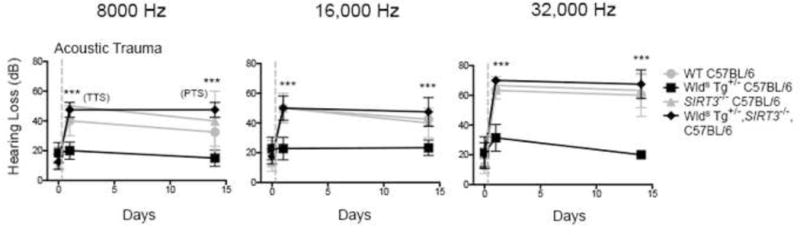
WT C57BL/6, WldSTg+/− C57BL/6, SIRT3−/− C57BL/6, and WldSTg+/−;SIRT3−/− C57BL/6 animals were tested for hearing loss just prior to a 2 hr 90 dB noise exposure, 24 hr after noise exposure, and 14 days after noise exposure. SIRT3 was necessary for WldS protection against NIHL for both transient and permanent hearing losses. Statistical analysis was performed using a two-way ANOVA followed by a Bonferroni post-test (***P < 0.001). Error bars represent SD.
We also asked if SIRT3 is required for the protective effects of NR administration. While wild-type mice treated with NR were protected from both transient and permanent NIHL at all frequencies at 24 hr and 14 d, SIRT3−/− mice treated with NR showed significantly reduced protection from either transient or permanent hearing loss following noise exposure (Fig. 5). Thus, the protective effects of both WldS and NR require SIRT3.
Figure 5. Administration of NR prevents NIHL in a SIRT3-dependent fashion.
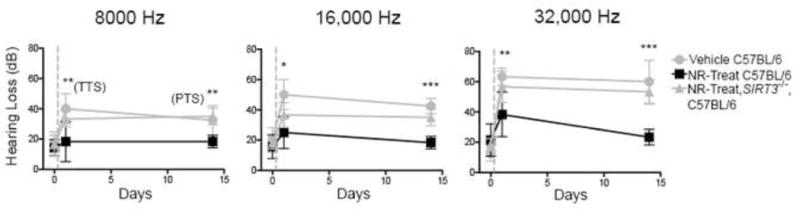
NR was administered to C57BL/6 animals at 1,000 mg/kg by intraperitoneal injection twice daily for 5 d prior to a noise exposure of 90 dB for 2 hr. SIRT3 was necessary for NR-mediated protection against transient hearing loss at 24 hr and permanent hearing loss at 14 d. Statistical analysis was performed using a two-way ANOVA followed by a Bonferroni post-test (*P < 0.05, **P < 0.01, ***P < 0.001). Error bars represent SD.
Although our focus is on NIHL, we asked if SIRT3 contributes to other forms of NAD+-dependent axon protection. Vinca alkaloids, such as vinblastine, induce neurite degeneration in cultured sensory neurons, which can be blocked by application of NAD+ (Araki et al., 2004; Conforti et al., 2007; Wang et al., 2005). We confirmed the axon-degeneration effect of vinblastine in a slice culture model, which preserves axon interactions with supporting cells (Supplementary Fig. 4). Using mouse E10.5 limb bud slice cultures containing dorsal root ganglia sensory neurons and their axonal projections, we found that 100 nM vinblastine induces axon degeneration, while application of 5 mM NAD+ blocked this effect (Supplementary Fig. 4). However, this protective effect was lost in SIRT3−/− slice cultures (Supplementary Fig. 4). These data suggest that the protective effects of SIRT3 might extend to other forms of axon degeneration.
NR prevents neurite retraction from inner hair cells in a SIRT3-dependent manner
NIHL is associated with retraction of spiral ganglia neurites from the inner hair cells (Spoendlin, 1975). To measure this, we examined inner hair cell innervation in the basal turn of the cochlea, the region most affected by acoustic trauma (Wang et al., 2002). Immunofluorescence labeling of cochlear sections using an NF-200 antibody to label spiral ganglia neurites provides an assessment of whether they are in contact with inner hair cells. The hair cells are readily identified by their basally located nucleus. In animals not exposed to noise, spiral ganglia neurites terminate near the base of the inner hair cells (Fig. 6a).
Figure 6. NR prevents neurite retraction from inner hair cells in a SIRT3-dependent manner.
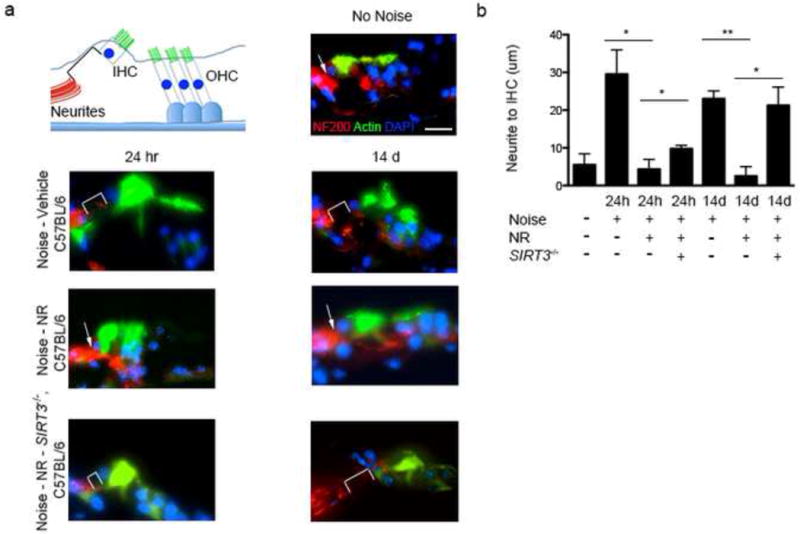
(a) Neurite retraction from inner hair cells is reduced following noise exposure in mice treated with NR. To determine whether NR treatment also reduced spiral ganglia neurite degeneration, C57BL6 mice were treated for 5 d with NR (1,000 mg/kg by intraperitoneal injection, twice daily) or vehicle prior to noise exposure. Cochlea were then harvested either 24 hr or 14 d after noise exposure and neurite retraction from inner hair cells was measured by staining with an anti-NF200 antibody (red, spiral ganglia neurites) and actin (green, inner hair cells). DAPI staining (blue) was used to identify the nucleus of the inner hair cells, which defines the base of these cells. Representative immunofluorescence staining is shown for control mice that were not exposed to noise, noise-exposed mice treated with vehicle, noise-exposed mice treated with NR, and noise-exposed SIRT3−/− mice treated with NR. Neurite retraction (indicated with a white bracket) is seen in vehicle-treated mice both 24 hr and 14 d following noise exposure. Neurite retraction is considerably reduced in NR-treated mice at both time points (the junction between the neurites and hair cells is indicated with a white arrow). The protective effect of NR against neurite retraction is lost in SIRT3−/− animals. This suggests that NR stabilizes spiral ganglia neurites and prevents their retraction following noise exposure in a SIRT3-dependent manner. Scale bar, 25 μm.
(b) Quantification of results in (a). The distance between the tips of the spiral ganglia neurites to the nucleus at the base of the inner hair cell (IHC) was measured to determine the degree of neurite retraction. Reduced neurite retraction is seen in noise-exposed mice that were treated with NR compared to those treated with vehicle alone. This reduction in neurite retraction is dependent on SIRT3. Shown are the average measurements obtained from a minimum of 5 cochlea from 3 mice for each group. Statistical analysis was performed using a one-way ANOVA followed by T-test (*P < 0.05, **P < 0.01). Error bars represent SD.
We therefore asked if NR and SIRT3 function by blocking neurite retraction. As expected, in vehicle-treated, noise-exposed wild type animals the spiral ganglia neurites were retracted from inner hair cells at 24 hr (29.5 ± 12.9 μm) 24 hr following noise exposure (Fig. 6a,b). The neurites remained retracted (23 ± 3.6 μm) 14 d after noise exposure (Fig. 6a,b). The persistent retraction indicates a permanent loss of synaptic connectivity between hair cells and spiral ganglia neurites in vehicle-treated animals after noise exposure.
In NR-treated animals, acoustic trauma resulted in minimal neurite retraction after both 24 hr (4.3 ± 4.5 μm) and 14 d (2.5 ± 3.5 μm) (Fig. 6a,b). These data suggest NR prevents hearing loss by preventing the loss of contacts between the inner hair cells and spiral ganglia neurites.
We next examined whether NR prevents neurite retraction in noise-treated SIRT3−/− mice (Fig. 6a,b). In control mice, NR treatment resulted in pronounced protection from noise-induced neurite retraction at 14 d. In contrast, neurite retraction was readily evident in SIRT3−/− mice that were treated with NR at 14 d (21.3 ± 8.4 μm). These data indicate that NR blocks the permanent neurite retraction seen at 14 d through SIRT3. At the 24 h time point, the effects of NR were incompletely abolished in SIRT3−/− mice, suggesting that other neuroprotective mechanisms of NR may be utilized. Taken together, these data indicate that SIRT3 mediates the ability of NR to prevent the neurite retraction that characterizes permanent hearing loss.
DISCUSSION
Currently, there is no effective therapy to prevent hearing loss following noise exposure. In this study we show that noise exposure results in a drop in cochlear NAD+ levels, and pharmacologic augmentation of NAD+ using NR provides robust protection from acoustic trauma in a SIRT3-dependent manner. We show that NR preserves the synaptic contacts between the spiral ganglia neurites and hair cells, demonstrating the effects of NR reflect preserved cochlear circuitry. These studies point to a novel NAD+/SIRT3 pathway that can be induced by NR to prevent NIHL and spiral ganglia neurite degeneration.
Our data identify SIRT3 as a target of NR and WldS in NIHL. The mitochondrial sirtuin SIRT3 is likely to be particularly responsive to pharmacologically administered NR, since NR preferentially increases mitochondrial NAD+ levels (Canto et al., 2012). Mitochondrial proteins appear particularly prone to enzyme-inactivating acetylation on catalytic lysine residues (Ghanta et al., 2013). SIRT3 maintains enzyme function by removing these acetylation marks (Wagner and Payne, 2013). Indeed, impaired SIRT3 function leads to enhanced reactive oxygen species (ROS) generation (He et al., 2012) and lower levels of reduced glutathione (Someya et al., 2010). Importantly, both ROS and impaired glutathione levels are linked to increased susceptibility to NIHL (Someya et al., 2010). The reduced cochlear NAD+ levels seen after noise exposure may account for the increased mitochondria-derived reactive oxygen species that is seen following acoustic trauma (Ohlemiller et al., 1999). Thus, activation of SIRT3 by augmenting intracellular NAD+ levels provides a mechanism to counteract this central pathogenic mechanism in hearing loss.
The WldS allele ameliorates axonal degeneration in a variety of neurodegenerative models (Kaneko et al., 2006; Sajadi et al., 2004; Samsam et al., 2003). However, therapeutic strategies that mimic WldS are currently unavailable. Administration of NAD+ is not viable due to the poor solubility, cell impermeability, and serum instability of this compound (Pitkanen, 1971). The recent development of a synthetic method to produce NR at quantity sufficient for administration to animals (Yang et al., 2007), allows the testing of the physiologic effects of augmenting intracellular NAD+ levels. Our finding that NR mimics the NIHL resistance seen in WldS animals raises the possibility that NR might also be useful for preventing axonal degeneration in other disease models that exhibit responsiveness to WldS expression.
The data supporting the role of NAD+ in axon protection has been inconsistent. Although WldS animals show delayed axon degeneration for several weeks in a transection model (Conforti et al., 2009), application of NAD+ to transected axons in culture delays degeneration for only a few hours or days (Wang et al., 2005) and requires millimolar concentrations. Additionally, inconsistent results using cultured neurons have made it unclear whether NAD+ would function to inhibit axon degeneration in animals. Poor bioavailability has prevented testing the role of NAD+ in animals using disease-relevant axon degeneration models. Our data using NR demonstrate that NAD+ and an NAD+ target, SIRT3, control axon degeneration in animals. It will be important to perform thorough analysis of NR and SIRT3 in other forms of axon degeneration in animals to define which of these can be blocked by NR.
Although our data identifies a NAD+/SIRT3 pathway that influences neurite degeneration, other pathways are also likely to affect neurite degeneration. First, it is likely that other sirtuins are activated by increased NAD+ levels and contribute to the axon-protective effects of NAD+, possibly by enhancing SIRT3 transcription as has been described for SIRT1 (Amat et al., 2009). Additionally, transection axon injury results in a loss of the axonal NAD+ biosynthetic enzyme NMNAT-2 (Gilley and Coleman, 2010). This is expected to result in an increase in NMN levels (Gilley and Coleman, 2010). Expression of WldS may delay transection-induced degeneration by converting this accumulated NMN to NAD+. However, in addition to the loss of the protective effects of NAD+, loss of NMNAT-2 could lead to the accumulation of NAD+ metabolic precursors that may have their own, potentially toxic, effects in axons (Di Stefano et al., 2014). NR could have direct protective effects by being converted to mitochondrial NAD+ and subsequently to mitochondrial NADPH, which is required to regenerate reduced glutathione, a neuroprotective agent in NIHL (Bellomo et al., 1987; Yamasoba et al., 1998). NR or NAD+ may also influence CNS auditory pathways. Lastly, axon protection mediated by overexpression of NMNAT enzymes may be in part due to chaperone-like functions of these proteins. This activity may contribute to their axon-protective effects in addition to their NAD+ biosynthetic function (Zhai et al., 2008). Nevertheless, our data suggest that NR mediates its effects through SIRT3 to induce substantial protective effects even in the absence of these additional mechanisms.
It is currently unclear why there are marked person-to-person differences in susceptibility to NIHL (Davis et al., 2003). Polymorphisms exist in the SIRT3 gene that affects its activity (Hirschey et al., 2011), and may in turn contribute to susceptibility to NIHL. NR is used as a dietary supplement and is a natural component of milk and certain other foods (Bogan and Brenner, 2008), raising the possibility that variability in dietary NR consumption could also contribute to the susceptibility to hearing loss in humans. Since NR exhibits minimal toxicity (Yang et al., 2007; Bieganowski and Brenner, 2004; Canto et al., 2012), NR supplementation may be a valuable approach for reducing the prevalence of NIHL.
EXPERIMENTAL PROCEDURES
Chemicals
Nicotinamide riboside (NR) was synthesized as previously described (Yang et al., 2007). Other chemicals and reagents were purchased from Sigma except as indicated below.
Animals
8–10 week old animals were used for all experiments to eliminate the effects of aging in C57BL/6 animal backgrounds as previously described (Someya et al., 2010). C57BL/6, BalbC and CBA mice were purchased from Jackson Laboratories. WldS tg/+ mice were provided backcrossed on a C57BL/6 background and were further backcrossed a minimum of 5 generations on the C57BL/6 background (Jackson Laboratories). These mice were a kind gift of Dr. Michael Coleman (Babraham Institute, University of Cambridge).
For experiments using SIRT3 knockout mice, male and female SIRT3−/− mice were purchased from the Mutant Mouse Resource Centers at the University of North Carolina-Chapel Hill (Chapel Hill, NC). These mice were developed from embryonic stem (ES) cells (Omni bank No. OST341297) with a retroviral promoter trap that functionally inactivates one allele of the SIRT3 gene. SIRT3−/− mice were backcrossed 5 generations onto the C57BL/6 background.
The transgene SIRT3-overexpressing vector was constructed by introducing a C-terminal FLAGed cDNA encoding the mouse mitochondrial form of SIRT3 (Cooper et al., 2009) into the pCall2 vector (Lobe et al., 1999). The vector pCall2-mSIRT3 was double-digested with XmnI and SfiI, and the resulting 7.9 kb fragment was gel purified and used to microinject C57BL/6 mouse embryos oocytes (Gladstone Transgenic Gene-Targeting Core). Several independent mouse lines carrying the SIRT3 transgene were generated. First-generation offspring (F1) of the injected mice were crossed with wild type mice and analyzed for germline incorporation of the transgene. Several F1 mice with germline incorporation were preserved as founding SIRT3 transgenic mouse lines. The mouse line used in this manuscript is called line 28 and contains the transgene inserted in an intergenic region on chromosome 10 (NCBI Ref NT_039500.7).
All animal behavioral studies were conducted at the AAALAC-approved facility at Weill Cornell Medical College. Experiments were performed in accordance with protocols approved by the Weill Cornell Medical College Institutional Animal Care and Use Committee (New York, NY)
Cochlear histology/immunohistology
Mouse cochlea were prepared as previously described (Whitlon et al., 2001). Briefly, mouse cochlea were quickly dissected from the temporal bone following rapid decapitation. Once separated, the apex of the cochlea was gently fenestrated and the cochlea were immediately fixed in 4% paraformaldehyde overnight at 4°C. The cochlea were then washed with three changes of PBS and incubated in a decalcification solution (10% EDTA/PBS pH 7.4) under constant rotation at 4°C for 7 d. Decalcification solution was changed daily. Cochlea were then washed in 3 changes of PBS and then treated with progressively increasing sucrose concentrations from 10–30%. They were then incubated overnight in 30% sucrose at 4°C. The cochlea were then incubated an additional 24 hr in OCT compound (Tissue-Tek). Following this final incubation the cochlea were transferred to cryomolds, carefully aligning the modiolus parallel to the bottom of the mold and frozen over dry ice. Mid-modiolar samples were then cut at a 10 μm thickness and mounted on glass slides (VWR superfrost plus). Sections were then dried for 2 hr prior to staining.
Slides were then post-fixed with 1.5% paraformaldehyde for 5 min. Slides were washed and then incubated with 0.5% triton X-100/PBS for 15 min. They were again washed and then blocked with 2% BSA/PBS. Sections were then incubated with phalloidin-488 (Invitrogen) as per the manufacturer’s instructions for 20 min. Slides were then washed and then incubated with 1:1000 rabbit anti-heavy neurofilament antibody overnight at 4°C. Slides were then washed and incubated with 1:1000 Alexafluor-546 goat anti-rabbit antibody (Invitrogen) for 1 hr at room temperature. After a final wash, sections were mounted with ProLong Gold antifade reagent with DAPI (Life Technologies). Three-color epifluorescence imaging was then performed using a Nikon Eclipse Ti microscope with a Coolsnap HQ2 camera.
NAD+ Quantification
Mice were injected twice daily with NR (1000mg/kg) for 5 days prior to noise and injury and for 48 hours thereafter until they were sacrificed and cochlea harvested. Noise injury was performed as indicated below. Cochlea were isolated by microdissection from the otic capsule. Pairs of cochlea were homogenized with a micropestle and resuspended in 7% perchloric acid. Samples were incubated for 5 min on ice, and any remaining insoluble debris was pelleted and saved for protein quantification via Bicinchoninic acid (BCA) assay. The supernatant was neutralized (pH 7) with NaOH and 0.1 M phosphate buffer. Sample was added to a 96-well plate and mixed with cycling assay buffer (5 mM Tris-HCl, 5 mM MgCl2, 50 mM KCl, 54 M resazurin, 2.25 mM lactate, 0.4 U/mL lactate dehydrogenase). Finally, 0.5 U diaphorase was added to each well, and the production of resorufin was recorded (530 nm excitation, 580 nm emission) every 30 sec for 15 min. NAD+ concentration was calculated based on a standard curve, and the results were expressed as nmol NAD+ per mg of protein. Three technical replicates were performed for n=7 mice per condition. Because of experiment-to-experiment variation in sample processing, samples were processed in parallel and normalized to the before-noise sample. The normalized values from each experiment were used to obtain the presented average values.
Auditory Testing
Auditory brainstem response testing was performed as previously described (Willott, 2005). Animals were tested following sedation with ketamine/xylazine (40 mg/kg and 10 mg/kg respectively). Tone burst stimuli at 8,000, 16,000 and 32,000 kHz for 5 msec were used to elicit auditory evoked responses using an auditory brainstem recording system (Intelligent Hearing Systems, Miami, FL). An evoked response was determined by identifying waveforms at proper time intervals that grew increased in magnitude with increasing volume as described previously (Willott, 2005).
Noise Exposure
Animals were exposed to a 90 dB octave band for 2 hr in a cage placed in a soundproof chamber (MAC-2, Industrial Acoustics Company, Bronx NY). The mice were able to freely move throughout the cage. The octave band was generated using ToneGen software (NCH software, Greenwood Village, CO) routed through an Audiosource Amp100 amplifier driving two down-facing Fostex FT-96H speakers. The sound pressure level was confirmed at 0, 30, 60, and 90 min, and again just prior to completion of sound exposure using an Extech microphone 407736.
Supplementary Material
Response to the Reviewer’s Comments.
We are delighted that the reviewers have found our response to their critiques acceptable. As requested by reviewer #2, we added a new “in press” reference to the manuscript.
We would like to again thank the reviewers for volunteering their time and effort to provide ideas for enhancing our manuscript.
Acknowledgments
We thank members of the Jaffrey lab for helpful suggestions, and M. Coleman (Babraham Institute, University of Cambridge) for WldS mice. This work was supported by startup funds from Weill Cornell to K.B., the NYS DOH Spinal Cord Injury Fund (S.R.J. and A.A.S.), NIH grants NS56306 and CA176638 (S.R.J.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
K.D.B. and S.R.J. designed experiments and wrote the manuscript, K.D.B. and S.M. performed the experiments, K.D.B. prepared the figures, E.V., Y.P., J.H. generated and characterized SIRT3 transgenic mice, W.L., S.M., and W.H. performed NAD+ measurements, W.H. performed limb bud assays, A.A.S. provided NR and guidance on NR dosage.
References
- Amat R, Planavila A, Chen SL, Iglesias R, Giralt M, Villarroya F. SIRT1 controls the transcription of the peroxisome proliferator-activated receptor-gamma Co-activator-1α (PGC-1α) gene in skeletal muscle through the PGC-1α autoregulatory loop and interaction with MyoD. J Biol Chem. 2009;284:21872–21880. doi: 10.1074/jbc.M109.022749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Avery MA, Rooney TM, Pandya JD, Wishart TM, Gillingwater TH, Geddes JW, Sullivan PG, Freeman MR. WldS prevents axon degeneration through increased mitochondrial flux and enhanced mitochondrial Ca2+ buffering. Curr Biol. 2012;22:596–600. doi: 10.1016/j.cub.2012.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellomo G, Mirabelli F, DiMonte D, Richelmi P, Thor H, Orrenius C, Orrenius S. Formation and reduction of glutathione-protein mixed disulfides during oxidative stress. A study with isolated hepatocytes and menadione (2-methyl-1,4-naphthoquinone) Biochem Pharmacol. 1987;36:1313–1320. doi: 10.1016/0006-2952(87)90087-6. [DOI] [PubMed] [Google Scholar]
- Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell. 2004;117:495–502. doi: 10.1016/s0092-8674(04)00416-7. [DOI] [PubMed] [Google Scholar]
- Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Ann Rev Nutr. 2008;28:115–130. doi: 10.1146/annurev.nutr.28.061807.155443. [DOI] [PubMed] [Google Scholar]
- Bortell R, Moss J, McKenna RC, Rigby MR, Niedzwiecki D, Stevens LA, Patton WA, Mordes JP, Greiner DL, Rossini AA. Nicotinamide adenine dinucleotide (NAD) and its metabolites inhibit T lymphocyte proliferation: role of cell surface NAD glycohydrolase and pyrophosphatase activities. J Immunol. 2001;167:2049–2059. doi: 10.4049/jimmunol.167.4.2049. [DOI] [PubMed] [Google Scholar]
- Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi Y, Sauve AA. Nicotinamide riboside, a trace nutrient in foods, is a vitamin B3 with effects on energy metabolism and neuroprotection. Curr Opin Clin Nutr Metab Care. 2013;16:657–661. doi: 10.1097/MCO.0b013e32836510c0. [DOI] [PubMed] [Google Scholar]
- Coling DE, Yu KC, Somand D, Satar B, Bai U, Huang TT, Seidman MD, Epstein CJ, Mhatre AN, Lalwani AK. Effect of SOD1 overexpression on age- and noise-related hearing loss. Free Rad Biol Med. 2003;34:873–880. doi: 10.1016/s0891-5849(02)01439-9. [DOI] [PubMed] [Google Scholar]
- Conforti L, Fang G, Beirowski B, Wang MS, Sorci L, Asress S, Adalbert R, Silva A, Bridge K, Huang XP, et al. NAD(+) and axon degeneration revisited: Nmnat1 cannot substitute for WldS to delay Wallerian degeneration. Cell Death Differ. 2007;14:116–127. doi: 10.1038/sj.cdd.4401944. [DOI] [PubMed] [Google Scholar]
- Conforti L, Tarlton A, Mack TG, Mi W, Buckmaster EA, Wagner D, Perry VH, Coleman MP. A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (WldS) mouse. Proc Natl Acad Sci USA. 2000;97:11377–11382. doi: 10.1073/pnas.97.21.11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Wilbrey A, Morreale G, Janeckova L, Beirowski B, Adalbert R, Mazzola F, Di Stefano M, Hartley R, Babetto E, et al. WldS protein requires Nmnat activity and a short N-terminal sequence to protect axons in mice. J Cell Biol. 2009;184:491–500. doi: 10.1083/jcb.200807175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper HM, Huang JY, Verdin E, Spelbrink JN. A new splice variant of the mouse SIRT3 gene encodes the mitochondrial precursor protein. PLoS One. 2009;4:e4986. doi: 10.1371/journal.pone.0004986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RR, Kozel P, Erway LC. Genetic influences in individual susceptibility to noise: a review. Noise & Health. 2003;5:19–28. [PubMed] [Google Scholar]
- Di Stefano M, Nascimento-Ferreira I, Orsomando G, Mori V, Gilley J, Brown R, Janeckova L, Vargas ME, Worrell LA, Loreto A, et al. A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ. 2014 doi: 10.1038/cdd.2014.164. in press. Published online October 17, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz E, Ge Y, Yang YH, Loh KC, Serafini TA, Okazaki Y, Hayashizaki Y, Speed TP, Ngai J, Scheiffele P. Molecular analysis of gene expression in the developing pontocerebellar projection system. Neuron. 2002;36:417–434. doi: 10.1016/s0896-6273(02)01016-4. [DOI] [PubMed] [Google Scholar]
- Ghanta S, Grossmann RE, Brenner C. Mitochondrial protein acetylation as a cell-intrinsic, evolutionary driver of fat storage: chemical and metabolic logic of acetyl-lysine modifications. Crit Rev Biochem Mol Biol. 2013;48:561–574. doi: 10.3109/10409238.2013.838204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J, Coleman MP. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010;8:e1000300. doi: 10.1371/journal.pbio.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L. Calorie restriction and sirtuins revisited. Genes Dev. 2013;27:2072–2085. doi: 10.1101/gad.227439.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer MS, Swinburn TK, Neitzel RL. Environmental Noise Pollution in the United States: Developing an Effective Public Health Response. Environmental health perspectives. 2013 doi: 10.1289/ehp.1307272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Newman JC, Wang MZ, Ho L, Verdin E. Mitochondrial sirtuins: regulators of protein acylation and metabolism. Trends in Endocrin Metab. 2012;23:467–476. doi: 10.1016/j.tem.2012.07.004. [DOI] [PubMed] [Google Scholar]
- Hebert AS, Dittenhafer-Reed KE, Yu W, Bailey DJ, Selen ES, Boersma MD, Carson JJ, Tonelli M, Balloon AJ, Higbee AJ, et al. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol Cell. 2013;49:186–199. doi: 10.1016/j.molcel.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskovits AZ, Guarente L. Sirtuin deacetylases in neurodegenerative diseases of aging. Cell Res. 2013;23:746–758. doi: 10.1038/cr.2013.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, Stancakova A, Goetzman E, Lam MM, Schwer B, et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell. 2011;44:177–190. doi: 10.1016/j.molcel.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager W, Goiny M, Herrera-Marschitz M, Brundin L, Fransson A, Canlon B. Noise-induced aspartate and glutamate efflux in the guinea pig cochlea and hearing loss. Exp Brain Res. 2000;134:426–434. doi: 10.1007/s002210000470. [DOI] [PubMed] [Google Scholar]
- Kaneko S, Wang J, Kaneko M, Yiu G, Hurrell JM, Chitnis T, Khoury SJ, He Z. Protecting axonal degeneration by increasing nicotinamide adenine dinucleotide levels in experimental autoimmune encephalomyelitis models. J Neurosci. 2006;26:9794–9804. doi: 10.1523/JNEUROSCI.2116-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujawa SG, Liberman MC. Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. J Neurosci. 2009;29:14077–14085. doi: 10.1523/JNEUROSCI.2845-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HW, Furman AC, Kujawa SG, Liberman MC. Primary neural degeneration in the Guinea pig cochlea after reversible noise-induced threshold shift. J Assoc Res Otol. 2011;12:605–616. doi: 10.1007/s10162-011-0277-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobe CG, Koop KE, Kreppner W, Lomeli H, Gertsenstein M, Nagy A. Z/AP, a double reporter for cre-mediated recombination. Dev Biol. 1999;208:281–292. doi: 10.1006/dbio.1999.9209. [DOI] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian Degeneration does not Hinder Regeneration in Peripheral Nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- Magnifico S, Saias L, Deleglise B, Duplus E, Kilinc D, Miquel MC, Viovy JL, Brugg B, Peyrin JM. NAD+ acts on mitochondrial SirT3 to prevent axonal caspase activation and axonal degeneration. FASEB J. 2013;27:4712–4722. doi: 10.1096/fj.13-229781. [DOI] [PubMed] [Google Scholar]
- Mizutari K, Fujioka M, Hosoya M, Bramhall N, Okano HJ, Okano H, Edge AS. Notch inhibition induces cochlear hair cell regeneration and recovery of hearing after acoustic trauma. Neuron. 2013;77:58–69. doi: 10.1016/j.neuron.2012.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlemiller KK, Wright JS, Dugan LL. Early elevation of cochlear reactive oxygen species following noise exposure. Audiology & neuro-otology. 1999;4:229–236. doi: 10.1159/000013846. [DOI] [PubMed] [Google Scholar]
- Pitkanen E. The hydrolysis of nicotinamide adenine dinucleotide phosphate by serum alkaline phosphatase. Enzyme. 1971;12:226–234. doi: 10.1159/000459535. [DOI] [PubMed] [Google Scholar]
- Puel JL, Ruel J, Gervais d’Aldin C, Pujol R. Excitotoxicity and repair of cochlear synapses after noise-trauma induced hearing loss. Neuroreport. 1998;9:2109–2114. doi: 10.1097/00001756-199806220-00037. [DOI] [PubMed] [Google Scholar]
- Puel JL, Saffiedine S, Gervais d’Aldin C, Eybalin M, Pujol R. Synaptic regeneration and functional recovery after excitotoxic injury in the guinea pig cochlea. Comptes rendus de l’Academie des sciences Serie III, Sciences de la vie. 1995;318:67–75. [PubMed] [Google Scholar]
- Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, Schilling B, Mooney SD, Kahn CR, Verdin E, et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc Natl Acad Sci USA. 2013;110:6601–6606. doi: 10.1073/pnas.1302961110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruel J, Wang J, Pujol R, Hameg A, Dib M, Puel JL. Neuroprotective effect of riluzole in acute noise-induced hearing loss. Neuroreport. 2005;16:1087–1090. doi: 10.1097/00001756-200507130-00011. [DOI] [PubMed] [Google Scholar]
- Saab AS, Tzvetanova ID, Nave KA. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr Opinion Neurobiol. 2013;23:1065–1072. doi: 10.1016/j.conb.2013.09.008. [DOI] [PubMed] [Google Scholar]
- Sajadi A, Schneider BL, Aebischer P. Wlds-mediated protection of dopaminergic fibers in an animal model of Parkinson disease. Curr Biol. 2004;14:326–330. doi: 10.1016/j.cub.2004.01.053. [DOI] [PubMed] [Google Scholar]
- Samsam M, Mi W, Wessig C, Zielasek J, Toyka KV, Coleman MP, Martini R. The Wlds mutation delays robust loss of motor and sensory axons in a genetic model for myelin-related axonopathy. J Neurosci. 2003;23:2833–2839. doi: 10.1523/JNEUROSCI.23-07-02833.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Araki T, Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci. 2006;26:8484–8491. doi: 10.1523/JNEUROSCI.2320-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Vohra BP, Lund FE, Milbrandt J. Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J Neurosci. 2009;29:5525–5535. doi: 10.1523/JNEUROSCI.5469-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spoendlin H. Retrograde degeneration of the cochlear nerve. Acta oto-laryngologica. 1975;79:266–275. doi: 10.3109/00016487509124683. [DOI] [PubMed] [Google Scholar]
- Wagner GR, Payne RM. Widespread and Enzyme-independent N{epsilon}-Acetylation and N{epsilon}-Succinylation of Proteins in the Chemical Conditions of the Mitochondrial Matrix. J Biol Chem. 2013;288:29036–29045. doi: 10.1074/jbc.M113.486753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol. 2005;170:349–355. doi: 10.1083/jcb.200504028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Hirose K, Liberman MC. Dynamics of noise-induced cellular injury and repair in the mouse cochlea. J Assoc Res Otolaryngology. 2002;3:248–268. doi: 10.1007/s101620020028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlon DS, Szakaly R, Greiner MA. Cryoembedding and sectioning of cochleas for immunocytochemistry and in situ hybridization. Brain Res Brain Res Prot. 2001;6:159–166. doi: 10.1016/s1385-299x(00)00048-9. [DOI] [PubMed] [Google Scholar]
- Willott JF. Measurement of the Auditory Brainstem Response (ABR) to Study Auditory Sensitivity in Mice. Curr Prot Neurosci. 2005;8.21B:B1–B12. doi: 10.1002/0471142301.ns0821bs34. [DOI] [PubMed] [Google Scholar]
- Yamasoba T, Nuttall AL, Harris C, Raphael Y, Miller JM. Role of glutathione in protection against noise-induced hearing loss. Brain Res. 1998;784:82–90. doi: 10.1016/s0006-8993(97)01156-6. [DOI] [PubMed] [Google Scholar]
- Yan D, Zhu Y, Walsh T, Xie D, Yuan H, Sirmaci A, Fujikawa T, Wong AC, Loh TL, Du L, et al. Mutation of the ATP-gated P2X(2) receptor leads to progressive hearing loss and increased susceptibility to noise. Proc Natl Acad Sci USA. 2013;110:2228–2233. doi: 10.1073/pnas.1222285110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Chan NY, Sauve AA. Syntheses of nicotinamide riboside and derivatives: effective agents for increasing nicotinamide adenine dinucleotide concentrations in mammalian cells. J Med Chem. 2007;50:6458–6461. doi: 10.1021/jm701001c. [DOI] [PubMed] [Google Scholar]
- Zhai RG, Zhang F, Hiesinger PR, Cao Y, Haueter CM, Bellen HJ. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature. 2008;452:887–891. doi: 10.1038/nature06721. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.