

Abstract
Epigenetics is a dynamically expanding field of science entailing numerous regulatory mechanisms controlling changes of gene expression in response to environmental factors. Over the recent years there has been a great interest in epigenetic marks as a potential diagnostic and prognostic tool or future target for treatment of various human diseases. There is an increasing body of published research to suggest that epigenetic events regulate progression of chronic liver disease. Experimental manipulation of epigenetic signatures such as DNA methylation, histone acetylation / methylation and the activities of proteins that either annotate or interpret these epigenetic marks can have profound effects on the activation and phenotype of HSC, key cells responsible for onset and progression of liver fibrosis. This review presents recent advances in epigenetic alterations, which could provide mechanistic insight into the pathogenesis of chronic liver disease and provide novel clinical applications.
Keywords: epigenetics, hepatic stellate cells, liver fibrosis, DNA methylation, microRNAs, histone modyfications
Introduction
Chronic liver disease (CLD) is one of the leading causes of mortality worldwide that is still on the rise; the term includes wide-ranging liver diseases, from steatosis, fibrosis and cirrhosis to hepatocellular cacrinoma [1]. The current major causes of CLD include viral infections (HBV, HCV), xenobiotics (alcohol, prescription drugs), metabolic disease (obesity-associated hepatic steatosis), inherited disorders (haemochromatosis, Wilson’s disease) and autoimmune hepatitis [1]. Common to all of these injuries is a pathobiology that is triggered by hepatocellular damage which, if persistent, can establish a chronic inflammatory state. The majority of individuals do not progress beyond chronic hepatitis and compensate for lost tissue mass by the highly regenerative capacity of the liver. However, in a significant minority of people (10–20%) the ongoing cell death and hepatitis stimulate the net deposition of extracellular matrix that can lead to fibrosis. If unchecked, the fibrotic process becomes progressive and self-sustaining resulting in the disturbance of normal tissue architecture and hepatic functions. End-stage liver disease is characterized by the maturation of fibrosis into cirrhosis where the profound loss of liver structure and function becomes life threatening and the risk of liver cancer dramatically increases [2]. The molecular mechanisms underlying CLD are still incompletely understood, with liver transplantation remaining the only effective treatment for the end stage of this disease.
When the liver is injured a wound healing response mounted, this includes the generation of activated myofibroblasts which promote the formation of granulation tissue, a key intermediate step in the repair process [3]. It is now accepted that transdifferentiation of HSC is the major event responsible for production of hepatic myofibroblasts [4]. In normal liver, HSC are quiescent perisinusoidal cells located within the space of Disse where they function to store retinoid and lipid droplets [5]. In response to tissue damage the quiescent HSC undergoes a dramatic reprogramming of its epigenome and transcriptome to enable its transdifferentiation to an ECM-producing myofibroblast [6]. The fate of the HSC-derived myofibroblast is then dictated by subsequent repair and injury. In the case of an acute transient injury, myofibroblasts are either cleared by apoptosis or alternatively a proportion may reverse their phenotype to a more quiescent state [4, 7]. However, if there is repeated injury to the liver, as in chronic disease, then, HSC-derived myofibroblasts persist in the tissue and via both paracrine and autocrine pathways drive the formation of mature fibrotic matrix. In addition, new evidence suggests that the persistence of HSC-derived myofibroblasts may actively repress hepatocyte regeneration via their production of TGFβ1 [8].
Cell phenotype and gene expression are governed by epigenetic mechanisms including DNA methylation, histone modifications and noncoding RNA [9, 10]. The term “epigenetics” is defined as heritable changes in gene expression without alteration in DNA sequence. These alterations change the structure of chromatin, which is a complex of DNA associated with proteins called histones [11]. The smallest unit of chromatin is the nucleosome, which consists of 147bp of DNA wrapped around a core of eight histone molecules (two copies each of H2A, H2B, H3 and H4. The transcriptional state of chromatin is influenced by covalent modifications to either DNA or histones, which regulate gene expression [12]. Due to chromatin condensation DNA is tightly packed and poorly accessible to transcription factors or chromatin-associated proteins, which leads to transcriptional silencing [13]. Conversely, gene activation requires chromatin to be in unfolded state and as a result it is accessible to polymerases involved in gene transcription [14].
The role of epigenetic mechanisms in hepatic myofibroblast transdifferentiation has been previously demonstrated in studies showing that the methyl CpG binding protein (MeCP2) facilitates myofibroblast transdifferentiation by silencing peroxisome proliferator-activated receptor gamma (PPARgamma) gene, a master regulator of adipogenic phenotype of the quiescent HSC [15]. Moreover, the MeCP2-regulated histone methyltransferase ASH1 is a key transcriptional activator that upregulates the expression of profibrogenic genes by stimulating Histone 3 Lysine 4 methylation (H3K4me) and a transcriptionally permissive state of chromatin [16].
Cirrhosis, the final outcome of liver fibrosis is highly variable between patients, with only a minority progressing to this end stage, irrespective of disease aetiology [17–19]. Factors that contribute to variation in progression of CLD include age-of-onset of disease, sex, a multitude of lifestyle-associated factors (e.g. smoking, weight, alcohol consumption, co-morbidities etc), genetic influences and environmental factors that modulate gene expression via epigenetic mechanisms [20, 9, 10, 21]. Furthermore, it is possible that population variability may be influenced by environmentally-induced factors transmitted between generations via heritable epigenetic marks [22]. However, the exact mechanism for this type of transgenerational epigenetic transmission is still to be discovered. In this review we consider recent discoveries concerning the epigenetic mechanisms that regulate the HSC phenotype and the fibrogenic process and which may vary considerably between individuals to influence disease progression.
Overview of DNA methylation
DNA methylation at position five of the cytosine ring occurs at most CpG dinucleotides in the mammalian genome. DNA methylation of the promoter region is generally associated with repressed chromatin state and inhibition of gene expression, whereas methylation within gene body may be responsible for gene activation [23]. Methylation can inhibit gene expression by two mechanisms, including transcriptional repression through the interference with the binding of transcription factors or via the recruitment of methyl-binding proteins (MeCP2, MBD1-4), which interact with chromatin silencing complexes [23]. Two classes of DNA methyltransferases (DNMTs) are responsible for introducing cytosine methylation. The de novo methyltransferases DNMT3A and DNMT3B stimulate and catalyse the addition of methyl groups at previously unmethylated CpG sites, whereas DNMT1 is responsible for the maintenance of methylation patterns onto the daughter strand during DNA replication [24]. Until recently DNA methylation was considered as a stable mark, however the discovery of TET enzymes that convert methylCpG to hydroxymethylCpG has revealed the highly dynamic nature of this epigenetic mark [25].
DNA methylation is essential for embryonic viability and is involved in key cellular processes, including transcriptional repression, X-chromosome inactivation and imprinting. Alterations in DNA methylation have been associated with a variety of human diseases including imprinting disorders and cancers [26]. Both global hypomethylation and hypermethylation have been found to be linked with cancer. Global hypomethylation is responsible for genomic instability, whereas hypermethylation of CpGs islands leads to direct silencing of tumour suppressor genes [26].
DNA methylation in liver fibrosis
DNA methylation and its associated regulatory proteins are now considered to be mechanistically important in fibrosis, in particular there is strong evidence of a pivotal orchestrating role for MeCP2 in myofibroblast activation and fibrogenesis. MeCP2 is a methyl-DNA-binding protein which due to its association with Rett syndrome has been mostly extensively examined in neurons [27]. The most compelling evidence for a role for MeCP2 in fibrosis are the reports that MeCP2-deficient mice are protected from liver [15] and lung fibrosis [28]. Moreover, MeCP2 is functionally implicated in myofibroblast differentiation in the heart [29] and eye [30]. Nevertheless, there is only a basic knowledge available as to how MeCP2 controls the myofibroblast phenotype. MeCP2 is upregulated in the earliest phases of HSC transdifferentiation via post-transcriptionally mechanisms, it then binds to methylated-CpG sites in the HSC genome and can subsequently recruit protein complexes which remodel chromatin [15]. A key target of MeCP2 in HSC is the PPARγ, gene which must be transcriptionally silenced for the HSC to lose its quiescent phenotype and undergo transdifferentiation [31]. MeCP2 can also act as an activator of gene transcription in both neurons and HSC, however the mechanisms responsible for this effect are still unclear. In neurons, MeCP2 binds to 5-hydroxymethylcytosine (5hmC), a modification of DNA methylation that may be associated with transcriptionally active genes, however it is not understood how this combination of MeCP2 and 5hmC influences chromatin structure [32]. In HSC it is proposed that, MeCP2 positively regulates expression of the histone methyltransferase, ASH1, which is responsible for attachment of methyl group to histone H3 lysine 4, a signature of actively transcribed genes. During HSC transdifferentiation, ASH1 binds to the regulatory regions of alpha smooth muscle actin (αSMA), collagen I, tissue inhibitor of metalloproteinase-1 (TIMP1) and transforming growth factor beta1 (TGFβ1) genes, depletion of this methyltransferase causes decreased expression of these profibrogenic genes indicating it is likely to be a downstream mediator of MeCP2-dependent activation of transcription [16].
Altered patterns of DNA methylation during HSC activation have been found at specific loci by studying the methylated CpGs or by treatment with DNA demethylating agent, 5-aza-2′-deoxycytidine (5-azadC) [33]. 5-azadC is incorporated into DNA during repilication and inhibits DNA methyltransferases activity, therefore genes are demethylated and reactivated. 5azadC reverses epigenetic repression of the PPARgamma gene in hepatic myofibroblast, indicating an inhibitory effect on HSC activation [33]. 5-azadC also blocks HSC proliferation by preventing loss of expression of proliferation-associated genes including Ras GTPase activating-like protein 1 (RASAL1) [34], Patched 1 (PTCH1) [35] and phosphatase and tensin homologue deleted on chromosome 10 (PTEN) [36], which are hypermethylated and consequently decreased during HSC activation. Knockdown of MeCP2 increased the expression of RASAL1 and PTCH1 in myofibroblasts, suggesting that DNA methylation and MeCP2 may provide molecular mechanism for silencing these genes and a possible pathway by which myofibroblasts are activated [35, 34]. Decreased expressions of RASAL1 also contributed to renal fibrosis progression [37], suggesting RASAL1 as a potential diagnostic marker to predict fibrosis.
Medical plant extracts may have many potential antifibrotic effects, however their mechanisms of action are still to be determined. The Chinese herbal prescription Yang-Gan-Wan, which contains rosmarinic acid and baicalin as active ingredients, prevents and reverses HSC activation by stimulating re-expression of PPARgamma [38]. Both rosmarinic acid and baicalin inhibit MeCP2 binding to the PPARgamma promoter and suppression, expression of EZH2, a histone methyltransferase that controls the repressive H3K27me3 mark which is also involved in PPARγ gene silencing [38]. Curcumin, a polyphenol isolated from yellow pigment of the spice turmeric, is able to suppress liver fibrosis through upregulation of tensin homologue deleted on chromosome 10 (PTEN) [36]. PTEN is a tumour suppressor and a negative regulator of liver fibrosis, hypermethylation of the PTEN promoter is responsible for the loss of PTEN expression during HSC activation. Curcumin treatment increased expression of miR-29b, which is involved in the hypomethylation of PTEN through downregulation of DNMT3b, this leads to suppression of HSC activation [36].
DNA methylation is an established regulator of gene transcription, and an epigenetic mark which has an important role in liver fibrosis. Given that DNA methylation is a highly dynamic modification it is likely that functionally instructive alterations in the HSC methylome occur during transdifferentiation, however at present this concept has not been experimentally investigated. Genome-wide studies of changes in DNA methylation during HSC activation are now warranted as they would bring new insights into the molecular events underpinning fibrogenesis and may provide biomarkers for disease progression as well as potential drug targets.
Non-coding RNAs (ncRNAs)
Numerous microRNAs (miRNAs) are involved in a variety of biological processes relevant to fibrosis, such as proliferation, apoptosis, TGFβ signalling and extracellular matrix deposition [39, 40]. miRNAs are small (22 nucleaotides long) non-coding RNA molecules that inhibit gene expression by targeting the 3′-untranslated region (3′-UTR) of numerous mRNAs to affect their stability and translation. A single miRNA may bind to a number of mRNAs transcripts and it is estimated that miRNAs regulate the expression of more than one-third of all human genes [41]. Dysregulation of miRNAs is often associated with human disease [42, 43] and recent studies suggest that miRNAs are functional in HSC transdifferentiation and liver fibrosis [44]. Microarray assays have revealed 12 upregulated miRNAs (miR-874, miR-29C*, miR-501, miR-349, miR-325-5p, miR-328, miR-138, miR-143, miR-207, miR-872, miR-140, and miR-193) and nine that are downregulated (miR-341, miR-20b-3p, miR-15b, miR-16, miR-375, miR-122, miR-146a, miR-92b, and miR-126), during activation of rat HSCs [45]. However, potentially there are many more miRNA involved in control of fibrosis including miR-181b [46], miR-9, miR-125b, miR-128 [47], miR-21 [48, 49], miR-33a [50], miR-221/222 [51], miR-214-5p, miR-199a [52], miR-571 and miR-652 [53] which have been show to be expressed in HSC-derived myofibroblasts. By contrast miR-122 [54], miR-133a [55], miR-146a [56], miR-29 [57] and miR-19b [58] are thought to be associated with quiescent phenotype of HSC (Table 1).
Table 1.
List of microRNAs involved in liver fibrosis.
| microRNA in liver fibrosis | ||
|---|---|---|
| Upregulated miRNA | Target in liver fibrosis | References |
| miR-181b | p27 | Wang et al. (2012) |
| miR-9 | CXCR5; CXCL6 | Noetel et al. (2013) |
| miR-128 | CXCL2; CCL5; CXCL11; CCL4 | |
| miR-125b | CXCL2; CXCL1 | |
| miR-21 | SMAD 7, Correlation with MMP-2; PTEN | Zhang et al. (2013) Wei et al. (2013) |
| miR-33a | PPAR-α; correlation with Col1A1, α-SMA | Li et al. (2014) |
| miR-221/222 | p27; PTEN; correlation with Col1a1 | Ogawa et al. (2014) |
| miR-214-5p | Correlation with MMP-2; α-SMA, TGF-β1 | Iizuka et al. (2012) |
| miR-571 | CREBBP | Roderburg et al. (2012) |
| miR-652 | Not known | |
| Downregulated miRNA | Target in liver fibrosis | References |
| miR-122 | P4HA1 | Li et al. (2013) |
| miR-133a | Correlation with Collagens (Col1a1, Col5a3) | Roderburg et al. (2012) |
| miR-146a | SMAD4 | He et al. (2013) |
| miR-29 | PDGF-C; IGF-I; Col1a1; Col4a1, Correlation with phospho-FADD; cleaved caspase-3, 8; Bax, Bcl-2, PARP, NFκB |
Kwiecinski et al. (2012) Tiao et al. (2013) |
| miR-19b | TGFβRII | Lakner et al. (2012) |
| miR-126 | VEGF-A | Guo et al. (2013) |
Transforming growth factor-beta 1 (TGFβ1) is a key driver of HSC transdifferentiation and is a regulator of microRNA expression [59]. TGFβ stimulates miR-181b, a regulator of cell proliferation via its control of p27 [46]. HSC expression of miR-33a correlates with TGFβ1-induced expression of α1 (I) collagen (Col1A1) and α-SMA, and PPAR-α is a putative target of miR-33a [50]. Treatment of HSC with TGFβ1 significantly downregulates miR-133a and miR-146a, which leads to decreased levels of type I collagen [55, 56]. It has been predicted that SMAD4 is a target of miR-146a [56]. The microRNA miR-19b is a negative regulator of TGFβ signalling by targeting TGFβ receptor II [58]. These discoveries indicate complex regulation of TGFβ1 signalling by a network of upstream and downstream miRNAs that may provide new strategies for targeting TGFβ1-driven fibrosis. However, more likely is that miRNAs will be exploited for biomarker development and in this context both miR-181b and miR-133a are elevated in the serum of cirrhosis patients compared to healthy controls [46, 55]. This finding could be helpful to design new diagnostic markers, however, first it is important to study how expression level of these miRNAs is related to the stage of liver fibrosis.
It is now emerging that there are now many different types of non-coding RNAs including the long non-coding RNAs (lncRNAs) that are predicted to contribute to disease pathologies [60]. LncRNAs are transcripts of over 200bp in length that are involved in diverse biological processes such as epigenetic modifications of DNA by recruiting chromatin remodelling complexes to specific loci, regulation of chromatin accessibility in a process that involves histone modification enzymes and RNA polymerase or X-chromosome inactivation [60, 61]. The association between disruption of lncRNAs patterns and human disease has been already reported and in the future may prove to be of importance in liver fibrosis.
Histone modifications
Amino-terminal tails of histones contain residues that are subjected to a diverse number of posttranslational modifications, including acetylation, methylation, phosphorylation and ubiquitination among others [9, 10]. Epigenetic control of gene expression at the level of chromatin modification is highly complex and in part entails attachment of the aforementioned chemical groups to histones; therefore specific enzymes controlling histone modifications may play a crucial role in transdifferentation of HSC. Histone acetylation is almost always linked with gene activation, whereas histone methylation is more modulatory with the ability to influence both the silencing and activation of genes [62]. There are many known epigenetic enzymes which carry out the attachment (histone methyltransferases) or removal (histone demethylases) of methyl groups to different lysine residues within histones [62]. The effect of histone modifications on chromatin structure depends on the specific lysine residue modified. Enrichment of histone methylation at H3K4me, H3K36me, and H3K79me is associated with active gene transcription [63]. By contrast, increase in histone modifications at H3K9me, H3K20me, H3K27me are linked with gene silencing [64, 65].
As previously mentioned in this review, one of the ways in which MeCP2 promotes liver fibrosis is by silencing of PPARγ [15]. MeCP2 binds directly to the PPARγ promoter (Figure 1) where it facilitates methylation of H3K9, this modification in turn provides binding sites for heterochromatin protein 1α (HP1α) and promotes transcriptional silencing. MeCP2 also stimulates expression of EZH2, which is recruited to the downstream coding region of PPARγ gene where it enriches the H3K27me3 mark and mediates polycomb-regulated transcriptional silencing. These two MeCP2-regulated epigenetic mechanisms therefore combine to ensure shut-down of PPARγ expression by inhibiting both the initiation of transcription at the promoter and elongation of transcription along the gene body. In line with this, 3-deazaneplanocin (dZNep), a potent inhibitor of EZH2, irreversibly blocks HSC activation [33]. However, treatment of skin fibroblasts with dZNep induced the expression of collagen and skin fibrosis [66]. These apparently paradoxical effects of dZNep on fibrogenic mechanisms may be indicative of its potential for effects on several histone methyltransferases [67] or may reflect cell-specific functions for H3K27 methylation.
Figure 1.
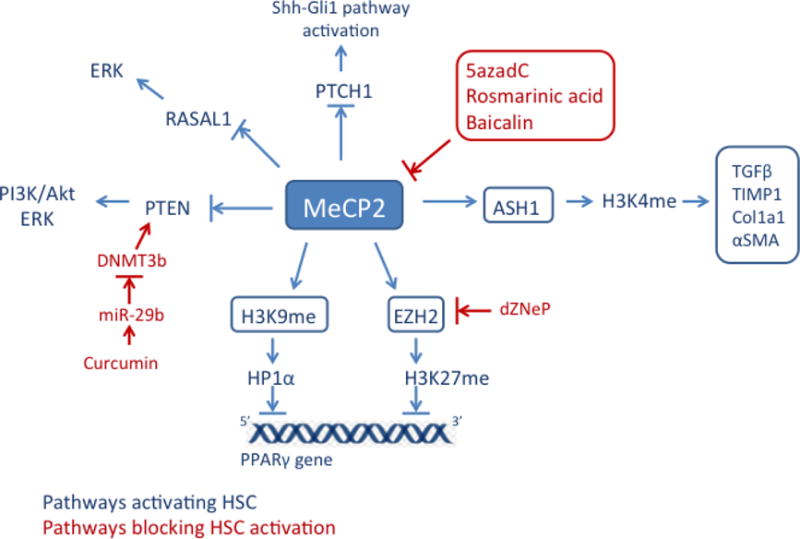
Overview of MeCP2 role in liver fibrosis.
MeCP2 upregulates expression of EZH2 resulting in silencing of the PPARgamma gene; MeCP2 also positively regulates expression of histone methyltransferase, ASH1, which is involved in activation of profibrogenic genes (TGFβ1, TIMP-1, Col1a1, αSMA) by trimethylating H3K4. In addition, MeCP2 promotes liver fibrosis by repressing expression of RASAL1, PTEN and PTCH1, which causes activation of ERK and Shh-Gli1 pathways and consequently leads to HSC activation.
Bile duct ligation (BDL), a experimental model of cholestatic liver disease, induces the hepatic expression of TGFβ1 and is linked with increased levels of active chromatin marks (H3K4me1, H3K4me2, and H3K4me3) and decreased levels of repressive marks (H3K9me2 and H3K9me3) at the TGFβ1 promoter [68]. BDL injury is also associated with increased expression of SET7/9, a H3K4 methyltransferase that is recruited to TGFβ1 promoter. SET7/9 gene knockdown significantly decreases BDL-induced TGFβ1 gene expression, serum enzymes and liver collagen content indicating a key role for this enzyme in fibrogenesis [68]. Retinoblastoma binding protein 2 (RBP2) is also an H3K4 demethylase and has been found in cirrhotic rat livers where its expression is stimulated by TGFβ. Depletion of RBP2 leads to reduced HSC proliferation and decreased levels of α-SMA and vimentin, suggesting that RBP2 may be a potential target in treatment of liver fibrosis [69].
Histone acetylation is closely associated with transcriptional activity and is controlled by two groups of enzymes histone acetyltransferases (HAT) and histone deacetylases (HDAC) [10]. HDACs are significantly upregulated in chronic liver disease and the histone deacytylase inhibitor trichostatine A suppresses HSC activation and proliferation which is associated with reduced expression of profibrogenic genes like SMA and collagen I [70, 71]. Additionally, MC1568, a compound with specificity towards class II HDACs, inhibits HSC proliferation and collagen secretion through the induction of microRNA-29, the latter having an antifibrotic function [72, 57]. HDAC inhibitors have become a centre of research interest, however their functions are not completely understood. In order to use them as therapeutics, it is necessary to further study their activities and substrates, and also to generate HDAC inhibitors with greater specificity and lower toxicity.
Conclusions
Epigenetics is a dynamically expanding field of science, not only in terms of basic biology of chromatin but also in clinical environment where scientists trying to discover changes underlying the pathobiology of their diseases of interest. There is an increasing proof that epigenetic events control HSC activation and progression of liver fibrosis. Most recent research on epigenetics presents studies on three systems including DNA methylation, microRNA and histone modifications. However, it is important to emphasize the role of interactions between these systems and take into consideration additional epigenetic influences such as transcription factors, histone remodelling complexes and other non-coding RNAs (long non-coding RNAs) as well as environmental interactions, which mould the phenotype of an individual. Therefore, in order to use epigenetics in clinical applications such as biomarkers, prognostic indicators or therapeutics, it is critical to study the crosstalk between these epigenetic elements. Enhanced knowledge of the complex epigenetic networks on cell phenotype and disease may introduce novel hypotheses relating to pathogenesis of liver fibrosis, could provide mechanistic insights into diagnosis, novel drug targets and provide a future treatment of many liver diseases.
Footnotes
Conflict of Interest
Agata Page, Derek A. Mann, and Jelena Mann declare they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
*Of importance
**Of major importance
- 1.Olaso E, Friedman SL. Molecular regulation of hepatic fibrogenesis. Journal of hepatology. 1998;29(5):836–47. doi: 10.1016/s0168-8278(98)80269-9. [DOI] [PubMed] [Google Scholar]
- 2.Fallowfield JA, Iredale JP. Targeted treatments for cirrhosis. Expert opinion on therapeutic targets. 2004;8(5):423–35. doi: 10.1517/14728222.8.5.423.. [DOI] [PubMed] [Google Scholar]
- 3.Pinzani M, Rombouts K, Colagrande S. Fibrosis in chronic liver diseases: diagnosis and management. Journal of hepatology. 2005;42(Suppl 1):S22–36. doi: 10.1016/j.jhep.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 4.Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nature communications. 2013;4:2823. doi: 10.1038/ncomms3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiological reviews. 2008;88(1):125–72. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rippe RA, Brenner DA. From quiescence to activation: Gene regulation in hepatic stellate cells. Gastroenterology. 2004;127(4):1260–2. doi: 10.1053/j.gastro.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 7.Kisseleva T, Cong M, Paik Y, Scholten D, Jiang C, Benner C, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(24):9448–53. doi: 10.1073/pnas.1201840109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ebrahimkhani MR, Oakley F, Murphy LB, Mann J, Moles A, Perugorria MJ, et al. Stimulating healthy tissue regeneration by targeting the 5-HT(2)B receptor in chronic liver disease. Nature medicine. 2011;17(12):1668–73. doi: 10.1038/nm.2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jenuwein T, Allis CD. Translating the histone code. Science (New York, NY) 2001;293(5532):1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 10.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 11.Bird A. Perceptions of epigenetics. Nature. 2007;447(7143):396–8. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 12.Gelato KA, Fischle W. Role of histone modifications in defining chromatin structure and function. Biological chemistry. 2008;389(4):353–63. doi: 10.1515/bc.2008.048. [DOI] [PubMed] [Google Scholar]
- 13.Talbert PB, Henikoff S. Spreading of silent chromatin: inaction at a distance. Nature reviews Genetics. 2006;7(10):793–803. doi: 10.1038/nrg1920. [DOI] [PubMed] [Google Scholar]
- 14.Richards EJ, Elgin SC. Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell. 2002;108(4):489–500. doi: 10.1016/s0092-8674(02)00644-x. [DOI] [PubMed] [Google Scholar]
- 15.Mann J, Chu DC, Maxwell A, Oakley F, Zhu NL, Tsukamoto H, et al. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology. 2010;138(2):705–14. 14 e1–4. doi: 10.1053/j.gastro.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perugorria MJ, Wilson CL, Zeybel M, Walsh M, Amin S, Robinson S, et al. Histone methyltransferase ASH1 orchestrates fibrogenic gene transcription during myofibroblast transdifferentiation. Hepatology (Baltimore, Md) 2012;56(3):1129–39. doi: 10.1002/hep.25754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116(6):1413–9. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 18.Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet. 1997;349(9055):825–32. doi: 10.1016/s0140-6736(96)07642-8. [DOI] [PubMed] [Google Scholar]
- 19.Teli MR, Day CP, Burt AD, Bennett MK, James OF. Determinants of progression to cirrhosis or fibrosis in pure alcoholic fatty liver. Lancet. 1995;346(8981):987–90. doi: 10.1016/s0140-6736(95)91685-7. [DOI] [PubMed] [Google Scholar]
- 20.Fischle W, Wang Y, Allis CD. Histone and chromatin cross-talk. Current opinion in cell biology. 2003;15(2):172–83. doi: 10.1016/s0955-0674(03)00013-9. [DOI] [PubMed] [Google Scholar]
- 21.Lavrov SA, Kibanov MV. Noncoding RNAs and chromatin structure. Biochemistry Biokhimiia. 2007;72(13):1422–38. doi: 10.1134/s0006297907130020. [DOI] [PubMed] [Google Scholar]
- 22**.Zeybel M, Hardy T, Wong YK, Mathers JC, Fox CR, Gackowska A, et al. Multigenerational epigenetic adaptation of the hepatic wound-healing response. Nature medicine. 2012;18(9):1369–77. doi: 10.1038/nm.2893. A new provocative research showing that development of liver fibrosis in male rats leads to epigenetic changes in the sperm, which transmit protection from liver fibrosis to their offsprings. The study suggests that population variability may be influenced by environmentally-induced factors transmitted between generations via heritable epigenetic marks. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bird A. DNA methylation patterns and epigenetic memory. Genes & development. 2002;16(1):6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 24.Cedar H, Bergman Y. Programming of DNA methylation patterns. Annual review of biochemistry. 2012;81:97–117. doi: 10.1146/annurev-biochem-052610-091920. [DOI] [PubMed] [Google Scholar]
- 25.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science (New York, NY) 2009;324(5929):930–5. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nan X, Bird A. The biological functions of the methyl-CpG-binding protein MeCP2 and its implication in Rett syndrome. Brain & development. 2001;23(Suppl 1):S32–7. doi: 10.1016/s0387-7604(01)00333-3. [DOI] [PubMed] [Google Scholar]
- 28.Hu B, Gharaee-Kermani M, Wu Z, Phan SH. Essential role of MeCP2 in the regulation of myofibroblast differentiation during pulmonary fibrosis. The American journal of pathology. 2011;178(4):1500–8. doi: 10.1016/j.ajpath.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng Y, Huang W, Wani M, Yu X, Ashraf M. Ischemic preconditioning potentiates the protective effect of stem cells through secretion of exosomes by targeting Mecp2 via miR-22. PloS one. 2014;9(2):e88685. doi: 10.1371/journal.pone.0088685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou P, Lu Y, Sun XH. Zebularine suppresses TGF-beta-induced lens epithelial cell-myofibroblast transdifferentiation by inhibiting MeCP2. Molecular vision. 2011;17:2717–23. [PMC free article] [PubMed] [Google Scholar]
- 31.Hazra S, She HY, Xiong SG, Rippe RA, Tangkjvanich P, Yee H, et al. PPAR gamma regulation of hepatic stellate cells. Hepatology (Baltimore, Md) 2001;34(4):444A-A. [Google Scholar]
- 32**.Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151(7):1417–30. doi: 10.1016/j.cell.2012.11.022. The paper presents a new potential role of MeCP2, which is a master epigenetic regulator in liver fibrosis. MeCP2 binds to 5-hydroxymethylcytosine (5hmC), a modification of DNA methylation that may be associated with transcriptionally active genes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mann J, Oakley F, Akiboye F, Elsharkawy A, Thorne AW, Mann DA. Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: implications for wound healing and fibrogenesis. Cell death and differentiation. 2007;14(2):275–85. doi: 10.1038/sj.cdd.4401979. [DOI] [PubMed] [Google Scholar]
- 34.Tao H, Huang C, Yang JJ, Ma TT, Bian EB, Zhang L, et al. MeCP2 controls the expression of RASAL1 in the hepatic fibrosis in rats. Toxicology. 2011;290(2–3):327–33. doi: 10.1016/j.tox.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 35.Yang JJ, Tao H, Huang C, Shi KH, Ma TT, Bian EB, et al. DNA methylation and MeCP2 regulation of PTCH1 expression during rats hepatic fibrosis. Cellular signalling. 2013;25(5):1202–11. doi: 10.1016/j.cellsig.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 36.Zheng J, Wu C, Lin Z, Guo Y, Shi L, Dong P, et al. Curcumin up-regulates phosphatase and tensin homologue deleted on chromosome 10 through microRNA-mediated control of DNA methylation–a novel mechanism suppressing liver fibrosis. The FEBS journal. 2014;281(1):88–103. doi: 10.1111/febs.12574. [DOI] [PubMed] [Google Scholar]
- 37.Bechtel W, McGoohan S, Zeisberg EM, Muller GA, Kalbacher H, Salant DJ, et al. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nature medicine. 2010;16(5):544–50. doi: 10.1038/nm.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang MD, Chiang YM, Higashiyama R, Asahina K, Mann DA, Mann J, et al. Rosmarinic acid and baicalin epigenetically derepress peroxisomal proliferator-activated receptor gamma in hepatic stellate cells for their antifibrotic effect. Hepatology (Baltimore, Md) 2012;55(4):1271–81. doi: 10.1002/hep.24792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen SL, Zheng MH, Shi KQ, Yang T, Chen YP. A new strategy for treatment of liver fibrosis: letting MicroRNAs do the job. BioDrugs: clinical immunotherapeutics, biopharmaceuticals and gene therapy. 2013;27(1):25–34. doi: 10.1007/s40259-012-0005-2. [DOI] [PubMed] [Google Scholar]
- 40.Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350–5. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 41.Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, et al. Combinatorial microRNA target predictions. Nature genetics. 2005;37(5):495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 42.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nature reviews Genetics. 2004;5(7):522–31. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 43.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nature reviews Cancer. 2006;6(11):857–66. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 44.Venugopal SK, Jiang J, Kim TH, Li Y, Wang SS, Torok NJ, et al. Liver fibrosis causes downregulation of miRNA-150 and miRNA-194 in hepatic stellate cells, and their overexpression causes decreased stellate cell activation. American journal of physiology Gastrointestinal and liver physiology. 2010;298(1):G101–6. doi: 10.1152/ajpgi.00220.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo CJ, Pan Q, Cheng T, Jiang B, Chen GY, Li DG. Changes in microRNAs associated with hepatic stellate cell activation status identify signaling pathways. The FEBS journal. 2009;276(18):5163–76. doi: 10.1111/j.1742-4658.2009.07213.x. [DOI] [PubMed] [Google Scholar]
- 46.Wang B, Li W, Guo K, Xiao Y, Wang Y, Fan J. miR-181b promotes hepatic stellate cells proliferation by targeting p27 and is elevated in the serum of cirrhosis patients. Biochemical and biophysical research communications. 2012;421(1):4–8. doi: 10.1016/j.bbrc.2012.03.025. [DOI] [PubMed] [Google Scholar]
- 47.Noetel A, Elfimova N, Altmuller J, Becker C, Becker D, Lahr W, et al. Next generation sequencing of the Ago2 interacting transcriptome identified chemokine family members as novel targets of neuronal microRNAs in hepatic stellate cells. Journal of hepatology. 2013;58(2):335–41. doi: 10.1016/j.jhep.2012.09.024. [DOI] [PubMed] [Google Scholar]
- 48.Wei J, Feng L, Li Z, Xu G, Fan X. MicroRNA-21 activates hepatic stellate cells via PTEN/Akt signaling. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2013;67(5):387–92. doi: 10.1016/j.biopha.2013.03.014. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Z, Gao Z, Hu W, Yin S, Wang C, Zang Y, et al. 3,3′-Diindolylmethane ameliorates experimental hepatic fibrosis via inhibiting miR-21 expression. British journal of pharmacology. 2013;170(3):649–60. doi: 10.1111/bph.12323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li ZJ, Ou-Yang PH, Han XP. Profibrotic effect of miR-33a with Akt activation in hepatic stellate cells. Cellular signalling. 2014;26(1):141–8. doi: 10.1016/j.cellsig.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 51.Ogawa T, Enomoto M, Fujii H, Sekiya Y, Yoshizato K, Ikeda K, et al. MicroRNA-221/222 upregulation indicates the activation of stellate cells and the progression of liver fibrosis. Gut. 2012;61(11):1600–9. doi: 10.1136/gutjnl-2011-300717. [DOI] [PubMed] [Google Scholar]
- 52.Iizuka M, Ogawa T, Enomoto M, Motoyama H, Yoshizato K, Ikeda K, et al. Induction of microRNA-214-5p in human and rodent liver fibrosis. Fibrogenesis & tissue repair. 2012;5(1):12. doi: 10.1186/1755-1536-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roderburg C, Mollnow T, Bongaerts B, Elfimova N, Vargas Cardenas D, Berger K, et al. Micro-RNA profiling in human serum reveals compartment-specific roles of miR-571 and miR-652 in liver cirrhosis. PloS one. 2012;7(3):e32999. doi: 10.1371/journal.pone.0032999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li J, Ghazwani M, Zhang Y, Lu J, Li J, Fan J, et al. miR-122 regulates collagen production via targeting hepatic stellate cells and suppressing P4HA1 expression. Journal of hepatology. 2013;58(3):522–8. doi: 10.1016/j.jhep.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roderburg C, Luedde M, Vargas Cardenas D, Vucur M, Mollnow T, Zimmermann HW, et al. miR-133a mediates TGF-beta-dependent derepression of collagen synthesis in hepatic stellate cells during liver fibrosis. Journal of hepatology. 2013;58(4):736–42. doi: 10.1016/j.jhep.2012.11.022. [DOI] [PubMed] [Google Scholar]
- 56.He Y, Huang C, Sun X, Long XR, Lv XW, Li J. MicroRNA-146a modulates TGF-beta1-induced hepatic stellate cell proliferation by targeting SMAD4. Cellular signalling. 2012;24(10):1923–30. doi: 10.1016/j.cellsig.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 57.Kwiecinski M, Elfimova N, Noetel A, Tox U, Steffen HM, Hacker U, et al. Expression of platelet-derived growth factor-C and insulin-like growth factor I in hepatic stellate cells is inhibited by miR-29. Laboratory investigation; a journal of technical methods and pathology. 2012;92(7):978–87. doi: 10.1038/labinvest.2012.70. [DOI] [PubMed] [Google Scholar]
- 58.Lakner AM, Steuerwald NM, Walling TL, Ghosh S, Li T, McKillop IH, et al. Inhibitory effects of microRNA 19b in hepatic stellate cell-mediated fibrogenesis. Hepatology (Baltimore, Md) 2012;56(1):300–10. doi: 10.1002/hep.25613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheng K, Yang N, Mahato RI. TGF-beta1 gene silencing for treating liver fibrosis. Molecular pharmaceutics. 2009;6(3):772–9. doi: 10.1021/mp9000469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nature reviews Genetics. 2009;10(3):155–9. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 61.Esteller M. Non-coding RNAs in human disease. Nature reviews Genetics. 2011;12(12):861–74. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 62.Santos-Rosa H, Caldas C. Chromatin modifier enzymes, the histone code and cancer. European journal of cancer (Oxford, England: 1990) 2005;41(16):2381–402. doi: 10.1016/j.ejca.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 63.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 64.Koch CM, Andrews RM, Flicek P, Dillon SC, Karaoz U, Clelland GK, et al. The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome research. 2007;17(6):691–707. doi: 10.1101/gr.5704207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nature reviews Molecular cell biology. 2007;8(12):983–94. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kramer M, Dees C, Huang J, Schlottmann I, Palumbo-Zerr K, Zerr P, et al. Inhibition of H3K27 histone trimethylation activates fibroblasts and induces fibrosis. Annals of the rheumatic diseases. 2013;72(4):614–20. doi: 10.1136/annrheumdis-2012-201615. [DOI] [PubMed] [Google Scholar]
- 67.Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, et al. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Molecular Cancer Therapeutics. 2009;8(6):1579–88. doi: 10.1158/1535-7163.mct-09-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sheen-Chen SM, Lin CR, Chen KH, Yang CH, Lee CT, Huang HW, et al. Epigenetic histone methylation regulates transforming growth factor beta-1 expression following bile duct ligation in rats. Journal of gastroenterology. 2013 doi: 10.1007/s00535-013-0892-0. [DOI] [PubMed] [Google Scholar]
- 69.Wang Q, Wang LX, Zeng JP, Liu XJ, Liang XM, Zhou YB. Histone demethylase retinoblastoma binding protein 2 regulates the expression of alpha-smooth muscle actin and vimentin in cirrhotic livers. Brazilian journal of medical and biological research = Revista brasileira de pesquisas medicas e biologicas / Sociedade Brasileira de Biofisica [et al] 2013;46(9):739–45. doi: 10.1590/1414-431x20132843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Niki T, Rombouts K, De Bleser P, De Smet K, Rogiers V, Schuppan D, et al. A histone deacetylase inhibitor, trichostatin A, suppresses myofibroblastic differentiation of rat hepatic stellate cells in primary culture. Hepatology (Baltimore, Md) 1999;29(3):858–67. doi: 10.1002/hep.510290328. [DOI] [PubMed] [Google Scholar]
- 71.Rombouts K, Niki T, Wielant A, Hellemans K, Geerts A. Trichostatin A, lead compound for development of antifibrogenic drugs. Acta gastro-enterologica Belgica. 2001;64(3):239–46. [PubMed] [Google Scholar]
- 72.Mannaerts I, Eysackers N, Onyema OO, Van Beneden K, Valente S, Mai A, et al. Class II HDAC inhibition hampers hepatic stellate cell activation by induction of microRNA-29. PloS one. 2013;8(1):e55786. doi: 10.1371/journal.pone.0055786. [DOI] [PMC free article] [PubMed] [Google Scholar]