

Abstract
Non-alcoholic fatty liver disease can result in changes to drug metabolism and disposition potentiating adverse drug reactions. Furthermore, arsenite exposure during development compounds the severity of diet-induced fatty liver disease. This study examines the effects of arsenite potentiated diet-induced fatty liver disease on hepatic transport in male mice. Changes were detected for Mrp2/3/4 hepatic transporter gene expression as well as for Oatp1a4/2b1/1b2. Plasma concentrations of Mrp and Oatp substrates were increased in arsenic exposure groups compared to diet-only controls. In addition, murine embryonic hepatocytes and adult primary hepatocytes show significantly altered transporter expression after exposure to arsenite alone: a previously unreported phenomenon. These data indicate that developmental exposure to arsenite leads to changes in hepatic transport which could increase the risk for adverse drug reactions during fatty liver disease.
Keywords: Fatty liver disease, arsenic, development, transport
Introduction
Non-alcoholic fatty liver disease (NAFLD) is the most prevalent chronic liver disease which ranges from steatosis to more advanced non-alcoholic steatohepatitis (NASH) which can progress to cirrhosis and hepatocellular carcinoma (1). The generally accepted prevalence of NAFLD in adults in the United States is approximately 30% (2). Previous studies have linked exposures to Arsenite at part per million (ppm) concentrations to the development of NAFLD (3–6); however, these exposure scenarios are not necessarily representative of more frequent levels of arsenite present in water (7). Our laboratory recently demonstrated that exposure to 100 part per billion (ppb) arsenite during development, in conjunction with a Western style diet, results in significantly worsened NAFLD and NASH in male Swiss Webster mice (8).
It has been demonstrated that NAFLD results in altered xenobiotic metabolism and disposition through a variety of different mechanisms. Rats and mice fed a methionine-choline-deficient (MCD) diet develop NASH, and it has been shown that these rodent models are similar to human NASH when examining multidrug resistance-associated protein (Mrp) and organic anion-transporting polypeptide (Oatp) transporters in the liver (9). In a rat model of MCD NASH, altered disposition of metabolites of acetaminophen occurred, likely as a result of detected increases in mRNA and protein of Mrp transporters involved in both biliary and sinusoidal efflux (10). Similarly, loss of membrane localization of the biliary efflux transporter Mrp2 occurs in NASH, which decreases the ability to eliminate xenobiotics and associated metabolites into the bile (11).
Alterations in xenobiotic disposition and metabolism contribute to increased rates of adverse drug reactions (ADRs) in populations suffering from NAFLD. In rats with NASH, there is increased susceptibility to hepatic and renal toxicity with administration of methotrexate (12). The large prevalence of NAFLD in the human population in conjunction with the role that arsenic exposure may play in predisposition to the disease later in life, suggests that arsenic-induced NAFLD may also contribute to ADRs as a result of altered disposition of xenobiotics and their metabolites. NASH also results in altered disposition of arsenic metabolites including increased liver accumulation of monomethylarsonous acid (13). The accumulation of this toxic metabolite in the liver in NAFLD likely plays a part in protracted liver damage. Lastly, to our knowledge, the direct effects of arsenic on transporter expression have not been examined.
The present study defines the effects of As (III) potentiated diet-induced NAFLD on transporter gene expression and the disposition of probable hepatic transporter substrates in male mice with developmental exposure to arsenite. In addition, cultured murine embryonic hepatocytes and adult primary hepatocytes are examined after exposure to arsenite in both the presence and absence of Intralipid (a soybean oil based 20% fat emulsion that contains free fatty acids [FFA]) to determine transporter expression independent of FFA induced triglyceride (TAG) accumulation. Intralipid, when added to culture media, induces steatosis in primary hepatocytes (14). The objectives of this study are to determine how As (III) potentiated diet-induced NAFLD affects hepatic transport and to gain insight into the relative contributions of increased hepatic FFAs and As (III) exposure to alterations in transporter gene expression in embryonic and adult hepatocytes.
Materials and Methods
Animals and Treatment
Animal tissue, plasma, and metabolomic information used in this study were derived from an initial investigation establishing effects of developmental As (III) exposure on susceptibility to diet-induced NAFLD in male Swiss Webster mice. (8). The most severe NAFLD was present in mice that were exposed to 100 ppb As (III) in the drinking water starting at embryonic day 5 through birth (IU) or through the end of the study (IU+13W). Sacrifice was at 13 weeks of age. Water and food were provided ad libitum (for all animals) and dams were provided standard chow (2019 Teklad Global 19% Protein Extruded Rodent Diet, Harlan Laboratories Inc) through weaning. All pups were provided Western diet (TestDiet) ad libitum after weaning and water was provided ad libitum with either 100 ppb As (III) as sodium arsenite (NaAsO2, Sigma) or 100 ppb sodium chloride in control animals (NaCl, VWR). The Arizona Laboratory for Emerging Contaminants (ALEC) verified stock arsenite concentrations and speciation by inductively coupled plasma mass spectrometry: ALEC also detected no arsenic species in the diet. Water was replaced and spiked with As (III) from frozen (−20°C) stocks weekly. All animal use and experimental protocols followed University of Arizona Institutional Animal Care and Use Committee (IACUC) regulations and remained in accordance with institutional guidelines ensuring that animals were treated humanely and with regard for alleviation of suffering. The methods for performing metabolomic analysis are provided in detail in the previous manuscript (8).
Reverse Transcription Real-Time Polymerase Chain Reaction (RT-qPCR)
TRIzol® was used to isolate RNA from flash frozen tissue samples, or cultured cells, according to the manufacturer’s protocol (Life Technologies). 1 μg RNA was used to generate first strand cDNA using the Transcriptor First Strand cDNA Synthesis kit (Roche). Real-time Polymerase Chain Reaction (qPCR) was performed using the TaqMan Master Primer-Probe System (Roche). 40S Ribosomal Protein 7 (Rps7) was used as a housekeeping gene for relative quantification of target mRNA. The genes of interest, the primer sequences, and the corresponding fluorescein probes from Roche’s Universal ProbeLibrary are listed in Table 1.
Table 1.
Primers and Probes for RT-qPCR.a
| Gene | Left (5′-3′) | Right (5′-3′) | Probe | NCBI RefSeq |
|---|---|---|---|---|
| Mrp2 | CAAATCCAATTCTCTACCTATGCAC | GCCTGCAGTGTTGGATCA | 92 | NM_013806.2 |
| Mrp3 | CTTCATGGTGGTTGTCTTGC | ACATAGAAGCGCTGCACAAA | 76 | NM_029600.3 |
| Mrp4 | CCACATGATTTACCGGAAGG | AGGTTAACTATCTGGCCTGTGG | 60 | NM_001033336.3 |
| Abcg2 | GCCTTGGAGTACTTTGCATCA | AAATCCGCAGGGTTGTTGTA | 100 | NM_011920.3 |
| Rps7 | AGCACGTGGTCTTCATTGCT | CTGTCAGGGTACGGCTTCTG | 101 | NM_011300.3 |
| Oatp1a1 | GGAAAACCTTGGAATCACTAAAGA | GGAGCACACTCGTAAGACTGAA | 84 | NM_013797.5 |
| Oatp1a4 | GGTGGCGATAGAAACTAACCA | ATAGGAGGGACTTGCATTGG | 22 | NM_030687.1 |
| Oatp1b2 | CCCGTGACTAATCCAACAACA | GCTTCTCAGAGACCATAGAAAACC | 51 | NM_020495.1 |
| Oatp2b1 | GCCACATCGCTTCCAGTTAT | CAGGATGCCAGGGTAGATTAAC | 41 | NM_175316.3 |
The examined genes are: ATP-Binding Cassette Sub-Family C Member 2/3/4 (Mrp2/3/4), ATP-Binding Cassette Sub-Family G Member 2 (Abcg2), 40S ribosomal protein S7 (Rps7), and Solute Carrier Organic Anion Transporter Family Member 1a1/1a4/1b2/2b1 (Oatp1a1/1a4/1b2/2b1).
Protein Isolation, Immunoblotting, and Densitometry
Total protein was isolated from the liver as previously described (15). Whole-cell lysate was combined with Laemmli Sample Buffer (Bio-Rad), separated by SDS-PAGE utilizing 7.5% polyacrylamide gels, and transferred to polyvinylidene difluoride membrane. The membrane was then blocked against non-specific binding with 5% nonfat dry milk (NFDM) in 1x PBS and 0.1% Tween 20. Primary antibodies Mrp3 (sc-5775; Santa Cruz Biotechnology [SCBT]), Mrp4 (ab15602; Abcam), Oatp1a1 (sc-47265; SCBT), Oatp1a4 (sc-18436; SCBT), and Oatp1b2 (sc-47270; SCBT) were diluted in 1% NFDM at 1:1000. Relative protein concentration was calculated utilizing ImageJ (NIH) with Erk2 (sc-154: 1:10000; SCBT) serving as a loading control and housekeeping protein.
Immunohistochemistry
Formalin fixed paraffin embedded serial liver sections were prepared at 7 μm and deparaffinized in xylene followed by rehydration in a series of ethanol solutions to distilled water. Following overnight antigen retrieval at 60 °C in Tris-EDTA Buffer (10mM Tris Base, 1mM EDTA Solution, 0.05% Tween 20, pH 9.0) an endogenous peroxidase block was performed with 3% H2O2 in PBS for 20 min. The sections were blocked in 5% serum (of the secondary antibody species) in PBS for 30 minutes. The sections were probed for Mrp3 (sc-5775, Santa Cruz Biotechnology) and Abcg2 (BXP-53, Kamiya Biomedical Company) diluted in 5% serum in PBS (donkey for Mrp3 and goat for Abcg2) for 2 hours at room temperature (RT). Sections were then probed with biotinylated secondary antibody for 30 min at RT (sc-2042 [Mrp3], sc-2041 [Abcg2]; Santa Cruz Biotechnology] in 5% serum in PBS. Sections were then incubated with Avidin D-HRP complex (sc-2053, Santa Cruz Biotechnology) for 30 minutes at RT. Sections were then developed with 3,3′-Diaminobenzidine (DAB) (D-5905, Sigma) and counterstained with hematoxylin (Thermo Scientific). 10 μm sections were stained with hematoxylin and eosin (H&E) (Thermo Scientific) to demonstrate previously established NAFLD (8). Images were captured using a bright field microscope.
Embryonic Liver Culture and Primary Hepatocytes for Gene Expression and Triglyceride Accumulation
Pregnant Swiss Webster mice (Harlan Laboratories Inc) were euthanized via CO2 euthanasia followed by cervical dislocation and tissues were surgically isolated from several E12.5 embryos. Observation of the vaginal plug was considered E0.5 and developmental stage was verified by somite number. The liver bud was surgically removed and rinsed in sterile phosphate buffered saline (PBS) multiple times prior to isolation of cells. Embryonic liver cultures were prepared in DMEM (Dulbecco’s modified Eagle’s medium) with serial passage through 18 and 25 gauge syringes: single cell suspensions were prepared and cells were allowed to adhere. All experiments were performed with cells at passage 5 or below. During passaging, cells were cultured in DMEM with 20% fetal bovine serum (FBS), antibiotics, and insulin–transferrin–selenium (Invitrogen, Carlsbad, CA). Primary hepatocytes were isolated as previously described (16) utilizing 2 male C57BL/6 mice.
During experiments, cells were cultured in DMEM with 10% FBS and antibiotics plus the relevant treatment. Cultures were incubated at 37°C with 5% CO2 and 95% humidity. Cells were treated with 100 ppb arsenite as sodium arsenite (NaAsO2, Sigma), 6% (by volume) Intralipid (Sigma), or both for 48 hr. There was no observed cytotoxicity for arsenite after 48 hr below 750 ppb via MTT assay (Vybrant MTT, Life Technologies) in embryonic liver cells (data not shown) or in primary hepatocytes as previously reported (17). 6% Intralipid causes marked steatosis in primary hepatocytes with no observed change in cell viability (14).
For TAG accumulation experiments, cells were freeze/thawed at −20 °C in PBS, scraped, sonicated, and extracted with 2:1 volume ratio of Chloroform and Methanol. The lipid fraction was resuspended with 1% Triton X-100 in 100% Ethanol. TAG was quantified using the TAG reagent set (Pointe Scientific) and normalized to total protein content as determined by BCA assay (Life Technologies).
Results
As (III) Potentiated Diet-induced NAFLD and Hepatic Transporter Gene Expression
Several hepatic transporters were examined to determine if As (III) potentiated diet-induced NAFLD affected a change in their gene expression (Figure 1). Increased expression of Mrp2, Mrp3, Mrp4, Oatp1a4, and Oatp2b1 was detected in the IU+13W animals with NASH compared to the control group (diet-induced steatosis). These detected changes were significant and no differences were detected for Oatp1a1. In the IU+13W and IU (borderline NASH) groups there were significant decreases in the expression of Oatp1b2 compared to control animals. No change in expression of Abcg2 was detected in any of the experimental groups (data not shown). Collectively, these data reveal changes in the expression of a number of hepatic transporters during As (III) potentiated diet-induced NAFLD with the most robust changes coinciding with severe NAFLD pathology: NASH.
Figure 1. As (III) Potentiated Diet-induced NAFLD Disrupts Hepatic Transporter mRNA Expression.
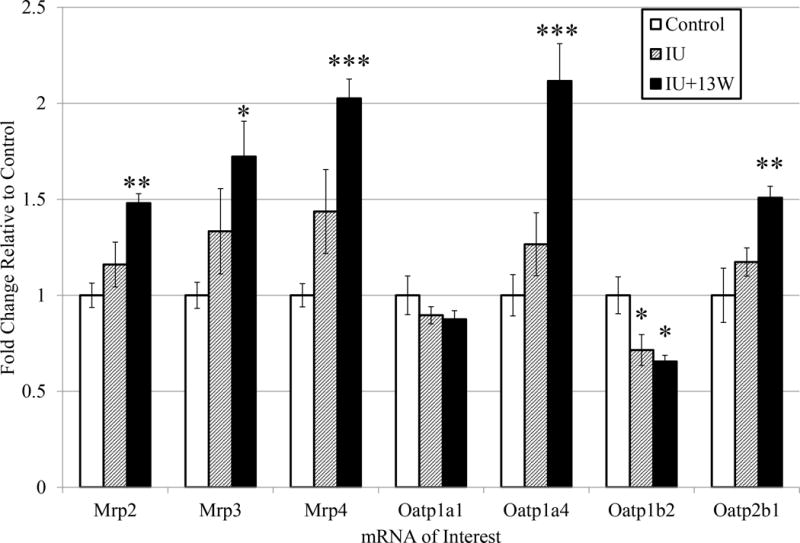
Transcript levels of select Mrp and Oatp transporter genes are shown as fold change compared to controls with error bars representing standard error. Analytical duplicates were used and a one-way ANOVA followed by a Dunnett’s multiple comparison test utilizing Prism6 (GraphPad) for comparisons between treatment groups and controls: the reported p-value is the multiplicity adjusted p-value corrected for multiple comparisons. (*** = p ≤ 0.001; ** = p ≤ 0.01 ; * = p ≤ 0.05 compared to controls). n=5.
Hepatic Transporter Protein Levels
Following the observed changes in mRNA levels, protein levels of the examined hepatic transporters were also determined. For the proteins where accurate detection was achieved, there was no significant alteration in protein level in any examined transporter (Figure 2). There was a modest increase in detection of Mrp3 in the IU animals compared to controls, but it did not reach statistical significance (p = 0.0615, n=4). This is consistent with the increased detection of mRNA for Mrp3 (Figure 1). In many other studies examining gene expression and protein quantity of hepatic transporters, there is not always a significant change detected at both the mRNA and protein level: in some cases, there is even robust change in one, but no change detected in the other.
Figure 2. Hepatic Transporter Protein Levels.
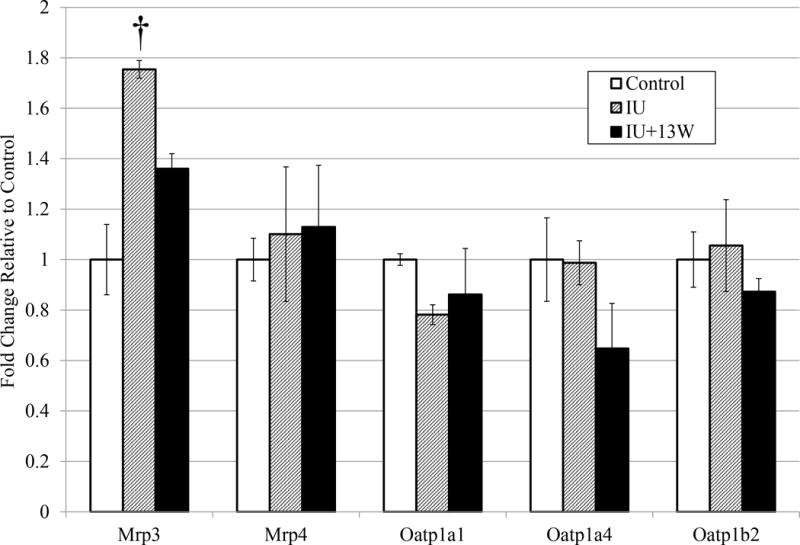
Mrp3, Mrp4, Oatp1a1, Oatp1a4, and Oatp1b2 protein levels were examined relative to Erk2 in liver lysate. Results representative from at least 2 analytical duplicates were subjected to densitometry analyses. The detected protein levels are shown as fold change compared to controls with error bars representing standard error. A one-way ANOVA followed by a Dunnett’s multiple comparison test utilizing Prism6 (GraphPad) for comparisons between treatment groups and controls was performed on representative blots from analytical replicates. (p ≤ 0.1 = † compared to controls). n=4.
Localization of Mrp3 and Abcg2 with As (III) Potentiated Diet-induced NAFLD
Despite the lack of statistically significant changes at the protein level, transporter localization could be disrupted and contribute to changes in substrate distribution. For example, Mrp2 can withdraw from the canalicular membrane during conditions of stress such as NASH (Hardwick et al. 2011). Therefore, immunohistochemistry (IHC) was utilized to determine if Mrp2 localization was altered in As (III) potentiated NAFLD, but detection proved unsuccessful with commercially available antibodies. In Figure 3 there does not appear to be any change in localization of Mrp3 along the sinusoidal membrane or for Abcg2 detection along the canalicular membrane in any of the treatment groups compared to controls. This is consistent with previous findings and is indicative of transporter specific effects (Hardwick et al. 2011). H&E staining of representative sections shows simple steatosis in the control animals, borderline NASH in the IU group, and definitive NASH present in the IU+ group as previously reported (Ditzel et al. 2015).
Figure 3. Localization of Mrp3 and Abcg2 with As (III) Potentiated Diet-induced NAFLD.
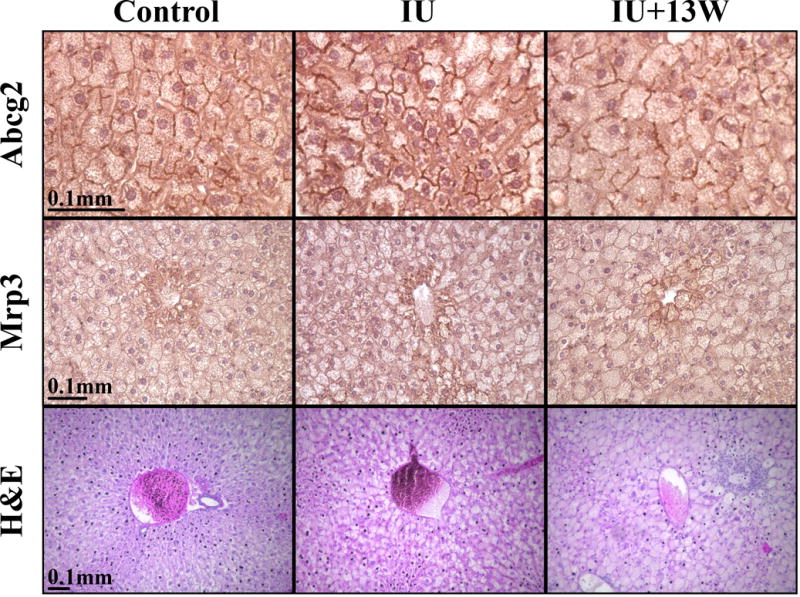
Liver sections were probed with Abcg2 and Mrp3. Mislocalization of Abcg2 to the canalicular membrane and Mrp3 to the sinusoidal membrane has not been reported in NALFD models and is not observed. H&E stained sections demonstrate the presence of NAFLD in IU and IU+13W treatment groups.
Metabolomic Analysis of Mrp and Oatp Substrates in Plasma
We speculated that disruption in Mrp and Oatp expression will coincide with changes in their substrates such as metabolites conjugated to glutathione, sulfate, or glucoronate and other known substrates (18). Therefore, we performed metabolomic analysis to detect substrates in blood plasma (Table 2). Two sulfated metabolites (possible Mrp substrates), catechol sulfate and 3-[3-(sulfooxy)phenyl]propanoic acid, were detected to be significantly increased in plasma of IU+13W mice. There was a slight increase in catechol sulfate detected in plasma of IU mice, but it did not reach statistical significance (p = 0.0587; n= 8 for IU and IU+13W and n=7 for controls). Finally, there was a modest increase in unconjugated bilirubin, an Oatp substrate, in plasma of IU+13W mice, but it did not reach statistical significance (p = 0.0767). These data provide further evidence that functional hepatic transport is altered during As (III) potentiated diet-induced NAFLD. However, it is important not to overextend the interpretation of this metabolomic data: differences in sulfotransferase activity and gene expression have been observed in NASH and the substrate specificity of the two proposed sulfated metabolites for Mrp’s was not determined (19).
Table 2.
Metabolomic Analysis of Mrp and Oatp Substrates.a
| Fold Change Relative to Control | Mean ± SD Relative to a Standard | ||||
|---|---|---|---|---|---|
| Metabolite | IU | IU+13W | Control | IU | IU+13W |
| catechol sulfate | 1.77† | 8.54* | 0.54 ± 0.23 | 0.95 ± 0.51 | 4.58 ± 2.38 |
| 3-[3-(sulfooxy)phenyl]propanoic acid | 1.64 | 9.11* | 0.45 ± 0.14 | 0.74 ± 0.61 | 4.13 ± 2.50 |
| bilirubin (E,E) | 1.01 | 1.82† | 0.52 ± 0.35 | 0.52 ± 0.25 | 0.94 ± 0.54 |
Plasma samples were subjected to metabolomic analyses. Mrp substrates were detected to be significantly increased in in utero arsenic exposure groups compared to controls. (IU and IU+13W n=8; Control n=7: * = p ≤ 0.05; p ≤ 0.1 = † compared to controls)
Effects of Intralipid and As (III) on Lipid Accumulation and Transporter Gene Expression in Embryonic Liver Culture and Primary Hepatocytes
TAG accumulation was determined in embryonic day 12.5 hepatocytes and adult primary hepatocytes that had been exposed to 6% Intralipid (to replicate increased FFA levels), 100 ppb As (III), or a combination of both. In embryonic cultures, there was no change in TAG accumulation detected with any of the treatments compared to controls (Figure S1A). Primary hepatocytes were examined under the same conditions and increased TAG was detected in the Intralipid and As (III) with Intralipid treated groups, but no significant difference between the two was detected (Figure S1B).
Transporter expression was also evaluated in both embryonic and adult liver culture under the indicated conditions to determine if there was a differential effect in the presences of As (III) compared to Intralipid alone. Expression of Oatp1a1 and Oatp1a4 was not detected in E12.5 hepatocytes, which is consistent with previous examinations of expression of hepatic transporters during development (20). There was no statistical difference between any of the treatment groups for expression of Mrp2 or Oatp1b2 (data not shown). In addition, expression of Mrp2 was low or undetectable within all groups and is consistent with previous findings that show expression to be markedly lower during development with a 75% increase in expression occurring two days before parturition (21). In contrast, significant changes were detected for Mrp3, Mrp4, and Oatp2b1 between various treatment groups (Figure 4). Mrp3 expression was significantly increased in both Intralipid and As (III) with Intralipid treated groups compared to control and As (III) alone; however, there is no significant difference between the Intralipid and As (III) with Intralipid groups (Figure 4A). When examining Mrp4 expression, there appears to be a significant additive increase in expression when Intralipid and As (III) are combined compared to either Intralipid or As (III) alone. There is a significant increase in expression of Mrp4 in the As (III) treatment group compared to controls, but no such increase was observed with Intralipid alone (Figure 4B). Finally, a robust and significant increase in expression in Oatp2b1 is detected in the Intralipid and As (III) co-treated group constituting a combinational effect, and no other treatment groups were significantly different than control (Figure 4C).
Figure 4. Effects of Intralipid and As (III) on Transporter Gene Expression in Embryonic Liver Culture.
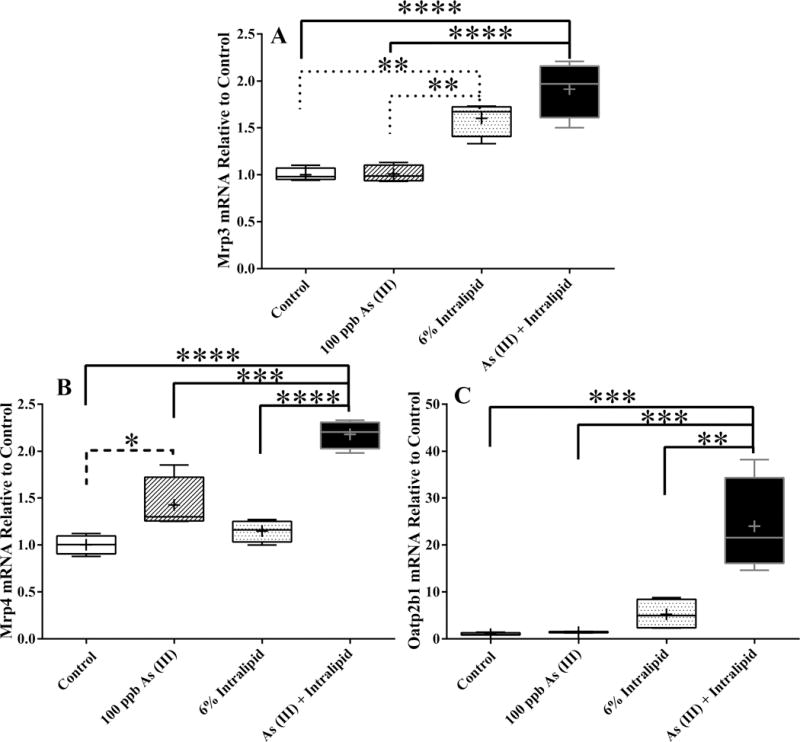
mRNA levels of select Mrp and Oatp transporter genes found to be statistically different than control with described treatments are shown as fold change compared to control. The box extends from 25th to 75th percentile and whiskers show the entire distribution: the horizontal line is the median and the + is the mean. Analytical duplicates were used and a one-way ANOVA followed by a Tukey’s multiple comparison test utilizing Prism6 (GraphPad) for comparisons between groups: the reported p-value is the multiplicity adjusted p-value corrected for multiple comparisons. (**** = p ≤ 0.0001; *** = p ≤ 0.001; ** = p ≤ 0.01 ; * = p ≤ 0.05 compared to controls). n=4.
The expression of the select hepatic transporters was also examined in adult primary hepatocytes subjected to the same treatments as the embryonic hepatocytes. Expression of Mrp2 was increased with As (III) exposure, decreased with Intralipid, and not statistically different from the combination of the two compared to controls (Figure 5A). The expression of Mrp3 was decreased with Intralipid alone and Intralipid and As (III) combined (Figure 5B). As (III) alone and As (III) with Intralipid caused increased expression of Mrp4 compared to controls (Figure 5C). Compared to controls, there was no statistically significant change in the expression of Oatp1a1 with any of the treatments, but there was a significant difference detected between the Intralipid and Intralipid with As (III) combination group (Figure 5D). The expression of Oatp1b2 was decreased with As (III) alone compared to controls (Figure 5E). Finally, the expression of Oatp2b1 was increased with Intralipid alone and Intralipid with As (III) compared to controls (Figure 5F). No significant difference in expression of Oatp1a4 was detected in any of the culture conditions (data not shown). These data show that in cultured hepatocytes (adult and embryonic) transporter expression is altered by exposure to As (III) alone, independent of TAG accumulation. It is important to note that the hepatocytes examined in these two groups of studies were derived from different strains of mice, so differences in their response may not be solely due to the comparison between adult and embryonic liver.
Figure 5. Effects of Intralipid and As (III) on Transporter Gene Expression in Primary Hepatocytes.
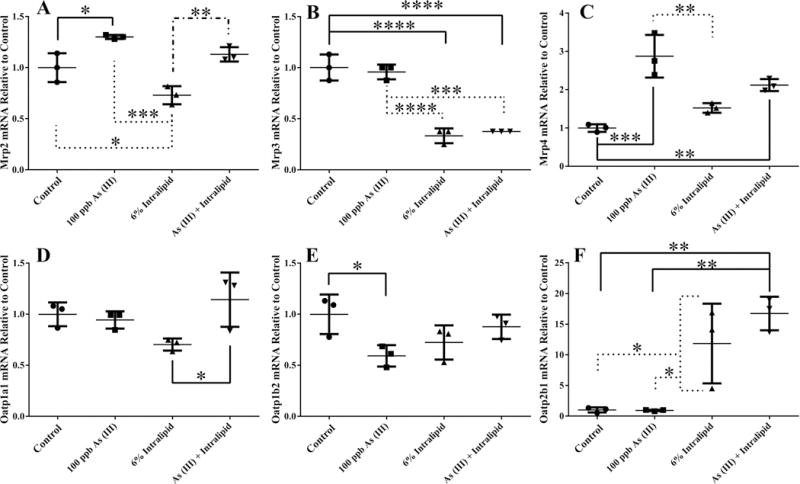
mRNA levels of select Mrp and Oatp transporter genes found to be statistically different in the indicated treatments compared to control. Fold change compared to control is shown for each target. The line is the mean and the error bars are the standard deviation. Analytical duplicates were used and a one-way ANOVA followed by a Tukey’s multiple comparison test utilizing Prism6 (GraphPad) for comparisons between groups: the reported p-value is the multiplicity adjusted p-value corrected for multiple comparisons. (**** = p ≤ 0.0001; *** = p ≤ 0.001; ** = p ≤ 0.01 ; * = p ≤ 0.05 compared to controls). Representative results are shown from a primary hepatocyte isolation with each treatment condition examined in triplicate. n=3.
Discussion
The in vivo alterations in the expression Mrps (Figure 1) are consistent with human and rodent models of NAFLD (9–11, 22). In Mrp2−/− mouse models, there is an increased detection in the expression of Mrp3 (23) and Mrp4 (24). Disruption of Mrp2 canalicular localization would result in similar functional defects to that of Mrp2−/− mice. Determining localization of Mrp2 in this model will be necessary to further elucidate potential mechanisms. Additionally, oxidative stress triggers Mrp2 internalization (25). FFA drives oxidative stress in NAFLD and hepatocytes treated with FFA undergo oxidative stress (26). As (III) also generates oxidative stress (27–29) and is capable of activating Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) (30). During oxidative stress, increased expression of Mrp3 and Mrp4 is partially dependent on activated Nrf2 (31). Thus, activation of Nrf2 by As (III) likely plays a role in the alteration of transporter expression.
The increase in Mrp substrates in plasma (Table 2) suggest that there is a functional change in metabolite disposition during As (III) potentiated diet-induced NAFLD. This is consistent with findings that link changes in Mrp function to altered xenobiotic disposition (10, 12, 15). In a genome wide evaluation of patients with NAFLD, there was a decrease in global hepatic Oatp expression (32). In mice, the effects are different. In multiple studies examining mouse models of NAFLD increased expression of Oatp1a4 was observed (9, 22, 33). In our mouse model, we detected increased expression of Oatp1a4 and Oatp2b1 (in both cell models as well), and decreased expression of Oatp1b2 in the IU+13W group (Figures 1, 4C, 5F). Decreased expression of Oatp transporters is associated with increased systemic exposure to xenobiotics in rat models of NASH (34, 35). The expression changes observed across our models and plasma concentration of unconjugated bilirubin (Table 2) suggest that As (III) mediated alterations of hepatic transport could affect Oatp mediated xenobiotic transport as well.
Male mice exposed to 100 ppb As (III) after weaning while on a Western diet do not develop NASH (8). Increased TAG accumulation was not detected in adult primary hepatocytes from combination exposure to Intralipid and As (III) compared to Intralipid exposure alone (Figure S1B). This highlights that developmental exposures are necessary for As (III)-induced NAFLD. TAG accumulation was undetected in E12.5 hepatocytes, but that is possibly due to the incomplete differentiation at that stage (Figure S1A). Phase I metabolism (and the examined transporters) reach adult expression and functionality at different stages of development (20, 21, 36). This gradient of transport and metabolizing competence could be one of the reasons for the differences observed between embryonic and adult primary hepatocytes exposed to identical conditions.
These data represent the first instance where low-level As (III) exposure in cultured hepatocytes result in alterations of transporter expression. If these effects translate to populations exposed to As (III), they represent a non-traditional endpoint that could have significant health effects. Additional in vivo studies on the effects of As (III) during embryogenesis and through development would be needed to verify if exposure to arsenite can alter hepatic transport independently of NAFLD progression and determine susceptibility to these alterations at different life stages. These finding suggest that exposure to As (III) could contribute to ADRs as a result of altered disposition of xenobiotics and their metabolites through direct As (III) mediated changes in transporter expression. The capability of As (III) to potentiate the severity of diet-induced NAFLD likely results in altered transport by virtue of creating a more severe NASH phenotype, but As (III) may be contributing through overlapping or independent mechanisms.
Supplementary Material
Acknowledgments
Grant Information: We appreciate the assistance by J. Clarke Ph.D., A. Dzierlenga, and D. Klein Ph.D. (University of Arizona) during IHC and immunoblotting optimizations. We appreciate helpful discussions with W. Waldorf. Funding was generously provided by: Superfund Research Program (NIH ES 04940); SWEHSC (P30ES006694); NIEHS Toxicology Training Grant (ES007091).
Footnotes
Competing Financial Interests: The authors declare they have no actual or potential competing financial interests, with the exception of N. Cherrington who has filed a broadly related patent for: Glucuronidated Acetaminophen as a Marker of Hepatic Disorders (US 20110220500 A1).
References
- 1.Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274–285. doi: 10.1111/j.1365-2036.2011.04724.x. [DOI] [PubMed] [Google Scholar]
- 2.Levene AP, Goldin RD. The epidemiology, pathogenesis and histopathology of fatty liver disease. Histopathology. 2012;61:141–152. doi: 10.1111/j.1365-2559.2011.04145.x. [DOI] [PubMed] [Google Scholar]
- 3.Arteel GE, Guo L, Schlierf T, Beier JI, Kaiser JP, Chen TS, Liu M, Conklin DJ, Miller HL, von Montfort C, States JC. Subhepatotoxic exposure to arsenic enhances lipopolysaccharide-induced liver injury in mice. Toxicol Appl Pharmacol. 2008;226:128–139. doi: 10.1016/j.taap.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reilly MP, Saca JC, Hamilton A, Solano RF, Rivera JR, Whitehouse-Innis W, Parsons JG, Dearth RK. Prepubertal exposure to arsenic(III) suppresses circulating insulin-like growth factor-1 (IGF-1) delaying sexual maturation in female rats. Reprod Toxicol. 2014;44:41–49. doi: 10.1016/j.reprotox.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shi X, Wei X, Koo I, Schmidt RH, Yin X, Kim SH, Vaughn A, McClain CJ, Arteel GE, Zhang X, Watson WH. Metabolomic analysis of the effects of chronic arsenic exposure in a mouse model of diet-induced Fatty liver disease. J Proteome Res. 2014;13:547–554. doi: 10.1021/pr400719u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tan M, Schmidt RH, Beier JI, Watson WH, Zhong H, States JC, Arteel GE. Chronic subhepatotoxic exposure to arsenic enhances hepatic injury caused by high fat diet in mice. Toxicol Appl Pharmacol. 2011;257:356–364. doi: 10.1016/j.taap.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smedley PL, Kinniburgh DG. A review of the source, behaviour and distribution of arsenic in natural waters. Appl Geochem. 2002;17:517–568. [Google Scholar]
- 8.Ditzel EJ, Nguyen T, Parker P, Camenisch TD. Effects of Arsenite Exposure during Fetal Development on Energy Metabolism and Susceptibility to Diet-Induced Fatty Liver Disease in Male Mice. Environ Health Perspect. 2015 doi: 10.1289/ehp.1409501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Canet MJ, Hardwick RN, Lake AD, Dzierlenga AL, Clarke JD, Cherrington NJ. Modeling human nonalcoholic steatohepatitis-associated changes in drug transporter expression using experimental rodent models. Drug Metab Dispos. 2014;42:586–595. doi: 10.1124/dmd.113.055996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lickteig AJ, Fisher CD, Augustine LM, Aleksunes LM, Besselsen DG, Slitt AL, Manautou JE, Cherrington NJ. Efflux transporter expression and acetaminophen metabolite excretion are altered in rodent models of nonalcoholic fatty liver disease. Drug Metab Dispos. 2007;35:1970–1978. doi: 10.1124/dmd.107.015107. [DOI] [PubMed] [Google Scholar]
- 11.Hardwick RN, Fisher CD, Canet MJ, Scheffer GL, Cherrington NJ. Variations in ATP-binding cassette transporter regulation during the progression of human nonalcoholic fatty liver disease. Drug Metab Dispos. 2011;39:2395–2402. doi: 10.1124/dmd.111.041012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hardwick RN, Clarke JD, Lake AD, Canet MJ, Anumol T, Street SM, Merrell MD, Goedken MJ, Snyder SA, Cherrington NJ. Increased susceptibility to methotrexate-induced toxicity in nonalcoholic steatohepatitis. Toxicol Sci. 2014;142:45–55. doi: 10.1093/toxsci/kfu156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Canet MJ, Hardwick RN, Lake AD, Kopplin MJ, Scheffer GL, Klimecki WT, Gandolfi AJ, Cherrington NJ. Altered arsenic disposition in experimental nonalcoholic fatty liver disease. Drug Metab Dispos. 2012;40:1817–1824. doi: 10.1124/dmd.112.046177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans ZP, Palanisamy AP, Sutter AG, Ellett JD, Ramshesh VK, Attaway H, Schmidt MG, Schnellmann RG, Chavin KD. Mitochondrial uncoupling protein-2 deficiency protects steatotic mouse hepatocytes from hypoxia/reoxygenation. Am J Physiol Gastrointest Liver Physiol. 2012;302:G336–42. doi: 10.1152/ajpgi.00049.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dzierlenga AL, Clarke JD, Hargraves TL, Ainslie GR, Vanderah TW, Paine MF, Cherrington NJ. Mechanistic basis of altered morphine disposition in nonalcoholic steatohepatitis. J Pharmacol Exp Ther. 2015;352:462–470. doi: 10.1124/jpet.114.220764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang W, Sargis RM, Volden PA, Carmean CM, Sun XJ, Brady MJ. PCB 126 and Other Dioxin-Like PCBs Specifically Suppress Hepatic PEPCK Expression via the Aryl Hydrocarbon Receptor. PLoS One. 2012;7:e37103. doi: 10.1371/journal.pone.0037103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shinkai Y, Sumi D, Fukami I, Ishii T, Kumagai Y. Sulforaphane, an activator of Nrf2, suppresses cellular accumulation of arsenic and its cytotoxicity in primary mouse hepatocytes. FEBS Lett. 2006;580:1771–1774. doi: 10.1016/j.febslet.2006.02.031. [DOI] [PubMed] [Google Scholar]
- 18.Konig J, Nies AT, Cui Y, Leier I, Keppler D. Conjugate export pumps of the multidrug resistance protein (MRP) family: localization, substrate specificity, and MRP2-mediated drug resistance. Biochim Biophys Acta. 1999;1461:377–394. doi: 10.1016/s0005-2736(99)00169-8. [DOI] [PubMed] [Google Scholar]
- 19.Hardwick RN, Ferreira DW, More VR, Lake AD, Lu Z, Manautou JE, Slitt AL, Cherrington NJ. Altered UDP-glucuronosyltransferase and sulfotransferase expression and function during progressive stages of human nonalcoholic fatty liver disease. Drug Metab Dispos. 2013;41:554–561. doi: 10.1124/dmd.112.048439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moscovitz JE, Aleksunes LM. Establishment of metabolism and transport pathways in the rodent and human fetal liver. Int J Mol Sci. 2013;14:23801–23827. doi: 10.3390/ijms141223801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maher JM, Slitt AL, Cherrington NJ, Cheng X, Klaassen CD. Tissue distribution and hepatic and renal ontogeny of the multidrug resistance-associated protein (Mrp) family in mice. Drug Metab Dispos. 2005;33:947–955. doi: 10.1124/dmd.105.003780. [DOI] [PubMed] [Google Scholar]
- 22.More VR, Slitt AL. Alteration of hepatic but not renal transporter expression in diet-induced obese mice. Drug Metab Dispos. 2011;39:992–999. doi: 10.1124/dmd.110.037507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vlaming ML, Mohrmann K, Wagenaar E, de Waart DR, Elferink RP, Lagas JS, van Tellingen O, Vainchtein LD, Rosing H, Beijnen JH, Schellens JH, Schinkel AH. Carcinogen and anticancer drug transport by Mrp2 in vivo: studies using Mrp2 (Abcc2) knockout mice. J Pharmacol Exp Ther. 2006;318:319–327. doi: 10.1124/jpet.106.101774. [DOI] [PubMed] [Google Scholar]
- 24.Chu XY, Strauss JR, Mariano MA, Li J, Newton DJ, Cai X, Wang RW, Yabut J, Hartley DP, Evans DC, Evers R. Characterization of mice lacking the multidrug resistance protein MRP2 (ABCC2) J Pharmacol Exp Ther. 2006;317:579–589. doi: 10.1124/jpet.105.098665. [DOI] [PubMed] [Google Scholar]
- 25.Sekine S, Ito K, Horie T. Oxidative stress and Mrp2 internalization. Free Radic Biol Med. 2006;40:2166–2174. doi: 10.1016/j.freeradbiomed.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 26.Soardo G, Donnini D, Domenis L, Catena C, De Silvestri D, Cappello D, Dibenedetto A, Carnelutti A, Bonasia V, Pagano C, Sechi LA. Oxidative stress is activated by free fatty acids in cultured human hepatocytes. Metab Syndr Relat Disord. 2011;9:397–401. doi: 10.1089/met.2010.0140. [DOI] [PubMed] [Google Scholar]
- 27.Flora SJ. Arsenic-induced oxidative stress and its reversibility following combined administration of N-acetylcysteine and meso 2,3-dimercaptosuccinic acid in rats. Clin Exp Pharmacol Physiol. 1999;26:865–869. doi: 10.1046/j.1440-1681.1999.03157.x. [DOI] [PubMed] [Google Scholar]
- 28.Kitchin KT, Ahmad S. Oxidative stress as a possible mode of action for arsenic carcinogenesis. Toxicol Lett. 2003;137:3–13. doi: 10.1016/s0378-4274(02)00376-4. [DOI] [PubMed] [Google Scholar]
- 29.Pi J, Qu W, Reece JM, Kumagai Y, Waalkes MP. Transcription factor Nrf2 activation by inorganic arsenic in cultured keratinocytes: involvement of hydrogen peroxide. Exp Cell Res. 2003;290:234–245. doi: 10.1016/s0014-4827(03)00341-0. [DOI] [PubMed] [Google Scholar]
- 30.Lau A, Zheng Y, Tao S, Wang H, Whitman SA, White E, Zhang DD. Arsenic Inhibits Autophagic Flux, Activating the Nrf2-Keap1 Pathway in a p62-Dependent Manner. Mol Cell Biol. 2013;33:2436–2446. doi: 10.1128/MCB.01748-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maher JM, Aleksunes LM, Dieter MZ, Tanaka Y, Peters JM, Manautou JE, Klaassen CD. Nrf2- and PPAR alpha-mediated regulation of hepatic Mrp transporters after exposure to perfluorooctanoic acid and perfluorodecanoic acid. Toxicol Sci. 2008;106:319–328. doi: 10.1093/toxsci/kfn177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lake AD, Novak P, Fisher CD, Jackson JP, Hardwick RN, Billheimer DD, Klimecki WT, Cherrington NJ. Analysis of global and absorption, distribution, metabolism, and elimination gene expression in the progressive stages of human nonalcoholic fatty liver disease. Drug Metab Dispos. 2011;39:1954–1960. doi: 10.1124/dmd.111.040592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clarke JD, Hardwick RN, Lake AD, Lickteig AJ, Goedken MJ, Klaassen CD, Cherrington NJ. Synergistic interaction between genetics and disease on pravastatin disposition. J Hepatol. 2014;61:139–147. doi: 10.1016/j.jhep.2014.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clarke JD, Hardwick RN, Lake AD, Canet MJ, Cherrington NJ. Experimental nonalcoholic steatohepatitis increases exposure to simvastatin hydroxy acid by decreasing hepatic organic anion transporting polypeptide expression. J Pharmacol Exp Ther. 2014;348:452–458. doi: 10.1124/jpet.113.211284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fisher CD, Lickteig AJ, Augustine LM, Oude Elferink RP, Besselsen DG, Erickson RP, Cherrington NJ. Experimental non-alcoholic fatty liver disease results in decreased hepatic uptake transporter expression and function in rats. Eur J Pharmacol. 2009;613:119–127. doi: 10.1016/j.ejphar.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choudhary D, Jansson I, Schenkman JB, Sarfarazi M, Stoilov I. Comparative expression profiling of 40 mouse cytochrome P450 genes in embryonic and adult tissues. Arch Biochem Biophys. 2003;414:91–100. doi: 10.1016/s0003-9861(03)00174-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.