

Abstract
Abstract
Five new ent-kaurane diterpenoids, named mascaroside III–V (1–3), and 20-nor-cofaryloside I–II (4–5), together with seven known diterpenoids, were isolated from methanol extracts of the green coffee beans of Yunnan Arabica Coffee. Their chemical structures were elucidated by extensive spectroscopic analyses. Meanwhile, cytotoxicity assay against HL-60, A-549, SMMC-7721, MCF-7 and SW480 cell lines showed that they have not evident inhibition of cytotoxicity.
Graphical Abstract
Electronic supplementary material
The online version of this article (doi:10.1007/s13659-016-0099-1) contains supplementary material, which is available to authorized users.
Keywords: Coffea arabica L., Green coffee beans, Diterpenoids, Structural elucidation
Introduction
Coffea arabica L., commonly known as coffee and widely distributed throughout the world, involving Africa, Latin America and Asia, is a very popular hot drink around the world because of its attractive aroma and unique taste [1, 2]. Previous studies have shown that coffee beans are consisted of caffeine, chlorogenic acids, saccharides [3–5], as well as diterpenoids, although, taking a minor proportion in the chemical constituents of coffee. However, due to their broad spectrum of biological activities, such as cytotoxicity, antioxidant, anti-inflammatory, researchers have carried out work on diterpenoids components from coffee beans, and found nearly 90 diterpenoids [6–9]. Yunnan Arabica Coffee was the Coffea arabica which were planted in Yunnan province. Along with the planting scale expanded to 120 thousand hectares, Yunnan province became a well-known cultivation base of C. arabica in the world. In order to investigate the chemical constituents of Yunnan Arabica Coffee and find bioactivity diterpenoids, we took Yunnan Arabica Coffee green beans collected in Dehong as the subject, and discovered five new ent-kaurane diterpenoids, along with seven known diterpenoids (Fig. 1). Herein, the isolation, structural elucidation, and their relevant bioactivities were also described.
Fig. 1.
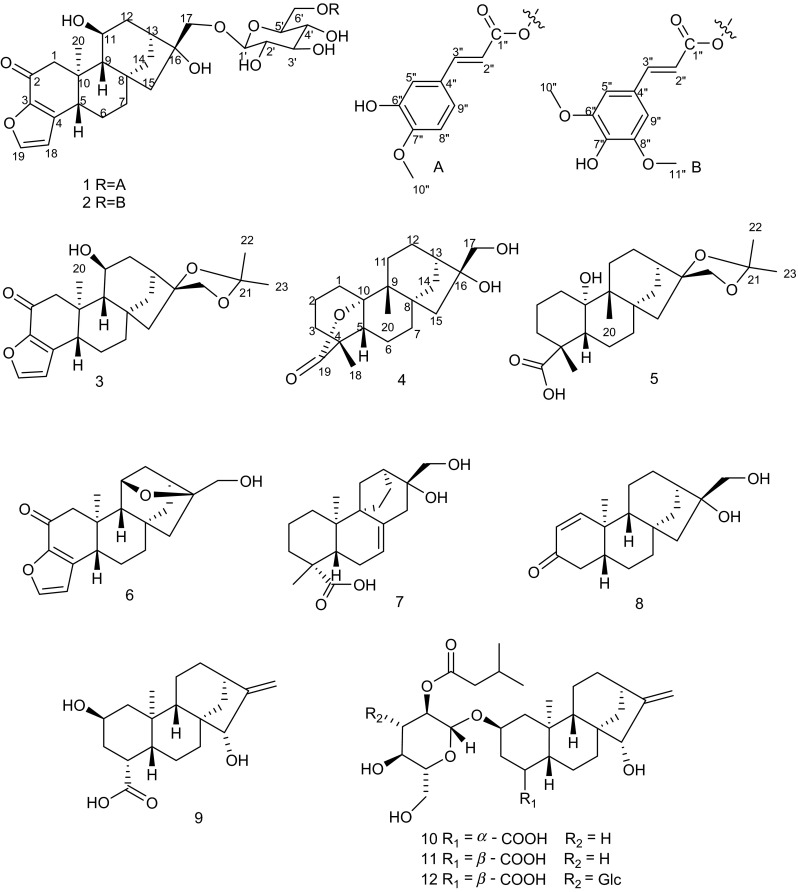
Structures of compounds 1–12
Results and Discussion
Mascaroside III (1) was isolated as a white amorphous powder. The molecular formula C36H44O13 was deduced from the molecular ion peak at m/z [M + Na]+ 707.2670 (calcd for C36H44O13Na, 707.2674) in HREIMS. The IR spectrum indicated that 1 possessed hydroxyl (3440 cm−1) and α,β-unsaturated ketone (1650 cm−1) groups. The 13C NMR (DEPT) data (Tables 2, 3) showed 36 carbon resonances, attributed to a monosaccharide, a cinnamic acids group and an aglycone moiety, and the aglycone moiety were classified as one methyl (δC 15.7), 7 methylenes (including one oxygenated), 6 methines (including one oxygenated, two olefinic), and 6 quaternary carbons (including one oxygenated, two olefinic, and one carbonyl). These data (Tables 1, 2, 3) were similar to those of mascaroside I [10, 11] except for 10 additional signals for a cinnamic acids group. The coupling constant (J2″, 3″ = 15.9 Hz) suggested the double bond of the cinnamic acid group was trans. Besides, δH 6.80(d, J = 8.2 Hz), 7.10(d, J = 8.2 Hz) were due to ortho-aromatic hydrogen suggested the two oxygenated sp2 quaternary carbons were at C-6″ and C-7″, along with CH3-10″ linked to OH-7″ confirmed by the HMBC correlations from H3-10″ (δH 3.88) to C-7″. Further, the OH-6′ in the glucose and COOH-1″ in trans-cinnamic acids group formed into ester were confirmed by HMBC correlations of H-6′ (δH 4.29, 4.69) to C-1″ (δC 169.5). The relative configuration of glucose anomeric proton was confirmed as β on the basis of the coupling constant (J1′, 2′ = 7.8 Hz). Furthermore, the glucose was identified as D-form by GC analysis comparing with a standard after acid hydrolysis [12, 13]. The relative configuration of 1 was same with mascaroside I by comparison of the NMR data. Furthermore, owing to the greatly predominant occurrence of this enantiomeric form in nature world, and until now, all the kaurene skeleton diterpenoids have been isolated from Coffea arabica were ent-kaurene series. Therefore, compound 1 was confirmed as an ent-kauranoid with the negative specific rotation value (−126.67) confirmed it further. ent-Kauranoid with the configuration of C-20 being α-orientated and H-5, H-9 being β-orientated. The key ROESY correlations of H-11 (δH 3.86)/H-20 (δH 0.78), and H-5 (δH 2.83)/H-9 (δH 1.61), H-9/H-15b (δH 2.12), and H-15b/H2-17 (δH 3.59, 4.71) allowed to assign H-11 as α-orientated, and CH2OH-17 as β-orientated, separately [14]. Hence, the structure of 1 was determined and named as mascaroside III (Fig. 2).
Table 2.
13C NMR spectral data of compounds 1–5 [δ in ppm]
| Position | 1 a | 2 a | 3 b | 4 a | 5 b |
|---|---|---|---|---|---|
| 1 | 54.3 (t) | 54.3 (t) | 53.4 (t) | 31.8 (t) | 29.7 (t) |
| 2 | 187.7 (s) | 187.6 (s) | 185.2 (s) | 19.8 (t) | 28.1 (t) |
| 3 | 148.0 (s) | 147.9 (s) | 146.6 (s) | 35.8 (t) | 37.6 (t) |
| 4 | 144.8 (s) | 144.8 (s) | 142.1 (s) | 50.1 (s) | 49.5 (s) |
| 5 | 45.8 (d) | 45.8 (d) | 44.8 (d) | 52.2 (d) | 50.1 (d) |
| 6 | 23.4 (t) | 23.3 (t) | 22.4 (t) | 21.6 (t) | 21.8 (t) |
| 7 | 37.1 (t) | 37.0 (t) | 39.6 (t) | 41.4 (t) | 39.3 (t) |
| 8 | 45.2 (s) | 45.2 (s) | 42.3 (s) | 43.5 (s) | 43.8 (s) |
| 9 | 62.1 (d) | 62.1 (d) | 61.0 (d) | 45.9 (s) | 43.8 (s) |
| 10 | 43.8 (s) | 43.8 (s) | 43.9 (s) | 90.6 (s) | 77.1 (s) |
| 11 | 66.1 (d) | 66.1 (d) | 65.2 (d) | 34.4 (t) | 36.2 (t) |
| 12 | 37.5 (t) | 37.5 (t) | 36.7 (t) | 24.9 (t) | 19.0 (t) |
| 13 | 46.5 (d) | 46.5(d) | 44.7 (d) | 44.8 (d) | 44.2 (d) |
| 14 | 41.3 (t) | 41.2 (t) | 38.0 (t) | 33.5 (t) | 32.3 (t) |
| 15 | 52.0 (t) | 52.0 (t) | 55.1 (t) | 50.9 (t) | 50.2 (t) |
| 16 | 82.4 (s) | 82.3 (s) | 89.3 (s) | 84.9 (s) | 89.2 (s) |
| 17 | 75.4 (t) | 75.4 (t) | 70.5 (t) | 66.2 (t) | 69.7 (t) |
| 18 | 111.4 (d) | 111.4 (d) | 110.0 (d) | 17.5 (q) | 29.1 (q) |
| 19 | 150.4 (d) | 150.4 (d) | 148.2 (d) | 183.0 (s) | 183.2 (s) |
| 20 | 15.7 (q) | 15.7 (q) | 15.3 (q) | 19.3 (q) | 17.5 (q) |
| 21 | – | – | 108.1 (s) | – | 104.2 (s) |
| 22 | – | – | 26.8 (q) | – | 26.9 (q) |
| 23 | – | – | 26.6 (q) | – | 26.8 (q) |
aData were measured at 150 MHz in CD3OD
bData were measured at 150 MHz in CDCl3
Table 3.
1H NMR and 13C NMR of the glucose and cinnamic acids group [δ in ppm, J in Hz]
| 1H NMR | 13C NMR | |||
|---|---|---|---|---|
| 1 | 2 | 1 | 2 | |
| 1′ | 4.32 (d, 7.8) | 4.34 (d, 7.5) | 104.8 (d) | 104.8 (d) |
| 2′ | 3.22 (m) | 3.26 (t, 8.3) | 75.4 (d) | 75.4 (d) |
| 3′ | 3.40 (m) | 3.43 (m) | 77.6 (d) | 77.6 (d) |
| 4′ | 3.40 (m) | 3.43 (m) | 71.5 (d) | 71.5 (d) |
| 5′ | 3.50 (m) | 3.61 (d, 10.1) | 75.5 (d) | 75.5 (d) |
| 6′ | 4.29 (m) | 4.32 (d, 4.7) | 63.9 (t) | 63.8 (t) |
| 4.69 (m) | 4.73 (m) | – | – | |
| 1″ | – | – | 169.5 (s) | 169.5 (s) |
| 2″ | 6.42 (d, 15.9) | 6.49 (d, 15.9) | 115.2 (d) | 115.7 (d) |
| 3″ | 7.65 (d, 15.9) | 7.67 (d, 15.9) | 147.4 (d) | 147.6 (d) |
| 4″ | – | – | 127.6 (s) | 126.9 (s) |
| 5″ | 7.20 (s) | 6.95 (s) | 111.8 (d) | 107.0 (d) |
| 6″ | – | – | 150.8 (s) | 149.5 (s) |
| 7″ | – | – | 149.4 (s) | 139.8 (s) |
| 8″ | 6.80 (d, 8.2) | – | 116.5 (d) | 149.5 (s) |
| 9″ | 7.10 (d, 8.2) | 6.95 (s) | 124.3 (d) | 107.0 (d) |
| 10″ | 3.88 (s) | 3.89 (s) | 56.5 (q) | 56.9 (q) |
| 11″ | – | 3.89 (s) | – | 56.9 (q) |
δ H Data were measured at 600 MHz in CD3OD
δ C Data were measured at 150 MHz in CD3OD
Table 1.
1H NMR spectral data of compounds 1–5 [δ in ppm, J in Hz]
| Position | 1 a | 2 a | 3 b | 4 a | 5 b |
|---|---|---|---|---|---|
| 1 | 2.39 (d, 16.3) | 2.42 (d, 16.3) | 2.33 (d, 16.1) | 1.28 (dd, 13.6, 4.4) | 0.94 (m) |
| 2.74 (d, 16.3) | 2.77 (d, 16.3) | 2.83 (d, 16.1) | 1.81 (m) | 2.01 (dd, 14.8, 5.9) | |
| 2 | – | – | – | 1.19 (m) | 1.52 (m) |
| – | – | – | 1.72 (m) | 1.64 (m) | |
| 3 | – | – | – | 1.58 (m) | 1.01 (m) |
| – | – | – | 1.65 (m) | 2.10 (m) | |
| 5 | 2.83 (br d, 14.2) | 2.85 (br d, 12.6) | 2.77 (dd, 12.4, 2.4) | 1.94 (dd, 14.0, 4.8) | 1.53 (m) |
| 6 | 1.61 (m) | 1.63 (m) | 1.62 (m) | 1.57 (m) | 1.84 (m) |
| 2.01 (m) | 2.03 (m) | 1.99 (m) | 1.86 (m) | 1.89 (m) | |
| 7 | 1.72 (m) | 1.73 (m) | 1.78 (m, 2H) | 1.77 (m) | 1.64 (m) |
| 2.04 (m) | 2.06 (m) | 1.90 (m) | 2.22 (d, 12.1) | ||
| 9 | 1.61 (m) | 1.63 (m) | 1.61 (m) | – | – |
| 11 | 3.86 (m) | 3.89 (m) | 3.93 (m) | 1.72 (m) | 1.44 (m) |
| – | – | – | 2.20 (dd, 13.8, 5.8) | 1.75 (m) | |
| 12 | 1.72 (m) | 1.73 (m) | 1.81 (m) | 1.61 (m) | 1.45 (m) |
| 1.78 (m) | 1.78 (m) | 1.95 (m) | 1.63 (m) | 1.51 (m) | |
| 13 | 2.10 (m) | 2.13 (m) | 2.15 (m) | 1.90 (m) | 2.11 (m) |
| 14 | 1.74 (m) | 1.73 (m) | 1.57 (m) | 1.77 (m) | 1.50 (m) |
| 1.77 (m) | 1.76 (m) | 1.85 (m) | 1.91 (m) | 1.65 (m) | |
| 15 | 1.37 (d, 14.3) | 1.40 (d, 14.4) | 1.85 (m) | 1.39 (d, 14.7) | 1.53 (m) |
| 2.12 (m) | 2.15 (d, 14.4) | 2.40 (m) | 2.02 (d, 14.7) | 2.44 (d, 15.0) | |
| 17 | 3.59 (d, 10.2) | 3.53 (d, 10.2) | 4.10 (d, 9.3) | 3.64 (d, 11.5) | 3.89 (d, 8.6) |
| 4.71 (d, 10.2) | 4.74 (d, 10.2) | 4.27 (d, 9.3) | 3.70 (d, 11.5) | 4.04 (d, 8.6) | |
| 18 | 6.58 (s) | 6.61 (s) | 6.42 (d, 1.4) | 1.06 (s) | 1.23 (s) |
| 19 | 7.78 (s) | 7.81 (s) | 7.60 (d, 1.4) | – | – |
| 20 | 0.78 (s) | 0.81 (s) | 0.87 (s) | 1.16 (s) | 1.08 (s) |
| 22 | – | – | 1.37 (s) | – | 1.39 (s) |
| 23 | – | – | 1.36 (s) | – | 1.36 (s) |
aData were measured at 600 MHz in CD3OD
bData were measured at 600 MHz in CDCl3
Fig. 2.

Key correlations in 2D NMR spectra of compound 1
Mascaroside IV (2) had the molecular formula of C37H46O14 according to the HRESIMS analysis at m/z [M + Na]+ 737.2786 (calcd for C37H46O14Na, 737.2780). The 1D NMR data (Tables 1, 2, 3) of 2 was identical to that of 1, except that the cinnamic acids group in 2 was substituted by one more oxygenated methyl, which was further verified by the HMBC correlations from H-10″, and H-11″ (δH 3.89) to C-6″, and C-8″ (δC 149.5). The chiral centers of 2 were same with those of compound 1. Therefore, the structure of 2 was elucidated as shown and given the name mascaroside IV.
Mascaroside V (3) was isolated as white powder. The HRESIMS of 3 showed an ion peak at m/z [M + Na]+ 409.1987 (calcd for 409.1985) suggesting a molecular formula C23H30O5. The 1D NMR spectroscopic features showed it similar to the aglycone moiety of 1 with the differences being that three more carbon atoms (δC 108.1 s, 26.8 q, 26.6 q). HSQC together with HMBC spectral signals, showed that CH3-22 (δC 26.8), CH3-23 (δC 26.6) were located at the same sp3 quaternary carbon (C-21 δC 108.1 s) which was confirmed by correlations from H-22, H-23 to C-21. This three carbons group was substituted at 16-OH, 17-OH to form a ketal ring [15] based on HMBC correlations from H2-17 (δH 4.10, 4.27) to C-21.The ROESY correlations were similar to compound 1, consequently, the structure of 3 was confirmed as mascaroside V.
20-nor-cofaryloside I (4) a white amorphous powder, displayed a [M + Na]+ 357.2032 (calcd for 357.2036) in HREIMS, consistent with the molecular formula of C20H30O4, indicating 6 degrees of unsaturation. The compound 4 displayed similar characteristic signals to 10α,16α,17-trihydroxy-9α-methyl-15-oxo-20-nor-kauran-19-oic acid γ-lactone (A) [16, 17] except for the absence of the carbonyl groups at C-15. This was confirmed by the chemical shift at C-15 (δC 50.9) in 4 which was upfield shifted comparing to that in A (δC 224.0). Besides, the HMBC correlations from H2-15 (δH 1.39, 2.02) to C-16 (δC 84.9) and C8 (δC 43.5) also supported these change. On biogenetic grounds, compound 4 as an ent-kaurane with the configuration of H-5 and H-9 being β-orientated, while OH-10 being α-orientated. Its ROESY correlations showed cross peaks of H-5 (δH 1.94)/Me-18 (δH 1.06), H-5/Me-20 (δH 1.16), Me-20/H-15b (δH 2.02), H-15b/H2-17 (δH 3.64, 3.70), revealing that the orientation of C-18, C-20 and C-17 was β-orientated (Fig. 3). Thereupon, the structure of 4 was identified as 10α,16α,17-trihydroxy-9β-methyl-20-nor-ent-kauran-19-oic acid γ-lactone, and named 20-nor-cofaryloside I.
Fig. 3.

Key correlations in 2D NMR spectra of compound 4
20-nor-cofaryloside II (5) possessed the molecular formula of C23H36O5, according to the HRESIMS analysis at m/z [M − H]− 391.2488 (calcd for 391.2490). Analyses of 5′s 1H and 13C NMR data indicated the existence of 23 carbon resonances, and C-21 (δC 104.2 s), C-22 (δC 26.9 q) and C-23 (δC 26.9 q) suggesting that the structure of compound 5 had the same ketal ring as compound 3 and this deduction was further supported by HMBC correlations from H2-17, H3-22 and H3-23 to C-21. The other 20 carbon signals showed that compound 5 was similar to 4 while the chemical shift of the oxygenated sp3 quaternary carbon C-10 (δC 77.1) of 5 was upfield shifted comparing to that of 4 (δC 90.6), and the molecular weight of 5 was 18 units more than that of 4 along with the degrees of unsaturation decrease by one. Therefore, the lactone linkage which assigned between COOH-19 and OH-10 was ring opened in 5. The relative configuration of 5 (Fig. 4) was same with 4. Thus, the structure was defined as 10α-hydroxy- 16α,17-[(1-methylethylidene)bis(oxy)]-9β-methyl-20-nor-ent-kauran-19-oic acid, and named 20-nor- cofaryloside II.
Fig. 4.

Key correlations in 2D NMR spectra of compound 5
Seven known diterpenoids were also obtained from this genus, bengalensol [18], villanovane [10], tricalysione A [14], 2β,16α,17-trihydroxy-ent-kauran-19α-oic acid [19], 2-O-(2-O-isovaleryl-β- d-gluco-pyranosyl)-4α-atractyligenin [20], 2-O-(2-O-isovaleryl-β-d-gluco-pyranosyl)-4β-atrac- tyligenin [20], 3-O-β-d-glucopyranosyl-2-O-(2-O-isovaleryl-β-d-gluco-pyranosyl)-4β-atracty ligenin [20]. Their structures were identified by comparison of their NMR data with literature data.
Compounds 1–5, 7, 8 were evaluated for cytotoxicity against HL-60, A-549, SMMC-7721, MCF-7 and SW480 cell lines. Unfortunately, they were inactive against all test cells (Electronic supplementary material, Table S1).
Experimental Section
General Experimental Procedures
1D and 2D NMR spectra were obtained on a Bruker Avance III 600 MHz spectrometer (Bruker Biospin GmbH, Karlsruhe, Germany). HREIMS was measured on Waters Xevo TQ-S and Waters Autospec Premier P776 mass spectrometers (Waters, Milford, MA, USA). HRESIMS were recorded on an Agilent 6200 Q-TOF MS system (Agilent Technologies, Santa Clara, CA, USA). UV spectra were recorded on a Shimadzu UV-2401PC (Shimadzu, Kyoto, Japan). Optical rotations were obtained on a JASCO P-1020 digital polarimeter (Horiba, Kyoto, Japan). IR spectra were detected on Bruker Tensor 27 FTIR (KBr pellets) spectrometers. Sephadex LH-20 (Amersham Biosciences, Upssala, Sweden) and silica gel (Qingdao Haiyang Chemical Co., Ltd) were used for column chromatography (CC). Preparative high performance liquid chromatography (prep-HPLC) was performed on an Agilent 1100 liquid chromatography system equipped with Zorbax SB-C18 columns (9.4 mm × 250 mm) and a DAD detector (Agilent Technologies, Santa Clara, CA, USA). Thin-layer chromatography was performed on precoated TLC plates (200–250 μm thickness, silica gel 60 F254, Qingdao Marine Chemical, Inc.), and spots were visualized by heating after spraying.
Plant Material
The green coffee beans of Coffea arabica L. were harvested in December 2014 and identified by Hong-bo Zhang, Dehong Institute of Tropical Agriculture. A voucher specimen of C. arabica was deposited in the Herbarium of Kunming Institute of Botany, Chinese Academy of Sciences (No. KCF 201412).
Extraction and Isolation
The air-dried and powdered green Dehong coffee beans (18 kg) were extracted with 95 % methanol three times, and then the combined filtrates were concentrated under reduced pressure to give a crude extract (5 kg). The crude extract was suspended in H2O and extracted with petroleum ether (PE), ethyl acetate (EtOAc), respectively. The EtOAc layer (160 g) was got rid of saccharides by D101 and then subjected to RP-18 column chromatography which eluted with MeOH–H2O (gradient from 15:85 to 100:0 v/v) to yield four fractions: fraction 1 (20 g), fraction 2 (19 g), fraction 3 (30 g), and fraction 4 (24 g). Fraction 4 was separated on silica gel CC using a CHCl3–MeOH gradient solvent system (80:0 → 1:1, v/v) to obtain eight subfractions (fraction 4–1 to 4–8). Then fraction 4–6 (6.5 g) was chromatographed on RP-18 CC (MeOH–H2O 10:90–100:0 v/v), Sephadex LH-20 (MeOH) and then purified by semipreparative HPLC (elute with CH3CN–H2O 15–75 %, 30 min) to afford 1 (5 mg), 2 (7 mg), 10 (2 mg), 11 (8 mg), 12 (40 mg). In the same way, 5 (14 mg), 6 (4 mg), 7 (17 mg) were isolated from fraction 4-2 (370 mg) and 3 (2 mg), 4 (5 mg), 8 (3 mg), 9 (6 mg) were isolated from fraction 4-3 (2.7 g).
Mascaroside III (1)
White amorphous powder; −126.67 (c 0.150, MeOH); UV (MeOH) λmax (log ε) 329 (4.87), 281 (4.14), 240 (3.94), 216 (3.97), 202 (4.05) nm; IR (KBr) νmax 3439, 2923, 2878,1703, 1657, 1515, 1437, 1270, 1165, 1126, 1033 cm−1; 1H (600 MHz, CD3OD) and 13C NMR (150 MHz, CD3OD) data, Tables 1, 2 and 3; ESIMS m/z 707 [M + Na]+; HREIMS m/z [M + Na] + 707.2670 (calcd for C36H44O13Na, 707.2674).
Mascaroside IV (2)
White amorphous powder; −107.20 (c 0.213, MeOH); UV (MeOH) λmax (log ε) 332 (4.00), 279 (4.03), 241 (4.03), 202 (4.12) nm; IR (KBr) νmax 3439, 2924, 2855, 1706, 1663, 1513, 1436, 1258, 1155, 1113, 1042 cm−1; 1H (600 MHz, CD3OD) and 13C NMR (150 MHz, CD3OD) data, Tables 1, 2, and 3; ESIMS m/z 737 [M + Na]+; HREIMS m/z [M + Na]+ 737.2786 (calcd for C37H46O14Na, 737.2780).
Mascaroside V (3)
White amorphous powder; −132.81 (c 0.243, CHCl3); UV (CHCl3) λmax (log ε) 276 (4.05), 232 (3.71), 208 (3.67), 198 (3.64) nm; IR (KBr) νmax 3492, 2925, 2866, 1665, 1436, 1368, 1208, 1128, 1049 cm−1; 1H (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data, Tables 1,2; ESIMS m/z 409 [M + Na]+; HREIMS m/z [M + Na]+ 409.1987 (calcd for C23H30O5Na, 409.1985).
20-Nor-cofaryloside I (4)
white amorphous powder; −7.77 (c 0.206, MeOH); UV (MeOH) λmax (log ε) 274 (1.85), 206 (2.59) nm; IR (KBr) νmax 3439, 2931, 2870, 1759, 1632, 1443, 1382, 1174, 1136, 1148, 935 cm−1; 1H (600 MHz, CD3OD) and 13C NMR (150 MHz, CD3OD) data, Tables 1,2; ESIMS m/z 737 [M + Na]+; HREIMS m/z [M + Na]+ 357.2032 (calcd for C20H30O4Na, 357.2036).
20-Nor-cofaryloside II (5)
White amorphous powder; −51.01 (c 0.105, CHCl3); UV (CHCl3) λmax (log ε) 280 (2.85), 241 (2.96), 229 (2.83), 192 (1.77) nm; IR (KBr) νmax 3442, 2936, 2872, 1718, 1699, 1464, 1373, 1252, 1213, 1052, 970 cm−1; 1H NMR (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data, Tables 1 and 2; ESIMS m/z 391 [M − H]−; HREIMS m/z [M – H]− 391.2488 (calcd for C23H35O5, 391.2490).
Cytotoxicity Assay
The cytotoxicity against HL-60, A-549, SMMC-7721, MCF-7 and SW480 cell lines of compounds 1–5, 7, 8 were tested by using MTS method. MTS [3-(4,5-dimethylthiazol-2-yl)-5(3-carboxymethoxyphenyl)-2-(4-sulfopheny)-2H-tetrazolium] is an analogue of MTT [21], which can be reduced into soluble formazan by succinate dehydrogenase in mitochondria of living cells. Moreover, the optical density value of formazan (490 nm) is proportional to the number of living cells.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported financially by Programme of Key New Productions of Yunnan Province, Centre of CHINA (No. 2015BB002), The STS Programme of Chinese Academy of Sciences (KFJ-SW-STS-143-8), as well as Foundation of State Key Laboratory of Phytochemistry and Plant Resources in West China (P2015-ZZ09).
Compliance with Ethical Standards
Conflict of Interest
All authors declare no conflict of interest.
References
- 1.Wei FF, Furihata K, Koda M, Hu FY, Miyakawa T, Tanokura M. J. Agric. Food Chem. 2012;60:1005–1012. doi: 10.1021/jf205315r. [DOI] [PubMed] [Google Scholar]
- 2.Cagliani LR, Pellegrino G, Giugno G, Consonni R. Talanta. 2013;106:169–173. doi: 10.1016/j.talanta.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Naidu MM, Sulochanamma G, Sampathu SR, Srinivas P. Food Chem. 2008;107:377–384. doi: 10.1016/j.foodchem.2007.08.056. [DOI] [Google Scholar]
- 4.Hughes WJ, Thorpe TM. J. Food Sci. 1987;52:1078–1083. doi: 10.1111/j.1365-2621.1987.tb14280.x. [DOI] [Google Scholar]
- 5.Fisk ID, Kettle A, Hofmeister S, Virdie A, Kenny JS. Flavor. 2012;1:1–9. doi: 10.1186/2044-7248-1-1. [DOI] [Google Scholar]
- 6.Li X, Xiao WL, Pu JX, Ban LL, Shen YH, Weng ZY, Li SH, Sun HD. Phytochemistry. 2006;67:1336–1340. doi: 10.1016/j.phytochem.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 7.Lee KJ, Choi JH, Jeong HG. Food Chem. Toxicol. 2007;45:2118–2125. doi: 10.1016/j.fct.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 8.Cardenas C, Quesada AR, Medina MA. PLoS One. 2011;6:1–9. doi: 10.1371/annotation/38262cc6-07cc-4074-8ce7-2181d4d0fbdc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kurzrock T, Speer K. Food Rev. Int. 2001;17:433–450. doi: 10.1081/FRI-100108532. [DOI] [Google Scholar]
- 10.Shu Y, Liu JQ, Peng XR, Wan LS, Zhou L, Zhang T, Qiu MH. J. Agric. Food Chem. 2014;62:2631–2637. doi: 10.1021/jf500788t. [DOI] [PubMed] [Google Scholar]
- 11.Roland P, Armin G. Phytochemistry. 1990;29:990–992. doi: 10.1016/0031-9422(90)80065-O. [DOI] [Google Scholar]
- 12.Zhao JQ, Wang YM, Lv JJ, Zhu HT, Wang D, Yang CR, Xu M, Zhang YJ. J. Braz. Chem. Soc. 2014;25:1446–1454. [Google Scholar]
- 13.Russell BW, Gary RE, Courtney MS. J. Nat. Prod. 2013;76:1592–1597. doi: 10.1021/np400501u. [DOI] [PubMed] [Google Scholar]
- 14.Nishimura K, Hitotsuyanagi Y, Sakakura K, Fujita K, Tachihara S, Fukaya H, Aoyagi Y, Hasuda T, Kinoshita T, Takeya K. Tetrahedron. 2007;63:4558–4562. doi: 10.1016/j.tet.2007.02.113. [DOI] [Google Scholar]
- 15.Wang R, Chen WH, Shi YP. J. Nat. Prod. 2010;73:17–21. doi: 10.1021/np9005579. [DOI] [PubMed] [Google Scholar]
- 16.Braca A, Armenise AI, Mendez J, Mi Q, Chai HB. Plant. Med. 2004;70:540–550. doi: 10.1055/s-2004-827155. [DOI] [PubMed] [Google Scholar]
- 17.Cai XF, Seon LI, Tien DN, Shen G, Ho KY. Phytother. Res. 2004;18:677–680. doi: 10.1002/ptr.1523. [DOI] [PubMed] [Google Scholar]
- 18.Choudhury MH, Quamrul H. Nat. Prod. Lett. 1994;5:55–60. doi: 10.1080/10575639408043935. [DOI] [Google Scholar]
- 19.Emika O, Satoshi K. Phytochemistry. 2004;65:885–890. doi: 10.1016/j.phytochem.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 20.Liu J, Feng L, Li HD, Dong QL, Chen R. Helv. Chim. Acta. 2012;95:221–226. doi: 10.1002/hlca.201100272. [DOI] [Google Scholar]
- 21.Mosmann T. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.