

Abstract
Bromine (Br2) gas inhalation poses an environmental and occupational hazard resulting in high morbidity and mortality. In this review, we underline the acute lung pathology (within 24 hours of exposure) and potential therapeutic interventions that may be utilized to mitigate Br2-induced human toxicity. We will discuss our latest published data, which suggests that an increase in heme-dependent tissue injury underlies the pathogenesis of Br2 toxicity. Our study was based on previous findings that demonstrated that Br2 upregulates the heme-degrading enzyme heme oxygenase-1 (HO-1), which converts toxic heme into billiverdin. Interestingly, following Br2 inhalation, heme levels were indeed elevated in bronchoalveolar lavage fluid, plasma, and whole lung tissue in C57BL/6 mice. High heme levels correlated with increased lung oxidative stress, lung inflammation, respiratory acidosis, lung edema, higher airway resistance, and mortality. However, therapeutic reduction of heme levels, by either scavenging with hemopexin or degradation by HO-1, improved lung function and survival. Therefore, heme attenuation may prove a useful adjuvant therapy to treat patients after Br2 exposure.
Keywords: Bromine, heme, heme oxygenase, hemopexin, lung injury
Exposure to Br2 presents a significant threat to public health
A study conducted on human volunteers demonstrated that a small exposure to 0.9 ppm bromine (Br2) for 5 min results in cough, headache, and irritation of the eyes, nose, and upper respiratory tract.1 Accidental exposure to higher doses of Br2 and its ensuing respiratory complications can lead to severe morbidity and mortality, yet there is a lack of human and animal data available on Br2 toxicity. A toxic industrial chemical, Br2 is a fuming liquid that targets the lungs, eyes, central nervous system, skin,2 and respiratory tract.3 World production of Br2 exceeds 300,000 tons per year; the three main producing countries are the United States, the United Kingdom, and Israel. Br2 is widely used in the manufacture of medicinal compounds, flame retardants, agricultural chemicals, gasoline additives, dyes, photographic chemicals, bleaching agents, and water disinfectants3,4 and is transported from manufacturing facilities in specially reinforced tank cars or trucks. A major accident recently occurred during Br2 transport by train in the city of Chelyabinsk, Russia (population 1.1 million), resulting in 42 people being hospitalized and over 200 individuals seeking medical attention. In another notable environmental accident, approximately 25,000 residents were exposed to Br2 vapors during transfer between containers in a factory in Switzerland in 1988. Furthermore, a terrorist attempt to blow up Br2 storage tanks in Ashdod, Israel, which would have resulted in mass casualties, was barely averted.5 The U.S. Department of Homeland Security considers facilities producing Br2 as at high risk with respect to terrorist attacks. According to their estimates, such an attack would result in approximately 17,500 deaths, 100,000 hospitalizations, and damages of millions of dollars.6
Br2 exposure has no specific antidote, and current treatments for patients exposed to Br2 are mainly supportive, including administration of supplemental oxygen, bronchodilators, and antibiotics in cases of infection. Assisted or supported ventilation with intubation and positive-pressure ventilation may also be necessary. Therefore, there is a clear need for additional research to identify the basic mechanisms by which Br2 and its reactive intermediates cause toxicity. These findings will then better help us develop novel countermeasures to reduce morbidity and mortality.
Systemic effects of Br2 toxicity
Occupational exposure is the most frequent cause of accidental Br2-induced injury. Exposure to Br2 leads to systemic damage to several organs. Brief skin contact with Br2 vapors results in brownish discoloration, tissue necrosis, formation of skin vesicles, and pustules, while prolonged contact leads to deep, painful burns that ulcerate and are slow healing.7 Repeated contact of the skin with Br2 in liquid or vapor form may also cause dermatitis and halogen acne. Br2-based flame retardants may limit the availability of iodine to the thyroid and interfere with hormone production,8–11 thereby potentially leading to hypothyroidism, goiter, and hormonal imbalance. Children exposed to high levels of brominated compounds lag in neural and cognitive development,12 most likely secondary to hypothyroidism. Br2 exposure is also linked to increased risk of preterm delivery13 and birth defects.14 Chronic exposure to Br2 can lead to a condition known as bromism, which is characterized by chronic fatigue, headaches, loss of muscular coordination, and memory defects.15 In adults, exposure to Br2 is also linked to the development of mental illnesses, such as psychosis and schizophrenia.16 Potassium bromate (KBrO3), which is used in water disinfection, and the food and cosmetics industries, is a nephrotoxic compound and a carcinogen.17 In addition, depending upon the dose, the inhalation of Br2 gas can lead to cough, lung injury, pulmonary fibrosis, and death due to respiratory failure.3,18 In our recent study, we demonstrated that exposure to 600 ppm of Br2 for 30 min causes mortality in 50% of C57BL/6 mice within 6–7 days,19 which correlates to the study performed by Bitron and Aharonson in 1978.20
Lung toxicity after bromine inhalation
Inhalation of Br2 results in bronchospasm, airway hyperreactivity (a prelude to asthma), lung edema, and death from respiratory failure. Surviving patients, like those with any form of adult respiratory distress syndrome (ARDS), are at risk of developing pulmonary fibrosis.3 Br2 and hypobromous acid (HOBr), its hydrolysis product, are strong oxidants that react with antioxidants present in the lung epithelial lining fluid, such as ascorbate, urate, and glutathione. Once antioxidant stores are depleted, Br2 and HOBr react with targets on the plasma membranes of lung epithelial cells to form secondary, longer-lasting intermediates, such as brominated lipids, which are capable of transducing injury to distal sites. The resulting inflammatory response amplifies the initial pulmonary and systemic injury. In addition, systemic injury may further amplify lung damage because of the release of inflammatory mediators and possibly xanthine oxidase, which limits nitric oxide bioavailability, as is the case with other forms of systemic injury.21
Our current knowledge of the mechanisms by which Br2 causes toxicity is based on animal models, since the data exploring the effects of Br2 at higher concentrations (levels which are likely to be encountered near sites of industrial accidents) in humans is rare. Mice exposed to 600 ppm of Br2 gas inhalation for 30 min and then returned to room air show significant lung injury within 24 h postexposure, as demonstrated by increased protein extravasation, total cell infiltration into the bronchoalveolar lavage fluid, disruption of the airway parenchyma, and increased lung cellularity.19 In these mice, Br2 gas inhalation also increases lung protein oxidation, airway resistance following methacholine challenge, and lung edema, as indicated by an elevations in the lung wet-to-dry weight ratio.19 A similar increase in pulmonary edema was reported in a patient requiring endotracheal intubation and ventilation for 5 days and hospitalization for 41 days after inadvertently inhaling Br2 vapors.22 In another patient who was admitted to an emergency ward after accidental exposure to Br2 gas, irritation of the airways by Br2 resulted in coughing and transitory respiratory obstruction, eventually culminating in alveolar rupture and mediastinal emphysema.23 Br2 exposure has also been linked to the acute development of pneumonitis and delayed respiratory complications, such as pulmonary fibrosis, in a nonsmoking patient who accidentally inhaled Br2 vapors for 5–10 minutes.24
In our study, we also found that, after Br2 exposure, the average blood pH in C57BL/6 mice drops to 7.1, which corresponded to an increase in the pCO2 to approximately 60 mmHg. These results are consistent with the available data on patients showing respiratory acidosis with a drop in blood pH and a rise in the pCO2 to 52 mmHg immediately after Br2 exposure.23 However, it seems that this change in blood gas may be transient. In a similar patient 3 days after Br2 gas exposure, arterial blood pH was 7.45 and the pCO2 was 28 mmHg, but indices of respiratory function, such as total lung capacity (TLC) and minute forced expiratory volume (FEV1), remained abnormal.24 Furthermore, mice subjected to Br2 inhalation have high mortality. Interestingly, the analysis by Kaplan-Meier survival curve shows that Br2-induced mortality is biphasic (early versus late) in these mice.19 An immediate decline in survival within the first 2 days is followed by a period of 4–5 days with no deaths and a subsequent increase in mortality thereafter.19 This pattern of mortality contrasts with chlorine-induced toxicity, which results in only early mortality,20,25 and therefore suggests a different pattern of lung injury. Owing to bromine’s higher solubility, and therefore its higher potency,26 than chlorine, Br2 is able to penetrate deeper than chlorine into lung tissue and cause peribronchiolar abscess formation.25 Therefore, animals surviving the initial insult may have persistent bronchial spasm and delays in lung healing.25
Role of heme in Br2-induced lung injury
Heme (iron protoporphyrin IX) is essential for diverse biologic processes and serves as the functional group of various proteins, including hemoglobin, myoglobin, nitric oxide synthase, and cytochromes.27 However, these heme moieties are readily oxidized and are a major source of reactive oxygen species.28 Free heme is lipophilic and therefore intercalates into the plasma membrane,28 resulting in oxidative stress and, subsequently, cellular injury29 and impaired cellular integrity.28 During cell damage, heme from the mitochondria and other cellular denatured proteins can also be released and contribute to inflammation.30 These inflammatory responses in the lung lead to increased permeability, the development of edema, and the migration of inflammatory cells from the blood into the lung parenchyma.
There are three major sources of heme: (1) hemoglobin in red blood cells, (2) heme-containing proteins in live cells, and (3) heme released from dead cells into the extracellular space. Mice exposed to Br2 gas have elevated levels of heme in their plasma, bronchoalveolar lavage fluid, and peripheral lung tissue.19 The blood cells obtained from the mice after Br2 exposure are more susceptible to hemolysis compared to the air-exposed mice,19 probably due to the exposure to low pH and higher oxidative stress31 after Br2 inhalation. Some clinical reports also suggest a role for Br2-related compounds, such as bromate, a bromine-based oxoanion, in the development of hemolytic anemia.32 In addition, chlorine-related compounds have also been shown to cause anemia.33 The increases in heme in the bronchoalveolar lavage fluid are possibly derived from the death of inflammatory cells or the sloughing of epithelial cells lining the airways, as evident by the increased number of apoptotic and necrotic cells and elevations in lactate dehydrogenase (LDH) in the bronchoalveolar lavage fluid of exposed mice.19 Similar sloughing of airway epithelial cells has been previously shown after chlorine inhalation in rats,34 pigs,35 and mice.36
In our study, two experimental approaches were investigated to determine if attenuating heme levels can have therapeutic effects on Br2-induced toxicity: scavenging free heme and increasing free heme degradation. In order to scavenge heme, we utilized the heme-scavenging protein, hemopexin, and in order to amplify heme degradation, we utilized transgenic mice overexpressing the heme-degrading enzyme heme oxygenase-1 (HO-1). Hemopexin (Hx), an endogenous intravascular protein with high affinity for heme, effectively nullifies the deleterious effects of excessive plasma heme by transporting intravascular heme to the liver for degradation.37 Furthermore, Hx strongly attenuates heme-stimulated lipid peroxidation, a process not inhibited by the iron chelator desferrioxamine or by apotransferrin.29 Moreover, Hx is stable and not oxidized by heme via interactions with H2O2, while albumin and glutathione S-transferases, which also decrease heme-catalyzed lipid peroxidation, are subject to oxidation.38 Further, the heme–Hx complex at higher concentrations (2–10 μM) exhibits pleiotropic protective effects, increasing cell survival under heme stress39 and enhancing cell adaptation to stress conditions, such as hemolysis, trauma, and ischemia–reperfusion. Hx-deficient mice, when subject to acute hemolysis by an intraperitoneal injection of phenylhydrazine, have high levels of lipid peroxidation and recover more slowly than wild-type littermates.40 A concentration of 5 μM of Hx has been shown to attenuate free radical production in a rat model of reperfusion injury.41 Similarly, in our study, a single injection of Hx (3 μg/g body weight) decreased lung heme levels and lung oxidative stress, ameliorating the associated tissue damage and respiratory dysfunction after Br2 inhalation.19
The presence of elevated heme induces proteins that protect against oxidative stress, such as HO-1, metallothionein-1 (MT-1), and ferritin.42,43 HO-1 catalyzes the first and rate-limiting step in heme degradation into equimolar amounts of iron, carbon monoxide (CO), billiverdin, and bilirubin.44 These catabolic products of heme have been shown to have antioxidant properties. Ferritin, by sequestering iron released from heme, and MTs, by sequestering reactive metals and inactivating hydroxyl radicals and superoxide, also protect cells from toxic effects. Though MT-1 and ferritin levels were not measured, HO-1 protein levels were significantly elevated after Br2 inhalation, which correlate with other studies that show that HO-1 mRNA expression is elevated in pigs exposed to Br2 vapors.4 However, it seems that, even though HO-1 levels are elevated over 24 h after Br2 inhalation, as shown in our study, the increase is not enough to overcome the toxic effects of the initial Br2 insult. Interestingly, when we treat mice with Hx after Br2 exposure, the protein levels of HO-1 do not increase in peripheral lung tissue, suggesting that heme is vital in HO-1 regulation. In fact, some studies have also shown that HO-1 induction is transitory after stress. For example, HO-1 protein levels have been shown to increase within 6 h of cigarette smoke inhalation and eventually decline over 72 h owing to regulation by the mitogen-activated protein kinase (MAPK) pathway.45 Therefore, to determine the protective role of HO-1 in Br2 toxicity, we exposed mice constitutively overexpressing human HO-1 and mice lacking endogenous HO-1 to Br2. Our results suggest that, while HO-1−/− mice are more susceptible to Br2 toxicity, mice overexpressing human heme oxygenase-1 (hHO-1) are mostly protected and have lower oxidative and inflammatory injury, improved respiratory function, and reduced mortality. Surprisingly, even though heme scavenging by Hx did not attenuate the Br2-induced increase in neutrophils in the bronchoalveolar lavage fluid, HO-1 overexpression successfully prevented the increase in neutrophil levels. This result may be explained by the additional anti-inflammatory effects of CO, a catabolite of heme degradation by HO-1.46 HO-1 induction and CO administration have been shown to be protective against airway pathologies, such as asthma,47 pulmonary fibrosis,48 and hypoxia-induced inflammation and hypertension.49 However, some studies suggest that excessive production of HO-1 may have deleterious effects due to increased generation of reactive iron during heme metabolism.50,51 This reactive iron could counter the antioxidant effects of heme degradation by HO-1 and limit the protective effects of HO-1.
Conclusions
In conclusion, generation of heme appears to underlie the pathogenesis of Br2-induced airway injury and toxicity (Fig. 1). Thus, therapeutic approaches that reduce heme levels may prove beneficial countermeasures that can be exploited by emergency room physicians to treat patients in the immediate aftermath of Br2 gas exposure to reduce morbidity and mortality. In this regard, the scavenging of heme by Hx, and to some extent by albumin and heptaglobin, in addition to the modulation of HO-1 enzyme expression by pharmacological inducers, may show promising results (Fig. 1). However, further studies would be required to fully elucidate the complex pathology of Br2 toxicity.
Figure 1.
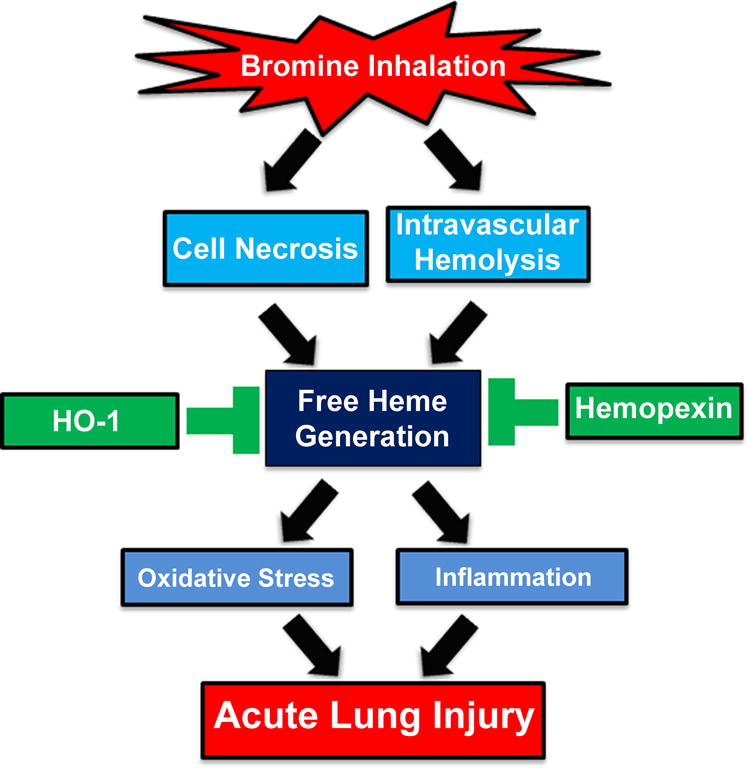
Proposed mechanism of bromine inhalation–induced lung injury. Bromine (Br2) gas inhalation increases intravascular hemolysis and death/necrosis of inflammatory cells and or epithelial cells lining the airways, resulting in elevated heme levels in plasma, bronchoalveolar lavage fluid, and peripheral lung tissue. The lipophilic heme intercalates into the plasma membrane, causing an acute increase in lung oxidative stress, inflammation, and cellular injury. This acute lung injury (ALI) increases lung permeability and impairs respiratory function. The treatment of animals with the heme-scavenging enzyme hemopexin or the induction/upregulation of the heme-degrading protein heme oxygenase-1 (HO-1) lowers lung and plasma heme levels and prevents the development of ALI.
Acknowledgments
Supported by the CounterACT Program, National Institutes of Health Office of the Director (NIH OD), the National Institute of Neurological Disorders and Stroke (NINDS), and the National Institute of Environmental Health Science. Grant Numbers: 5R21 ES024027 02, 1R21ES025423 01, and 1U01ES026458-01A1.
Footnotes
Conflicts of interest
The authors report no conflicts of interest.
References
- 1.Rupp H, Henschler D. Effect of low chlorine and bromine concentrations on man. Int Arch Arbeitsmed. 1967;23:79–90. [PubMed] [Google Scholar]
- 2.Morabia A, et al. Accidental bromine exposure in an urban population: an acute epidemiological assessment. Int J Epidemiol. 1988;17:148–152. doi: 10.1093/ije/17.1.148. [DOI] [PubMed] [Google Scholar]
- 3.Woolf A, Shannon M. Reactive airways dysfunction and systemic complaints after mass exposure to bromine. Environ Health Perspect. 1999;107:507–509. doi: 10.1289/ehp.99107507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rogers JV, et al. An assessment of transcriptional changes in porcine skin exposed to bromine vapor. J Biochem Mol Toxicol. 2011;25:252–262. doi: 10.1002/jbt.20383. [DOI] [PubMed] [Google Scholar]
- 5.Makarovsky I, et al. Bromine–the red cloud approaching. Isr Med Assoc J. 2007;9:677–679. [PubMed] [Google Scholar]
- 6.The Homeland Security Council. Planning Scenarios: Executive Summaries. 2004;004:8-1. https://www.facs.org/~/media/files/quality%20programs/trauma/disaster/wmd_dhs_planning.ashx. Accessed April 8, 2016. [Google Scholar]
- 7.Sagi A, et al. Burns caused by bromine and some of its compounds. Burns Incl Therm Inj. 1985;11:343–350. doi: 10.1016/0305-4179(85)90097-x. [DOI] [PubMed] [Google Scholar]
- 8.Pavelka S. Metabolism of bromide and its interference with the metabolism of iodine. Physiol Res. 2004;53(Suppl 1):S81–90. [PubMed] [Google Scholar]
- 9.Allain P, et al. Bromine and thyroid hormone activity. J Clin Pathol. 1993;46:456–458. doi: 10.1136/jcp.46.5.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Velicky J, et al. Long-term action of potassium bromide on the rat thyroid gland. Acta Histochem. 1998;100:11–23. doi: 10.1016/S0065-1281(98)80003-2. [DOI] [PubMed] [Google Scholar]
- 11.Velicky J, et al. The effect of bromide on the ultrastructure of rat thyrocytes. Ann Anat. 2004;186:209–216. doi: 10.1016/S0940-9602(04)80004-9. [DOI] [PubMed] [Google Scholar]
- 12.Herbstman JB, et al. Prenatal exposure to PBDEs and neurodevelopment. Environ Health Perspect. 2010;118:712–719. doi: 10.1289/ehp.0901340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horton BJ, et al. The effect of water disinfection by-products on pregnancy outcomes in two southeastern US communities. J Occup Environ Med. 2011;53:1172–1178. doi: 10.1097/JOM.0b013e31822b8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chisholm K, et al. Risk of birth defects in Australian communities with high levels of brominated disinfection by-products. Environ Health Perspect. 2008;116:1267–1273. doi: 10.1289/ehp.10980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horowitz BZ. Bromism from excessive cola consumption. J Toxicol Clin Toxicol. 1997;35:315–320. doi: 10.3109/15563659709001219. [DOI] [PubMed] [Google Scholar]
- 16.Levin M. Transitory schizophrenias produced by bromide intoxication. Am J Psychiatry. 1946;103:229–237. doi: 10.1176/ajp.103.2.229. [DOI] [PubMed] [Google Scholar]
- 17.Geter DR, et al. Kidney toxicogenomics of chronic potassium bromate exposure in f344 male rats. Transl Oncogenomics. 2006;1:33–52. [PMC free article] [PubMed] [Google Scholar]
- 18.Burns MJ, Linden CH. Another hot tub hazard. Toxicity secondary to bromine and hydrobromic acid exposure. Chest. 1997;111:816–819. doi: 10.1378/chest.111.3.816. [DOI] [PubMed] [Google Scholar]
- 19.Aggarwal S, et al. Heme Attenuation Ameliorates Irritant Gas Inhalation-Induced Acute Lung Injury. Antioxid Redox Signal. 2016;24:99–112. doi: 10.1089/ars.2015.6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bitron MD, Aharonson EF. Delayed mortality of mice following inhalation of acute doses of CH2O, SO2, Cl2, and Br2. Am Ind Hyg Assoc J. 1978;39:129–138. doi: 10.1080/0002889778507726. [DOI] [PubMed] [Google Scholar]
- 21.Weinbroum A, et al. Liver ischemia-reperfusion increases pulmonary permeability in rat: role of circulating xanthine oxidase. Am J Physiol. 1995;268:G988–996. doi: 10.1152/ajpgi.1995.268.6.G988. [DOI] [PubMed] [Google Scholar]
- 22.Inagaki N, et al. Case with bromine exposure leading to respiratory insufficiency. Chudoku Kenkyu. 2005;18:141–147. [PubMed] [Google Scholar]
- 23.Lossos IS, Abolnik I, Breuer R. Pneumomediastinum: a complication of exposure to bromine. Br J Ind Med. 1990;47:784. doi: 10.1136/oem.47.11.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kraut A, Lilis R. Chemical pneumonitis due to exposure to bromine compounds. Chest. 1988;94:208–210. doi: 10.1378/chest.94.1.208. [DOI] [PubMed] [Google Scholar]
- 25.Schlagbauer M, Henschler D. Toxicity of chlorine and bromine in single and repeated inhalation. Int Arch Arbeitsmed. 1967;23:91–98. [PubMed] [Google Scholar]
- 26.Snider TH, et al. A dynamic system for delivering controlled bromine and chlorine vapor exposures to weanling swine skin. Cutan Ocul Toxicol. 2014;33:161–167. doi: 10.3109/15569527.2013.806524. [DOI] [PubMed] [Google Scholar]
- 27.Ryter SW, et al. Heme oxygenase/carbon monoxide signaling pathways: regulation and functional significance. Mol Cell Biochem. 2002;234–235:249–263. doi: 10.1023/A:1015957026924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryter SW, Tyrrell RM. The heme synthesis and degradation pathways: role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic Biol Med. 2000;28:289–309. doi: 10.1016/s0891-5849(99)00223-3. [DOI] [PubMed] [Google Scholar]
- 29.Gutteridge JM, Smith A. Antioxidant protection by haemopexin of haem-stimulated lipid peroxidation. Biochem J. 1988;256:861–865. doi: 10.1042/bj2560861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wagener FA, et al. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood. 2001;98:1802–1811. doi: 10.1182/blood.v98.6.1802. [DOI] [PubMed] [Google Scholar]
- 31.Perrone S, et al. Oxidative injury in neonatal erythrocytes. J Matern Fetal Neonatal Med. 2012;25:104–108. doi: 10.3109/14767058.2012.715471. [DOI] [PubMed] [Google Scholar]
- 32.Gradus D, et al. Acute bromate poisoning associated with renal failure and deafness presenting as hemolytic uremic syndrome. Am J Nephrol. 1984;4:188–191. doi: 10.1159/000166804. [DOI] [PubMed] [Google Scholar]
- 33.de Oliveira RM, et al. Warning: an anemia outbreak due to chloramine exposure in a clean hemodialysis unit–an issue to be revisited. Ren Fail. 2009;31:81–83. doi: 10.1080/08860220802546503. [DOI] [PubMed] [Google Scholar]
- 34.Demnati R, et al. Histopathological effects of acute exposure to chlorine gas on Sprague-Dawley rat lungs. J Environ Pathol Toxicol Oncol. 1995;14:15–19. [PubMed] [Google Scholar]
- 35.Gunnarsson M, et al. Exposure to chlorine gas: effects on pulmonary function and morphology in anaesthetised and mechanically ventilated pigs. J Appl Toxicol. 1998;18:249–255. doi: 10.1002/(sici)1099-1263(199807/08)18:4<249::aid-jat507>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 36.Tian X, et al. Acute lung injury induced by chlorine inhalation in C57BL/6 and FVB/N mice. Inhal Toxicol. 2008;20:783–793. doi: 10.1080/08958370802007841. [DOI] [PubMed] [Google Scholar]
- 37.Vinchi F, et al. Hemopexin therapy improves cardiovascular function by preventing heme-induced endothelial toxicity in mouse models of hemolytic diseases. Circulation. 2013;127:1317–1329. doi: 10.1161/CIRCULATIONAHA.112.130179. [DOI] [PubMed] [Google Scholar]
- 38.Vincent SH, et al. The influence of heme-binding proteins in heme-catalyzed oxidations. Arch Biochem Biophys. 1988;265:539–550. doi: 10.1016/0003-9861(88)90159-2. [DOI] [PubMed] [Google Scholar]
- 39.Eskew JD, et al. Cellular protection mechanisms against extracellular heme. heme-hemopexin, but not free heme, activates the N-terminal c-jun kinase. J Biol Chem. 1999;274:638–648. doi: 10.1074/jbc.274.2.638. [DOI] [PubMed] [Google Scholar]
- 40.Tolosano E, et al. Defective recovery and severe renal damage after acute hemolysis in hemopexin-deficient mice. Blood. 1999;94:3906–3914. [PubMed] [Google Scholar]
- 41.Brass CA, et al. Hemopexin decreases spontaneous chemiluminescence of cold preserved liver after reperfusion. Biochem Biophys Res Commun. 1998;248:574–577. doi: 10.1006/bbrc.1998.9023. [DOI] [PubMed] [Google Scholar]
- 42.Alam J, Smith A. Receptor-mediated transport of heme by hemopexin regulates gene expression in mammalian cells. J Biol Chem. 1989;264:17637–17640. [PubMed] [Google Scholar]
- 43.Alam J, Smith A. Heme-hemopexin-mediated induction of metallothionein gene expression. J Biol Chem. 1992;267:16379–16384. [PubMed] [Google Scholar]
- 44.Tenhunen R, Marver HS, Schmid R. Microsomal heme oxygenase. Characterization of the enzyme. J Biol Chem. 1969;244:6388–6394. [PubMed] [Google Scholar]
- 45.Goven D, et al. Prolonged cigarette smoke exposure decreases heme oxygenase-1 and alters Nrf2 and Bach1 expression in human macrophages: roles of the MAP kinases ERK(1/2) and JNK. FEBS Lett. 2009;583:3508–3518. doi: 10.1016/j.febslet.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 46.Otterbein LE, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. 2000;6:422–428. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- 47.Chapman JT, et al. Carbon monoxide attenuates aeroallergen-induced inflammation in mice. Am J Physiol Lung Cell Mol Physiol. 2001;281:L209–216. doi: 10.1152/ajplung.2001.281.1.L209. [DOI] [PubMed] [Google Scholar]
- 48.Tsuburai T, et al. Adenovirus-mediated transfer and overexpression of heme oxygenase 1 cDNA in lung prevents bleomycin-induced pulmonary fibrosis via a Fas-Fas ligand-independent pathway. Hum Gene Ther. 2002;13:1945–1960. doi: 10.1089/10430340260355356. [DOI] [PubMed] [Google Scholar]
- 49.Minamino T, et al. Targeted expression of heme oxygenase-1 prevents the pulmonary inflammatory and vascular responses to hypoxia. Proc Natl Acad Sci U S A. 2001;98:8798–8803. doi: 10.1073/pnas.161272598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kvam E, et al. Heme oxygenase activity causes transient hypersensitivity to oxidative ultraviolet A radiation that depends on release of iron from heme. Free Radic Biol Med. 2000;28:1191–1196. doi: 10.1016/s0891-5849(00)00205-7. [DOI] [PubMed] [Google Scholar]
- 51.Suttner DM, Dennery PA. Reversal of HO-1 related cytoprotection with increased expression is due to reactive iron. FASEB J. 1999;13:1800–1809. doi: 10.1096/fasebj.13.13.1800. [DOI] [PubMed] [Google Scholar]