

Abstract
Hormonal regulation of gene expression by androgen receptor (AR) is tightly controlled by many transcriptional cofactors, including pioneer factors FOXA1 and GATA2, which, however, exhibit distinct expression patterns and functional roles in prostate cancer. Here, we examined how FOXA1, GATA2, and AR crosstalk and regulate hormone-dependent gene expression in prostate cancer cells. ChIP-seq analysis revealed that FOXA1 reprograms both AR and GATA2 cistrome by preferably recruiting them to FKHD-containing genomic sites. By contrast, GATA2 is unable to shift AR or FOXA1 to GATA motifs. Rather, GATA2 co-occupancy enhances AR and FOXA1 binding to nearby ARE and FKHD sites, respectively. Similarly, AR increases, but not re-programs, GATA2 and FOXA1 cistromes. Concordantly, GATA2 and AR strongly enhance the transcriptional program of each other, whereas FOXA1 regulates GATA2- and AR-mediated gene expression in a context-dependent manner due to its reprogramming effects. Taken together, our data delineated for the first time the distinct mechanisms by which GATA2 and FOXA1 regulate AR cistrome and suggest that FOXA1 acts upstream of GATA2 and AR in determining hormone-dependent gene expression in prostate cancer.
Keywords: ChIP-seq, transcriptional regulation, cistrome, androgen receptor, pioneer factor
INTRODUCTION
The hormonal transcription factor androgen receptor (AR) is critical for prostate epithelial cell differentiation. Chromatin binding and transcriptional regulation by AR dictates the prostate-specific gene expression pattern1, 2. This genomic action of AR is in turn regulated by a large number of collaborating transcription factors, among which is FOXA1, a pioneer factor that possesses the ability to engage with closed chromatin, move nucleosomes, and ultimately allow subsequent binding of other transcription factors3. FOXA1 defines prostate lineage-specific AR cistrome through binding to enhancers marked with H3K4 mono- and di-methylations2, 4. Recently, we and others have demonstrated that, due to its pioneering effects, FOXA1 is capable of reprogramming AR by altering local chromatin accessibility5–7. AR binding sites in the FOXA1-knockdown cells were enriched for ARE motifs, whereas AR binding in FOXA1-expressing cells were mediated by FKHD motifs6. FOXA1 thus appears to inhibit AR binding to the high-affinity ARE-containing regions by recruiting AR to form weaker binding at FKHD sites. Consequently, FOXA1 knockdown leads to stronger AR binding events, especially under androgen-depleted conditions, thereby contributing to prostate cancer progression to castration resistance6. Due to its dual roles in functioning as a collaborating factor as well as a re-programming factor of AR, FOXA1 has been shown to exhibit both oncogenic and tumor suppressive roles in prostate cancer, probably due to the different contexts under investigation6–10.
Similar to FOXA1, GATA family proteins have also been suggested to act as pioneer factors11, 12. GATA1, for instance, is important for differentiation of erythrocytes and megakaryocytes13–15, and has been shown capable of disrupting nucleosome structures and thus generating nuclease hypersensitive sites16. On the other hand, GATA4 has been demonstrated to have the ability of initiating chromatin opening in endoderm cells or precursors to the liver17. Among the six members of the GATA family, GATA2 was recently shown to have the highest expression in prostate cancer18, 19. GATA2 increases AR expression level and displays a strong positive correlation with AR level in prostate cancer18, 19. High GATA2 expression and transcriptional activity has been strongly linked to poor clinical outcome in prostate cancer patients18. In contrast to the arguable roles of FOXA1 in prostate cancer progressioin, GATA2 has been consistently shown to induce prostate tumorigenicity and chemotherapy resistance20.
It is however unclear how FOXA1 and GATA2, both thought to be AR pioneer factors, would exhibit such distinct effects on AR signaling and prostate cancer progression. In addition, the hierarchical regulatory network involving FOXA1, GATA2 and AR in determining hormone-dependent gene expression in prostate cancer is yet to be elucidated. Here, we demonstrated that, while FOXA1 is a pioneer factor that reprograms and inhibits AR cistrome, GATA2 functions as a transcription co-activator that enhances AR signaling. In addition, we found that FOXA1 is not only capable of reprogramming AR but also GATA2, thus acting as a pioneering factor for both. Taken together, our data suggest a hierarchical network of transcription regulation underpinned by FOXA1 that controls AR-mediated gene expression program in prostate cancer.
RESULTS
GATA2 enhances, but not reprograms, AR cistrome
Both FOXA1 and GATA2 were thought pioneer cofactors of AR. We and others have recently demonstrated that FOXA1 reprograms AR cistrome by recruiting AR from ARE-only sites to FKHD sites6, 7. However, how GATA2 regulates AR cistromes has not been carefully investigated. To address this, we first performed comparative genome-wide location analyses of all three transcription factors in LNCaP cells, and found that substantially more AR binding sites (ARBS) are co-occupied by FOXA1 than GATA2 (Figure 1A). Approximately 25% of ARBS (c+d sites in Figure 1A) were co-occupied by GATA2, whereas up to 75% of ARBS (b+d) were co-bound by FOXA1. Moreover, a majority (86%, d) of GATA2-AR co-occupied loci were also bound by FOXA1, leaving only 3% of ARBS (c) co-occupied by GATA2 alone. These data suggest that, unlike FOXA1, GATA2 alone is probably insufficient in recruiting AR. Concordantly, motif analysis demonstrated that ARE is only weakly enriched in ARBS co-occupied by FOXA1, either alone (15%, b) or together with GATA2 (14%, d), suggesting the strong ability of FOXA1 in recruiting AR to chromatin regions lacking full AREs (Figure 1B). By contrast, ARE remains highly prevalent, and thus may be necessary, in the ARBS co-occupied by GATA2 only (c sites). Our data thus suggest major differences between FOXA1 and GATA2 regulation of AR cistrome; unlike FOXA1, GATA2 may be unable to reprogram AR.
Figure 1. GATA2 enhances but not reprograms AR cistrome.
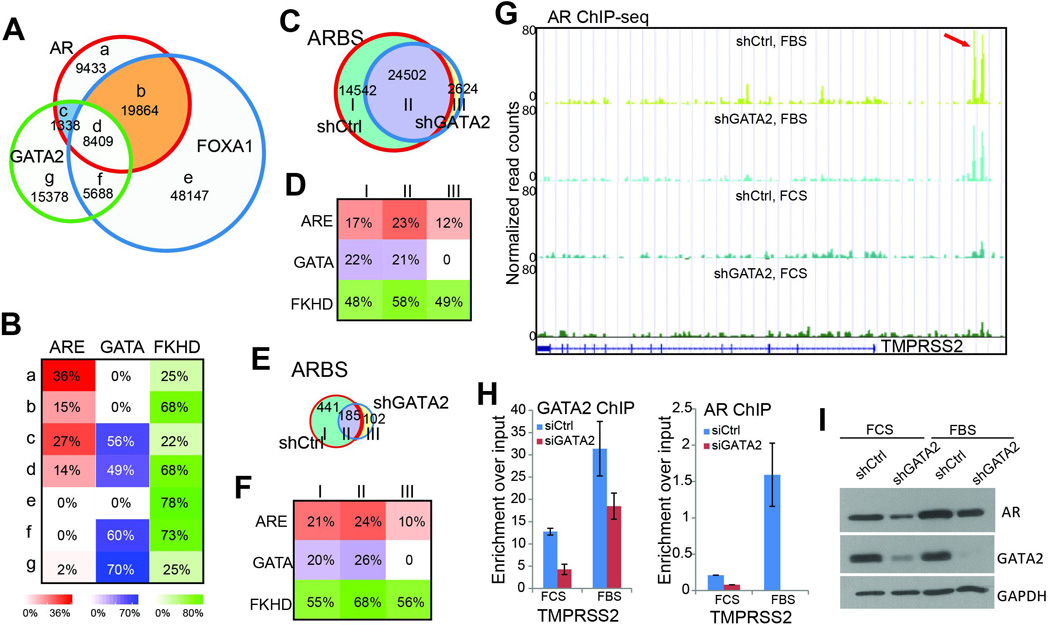
A. GATA2 co-occupies substantially less ARBS (only 25%) than FOXA1 (which occupies 75% of ARBS), and a majority of the GATA2-AR co-occupied regions are also bound by FOXA1. ChIP-seq of AR, FOXA1, and GATA2 were performed in LNCaP cells.
B. FOXA1, but not GATA2, plays a strong role in recruiting AR. Motif analysis showed that ARE motifs are the most enriched in the AR-only sites (a sites). ARE motifs are much less frequent within FOXA1-coocupied sites (b and d sites) than GATA2-coocupied sites (c sites).
C. Venn diagram showing overlapping ARBS identified in control (shCtrl) and GATA2 knockdown (shGATA2) LNCaP cells in the presence of androgen.
D. Motif enrichment in the three categories of ARBS identified in C.
E–F. Venn diagram (E) and motif analysis of ARBS identified in control (shCtrl) and GATA2 knockdown (shGATA2) LNCaP cells in the absence of androgen.
G. Genome browser view of AR occupancy around the TMPRSS2 gene in shCtrl and shGATA2 LNCaP cells with (FBS) or without (FCS) androgen stimulation. Red arrow indicates the position of the known TMPRSS2 enhancer11.
H. ChIP-PCR showed that AR-induced gene TMPRSS2 are less occupied by AR following GATA2 knockdown both in the presence and absence of androgen. Data shown are mean ± SEM in triplicate qPCR and is a representative of at least two independent experiments.
I. Western blot analysis of control and shGATA2 LNCaP cells in the presence and absence of androgen.
To understand how GATA2 regulates AR binding profile, we carried out GATA2 knockdown in LNCaP cells. ChIP-seq confirmed substantially reduced number of GATA binding sites (GTBS) following GATA2 depletion (Figure S1A). Importantly, we found that the number of ARBS was also significantly reduced (Figure 1C). There were very few gained ARBS, which contains no GATA motif and have reduced, rather than increased, enrichment for ARE, likely representing experimental variability (Figure 1D). This is in great contrast to the substantial amount of newly gained, ARE-mediated ARBS following FOXA1 KD as recently reported5–7. As we previously observed the greatest AR-reprogramming effects of FOXA1 under hormone-depleted conditions6, analogously we performed GATA2 knockdown in hormone-deprived LNCaP cells (Figure S1B). As expected, there were very few ARBS in the absence of androgen and, importantly, GATA2 knockdown further reduced more than 50% of them. Similarly, the few new ARBS were not enriched for ARE motif (Figure 1E–F). In contrast, we have previously shown that FOXA1 knockdown led to nearly 4 fold increase of ARBS, strongly enriched for ARE motif, even in the absence of androgen6.
To validate that GATA2 knockdown attenuates AR binding events, we examined AR binding peaks at the known enhancers of androgen-induced gene PSA and TMPRSS2. Genome browser view demonstrated that AR ChIP enrichment was the highest in the presence of both androgen and GATA and was decreased by GATA2 knockdown both in the presence (FBS) and absence (FCS) of androgen (Figure 1G & S1C). ChIP-qPCR further confirmed that GATA2 knockdown substantially reduced AR binding to TMPRSS2 and PSA enhancers in the presence and absence of androgen (Figure 1H & S1D). This striking decrease of AR binding events following GATA2 knockdown could not be fully explained by the relatively small amount of overlap between GTBS and ARBS. In addition, a study has very recently reported that GATA2 depletion decreases AR expression18. Indeed, western blot analysis demonstrated remarkable decrease in AR protein level following GATA2 knockdown (Figure 1I). Taken together, our data revealed distinct mechanisms by which GATA2 and FOXA1 regulate AR cistrome.
AR increases GATA2 and FOXA1 binding to the chromatin
As GATA2-mediated transcriptional regulation has recently been shown to play important roles in prostate tumorigenesis20, we next asked whether AR might also enhance GATA2 function, forming a potential feed-forward loop further contributing to prostate cancer progression. We first performed western blotting of LNCaP cells grown in the presence (FBS) or absence of androgen (FCS). Our data demonstrated that, as previously reported21, androgen stimulation drastically increased AR protein level in LNCaP cells. Interestingly, GATA2 protein level was slightly decreased in the presence of androgen (Figure 2A). ChIP-seq analysis showed that, as expected, there are many more AR binding events in the presence of androgen (Figure S2A). Importantly, despite the significant decrease of GATA2 protein, ChIP-seq analysis revealed an increase in total number of GTBS and a clear shift of GTBS to ARE-enriched regions upon androgen stimulation (Figure 2B–C). This suggests that AR occupancy on the chromatin might facilitate GATA2 binding to the same sites. Indeed, heatmap view of ChIP-seq data demonstrated that AR is much more enriched at conserved (category II) and gained GTBS (III) and AR enrichment were further enhanced under FBS conditions (Figure 2D).
Figure 2. AR co-occupancy potentiates GATA2 binding on the chromatin.
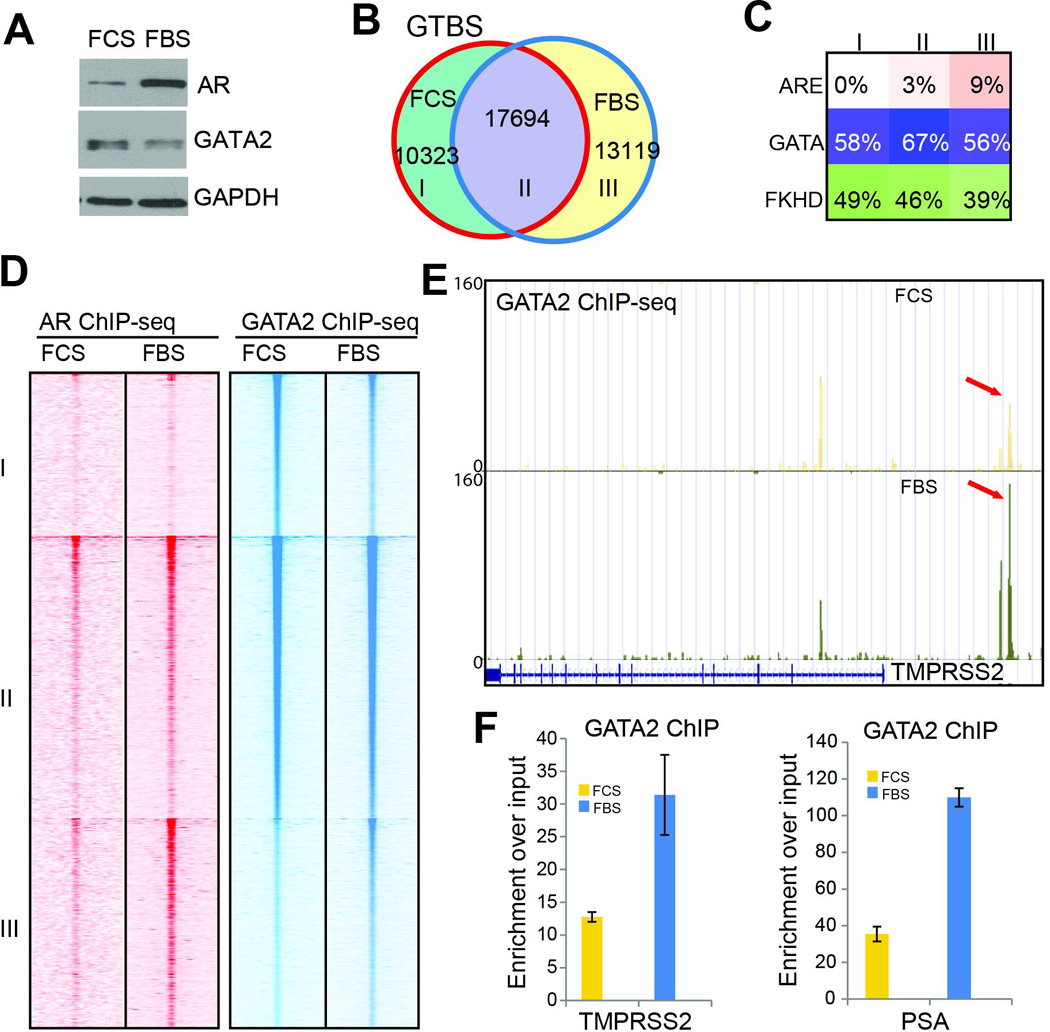
A. Western Blot analysis of GATA2 and AR in LNCaP cells in the presence (FBS) and absence (FCS) of androgen.
B. Venn diagram showing overlap between GATA binding sites in androgen-depleted and androgen-stimulated LNCaP cells.
C. Motif analysis of ARE, GATA and FKHD enrichment in the three categories of GTBS as shown in B.
D. Heatmap of AR and GATA2 ChIP-seq read intensity around the three categories of GTBS identified in B.
E. Genome browser view showing increased GATA2 binding at the TMPRSS2 gene enhancer in androgen-stimulated cells.
F. ChIP-PCR showing GATA2 enrichment at the TMPRSS2 and PSA enhancers in LNCaP cells in the absence (FCS) or presence of androgen (FBS). Data shown are mean ± SEM in triplicate qPCR and is a representative of at least two independent experiments.
To confirm the collaborative role of AR in regulating GATA2 binding events, we further examined the positive control gene TMPRSS2. Genome browser view of ChIP-seq peaks showed that, despite the decrease in total GATA2 protein level, its occupancy at the TMPRSS2 enhancers was increased by nearly two fold in FBS compared to FCS (Figure 2E). Moreover, ChIP-qPCR analysis confirmed that GATA2 binding at AR-induced genes such as TMPRSS2 and PSA was drastically enhanced by androgen (Figure 2F & S2B). These data strongly support that GATA2 binding on the genome could be enhanced by AR co-occupancy.
Next, we asked whether AR could similarly regulate FOXA1 binding on the chromatin. ChIP-seq analysis of FOXA1 showed that FOXA1 pre-occupies target genomic regions in the absence of androgen (Figure S2C), similar as previously reported2. Androgen stimulation shifted some FOXA1 to new regions and also slightly increased the number of FOXA1 binding sites (FXBS), as in the case of GATA2. In addition, motif analysis revealed that the gained FXBS are much more enriched for ARE (Figure S2D). To exclude the possibility that these changes are due to experimental variability, we performed another FOXA1 ChIP-seq under hormone-deprived conditions. Data analysis revealed that ~76% of FXBS was conserved between duplicate experiments (Figure S2E). Moreover, by comparing peaks identified in this replicate experiment with FXBS identified in hormone-replenished cells we again found that androgen treatment slightly increased the number of FXBS (Figure S2F). Therefore, similar to GATA2, FOXA1 genomic occupancy can also be enhanced by AR. This may be a general rule governing co-occupying transcription factors. Taken together, we have shown that GATA2 and AR act as collaborating factors that enhance chromatin occupancy of each other, forming a positive feedback loop.
Collaborative effects of GATA2 and AR in transcriptional regulation
Next, we asked whether the collaborative roles of AR and GATA2 lead to corresponding expressional regulation of their target genes. To determine whether GATA2 also enhances AR-mediated transcriptional program, we performed gene set enrichment analysis (GSEA) of androgen-induced and –repressed gene sets derived from LNCaP cells following androgen stimulation as previously described22. Through analysis of microarray data profiling gene expression in control and GATA2 knockdown LNCaP cells, we found that androgen-induced genes are markedly decreased upon GATA2 knockdown, whereas androgen-repressed genes increased (Figure S3A), which is consistent with findings in a recent study18. As we have found that depletion of GATA2 further reduced ARBS in androgen-deprived cells as shown in Figure 1D, we examined GATA2 regulation of AR signaling in the absence of androgen. GSEA demonstrated that androgen-induced genes were further down-regulated, while androgen-repressed genes increased following GATA2 knockdown in androgen-deprived LNCaP cells (Figure S3B). Therefore, being consistent with the ability of GATA2 in increasing AR protein level and enhancing AR chromatin binding, GATA2 positively regulates AR signaling both under androgen-depleted and androgen-replenished conditions. However, this is in great contrast to FOXA1, which we have recently shown to increase AR signaling in the presence of androgen, but inhibits it in the absence of androgen6.
Next, we asked how androgen regulates GATA2 and FOXA1-mediated transcriptional programs. To discern any context-dependent effects, we obtained GATA2-induced and –repressed gene sets through microarray analysis of GATA2-knockdown in the presence and absence of androgen. GSEA revealed that, regardless of the androgen environment, GATA2-induced genes were significantly enriched for up-regulation by androgen (Figure 3A–B), whereas GATA2-repressed genes were further down-regulated following androgen treatment (Figure S3C–D). By contrast, the genes induced by FOXA1 in androgen-depleted LNCaP cells were significantly enriched for down-regulation by androgen (Figure 3C), whereas those induced by FOXA1 in androgen-replenished cells were up-regulated by androgen (Figure 3D), being consistent with its context-dependent roles as a pioneer factor22. Heatmap view of GATA2-induced and –repressed genes further illustrated the collaborative roles of GATA2 and androgen in regulating gene regulation (Figure 3E). Moreover, qRT-PCR analysis confirmed that GATA2 depletion decreased the expression of AR-induced genes such as PSA and KLK2, while restoring AR-repressed gene NOV (Figure 3F). Therefore, FOXA1 and GATA2 play distinct roles in their regulation of AR-mediated transcriptional program; Unlike FOXA1, GATA2 acts as a collaborating transcription factor, rather than a reprogramming factor, of AR.
Figure 3. Collaborative effects of GATA2 and AR in transcriptional regulation.
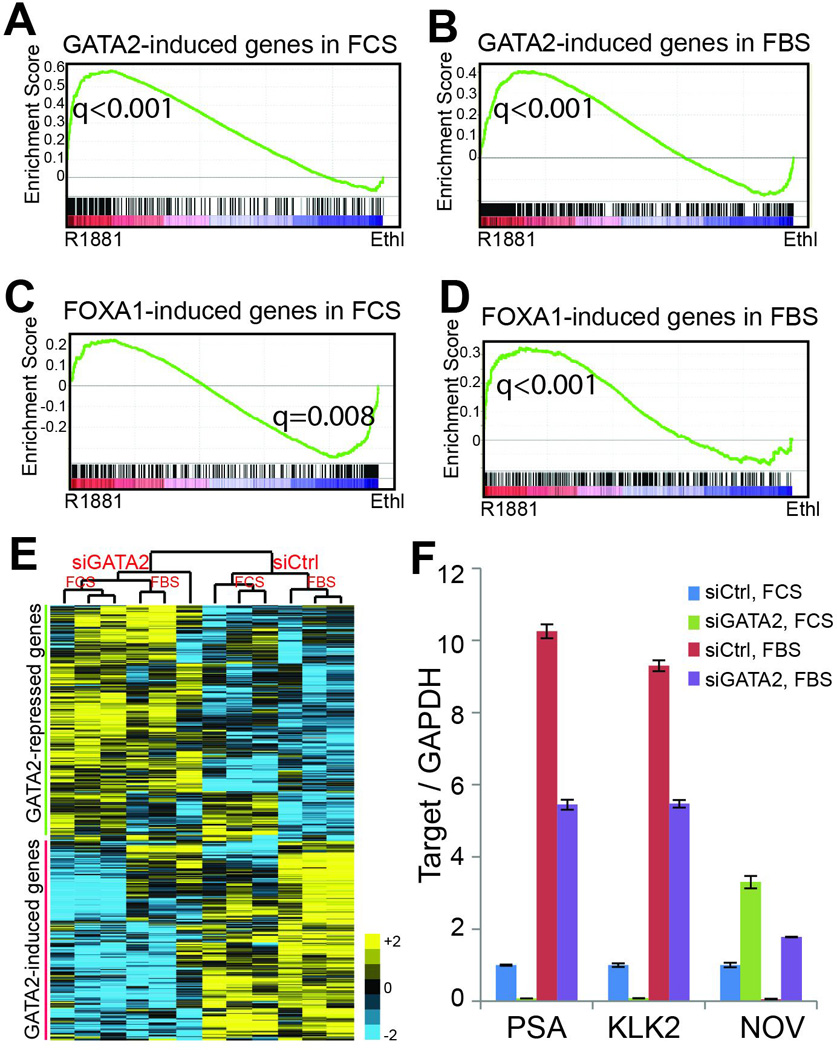
A–B. Genes induced by GATA2 in androgen-depleted (A) or androgen-stimulated cells (B) are up-regulated by androgen. GATA2-induced gene sets were obtained through microarray analysis of control and GATA2-knockdown LNCaP cells grown in the presence or absence of androgen. GSEA analysis was done in microarray dataset comparing gene expression in LNCaP cells with or without androgen stimulation.
C–D. Genes induced by FOXA1 in androgen-depleted LNCaP cells are significantly enriched for down-regulation by androgen (C), while FOXA1-induced genes in the presence of androgen tend to be up-regulated by androgen (D). FOXA1-induced gene sets were obtained through microarray analysis of control and FOXA1-knockdown LNCaP cells grown under androgen-depleted or –replenished conditions and subjected to GSEA analysis.
E. Hierarchical clustering showing that GATA2-induced genes in the absence of androgen are further induced by androgen, whereas GATA2-repressed genes are further repressed by androgen. GATA2-induced or –repressed gene sets were derived from microarray data of control and GATA2-knockdown LNCaP cells in the absence of androgen.
F. QRT-PCR confirming that GATA2 positively regulates AR activity under both androgen-depleted and –replenished conditions. LNCaP cells grown in the presence or absence of androgen were subjected to control or GATA2 knockdown and then qRT-PCR analysis. PSA and KLK2 are known AR-induced genes, whereas NOV has been reported to be an AR-repressed gene29. Data shown are mean ± SEM in triplicate qPCR and is a representative of at least two independent experiments.
FOXA1 acts as a pioneer factor that reprograms GATA2
The effect of FOXA1 as a pioneer factor appears unique, in that it alters chromatin accessibility to re-distribute AR, reduce AR binding to high-affinity ARE sites, and dilute AR across the genome, overall attenuating AR binding events6. We next asked whether FOXA1 might regulate other transcription factors such as GATA2 in a similar fashion. We first conducted RNA interference of FOXA1 in androgen-deprived and –replenished LNCaP cells. Western blot analysis confirmed successful FOXA1 knockdown and demonstrated a clear decrease of GATA2 protein level, mainly in the presence of androgen (Figure 4A). To determine how FOXA1 regulates GATA2 cistrome, we first performed GATA2 ChIP-seq in control and FOXA1-knockdown cells in the absence of androgen. Data analysis revealed remarkably increased number of GTBS following FOXA1 knockdown; 30,546 new GTBS were identified, while only 8,162 GTBS were lost (Figure 4B). Further, heatmap view of ChIP-seq read intensity showed greatly increased GATA2 enrichment at the gained as well as conserved GTBS (Figure 4C). Average read intensity plots demonstrated that the gained GTBS were in average much stronger than the lost ones and that the conserved GTBS were enhanced by nearly 2 fold (Figure 4D). To investigate what mediates each type of GATA2 binding events we performed motif analysis. Our data showed that the lost GTBS (category I) were strongly enriched for FKHD motif, whereas the conserved (II) and gained GTBS (III) were mediated largely by GATA2 motif (Figure 4E), supporting that FOXA1 depletion led to a shift of GTBS from FKHD- to GATA-containing regions.
Figure 4. FOXA1 acts as a pioneer factor that reprograms GATA2 cistrome.
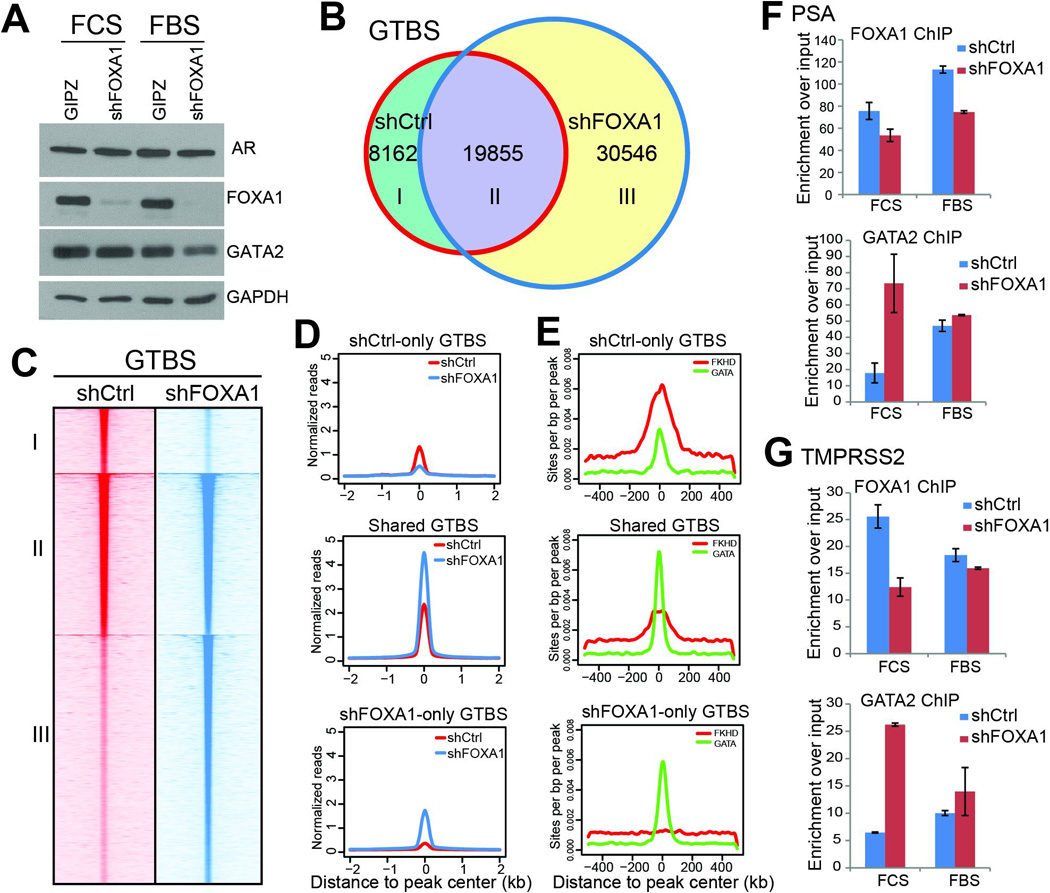
A. Western blot analysis showing a slight decrease of GATA2 protein level following FOXA1 knockdown, especially in the presence of androgen.
B. Venn diagram showing greatly increased GTBS following FOXA1 knockdown. Control and FOXA1-knockdown (shFOXA1) LNCaP cells were hormone-deprived and subjected to GATA2 ChIP-seq.
C–D. Heatmap view (C) and average intensity plot (D) of GATA2 ChIP-seq read intensity around the three categories of GTBS identified in B.
E. FKHD and GATA motif intensity around the three categories of GTBS identified in B.
F–G. ChIP-PCR confirming increased GATA2 binding to target genes PSA (F) and TMPRSS2 (G) following FOXA1 knockdown. Control and FOXA1-knockdown LNCaP cells in the absence or presence of androgen were subjected to FOXA1 and GATA2 ChIP followed by qPCR analysis. Data shown are mean ± SEM in triplicate qPCR and is a representative of at least two independent experiments.
We next tried to confirm this regulatory mechanism in cells maintained in the presence of androgen, albeit the regulatory pathway may be confounded by AR co-activating effects. Interestingly, although GATA2 protein level was dramatically decreased by FOXA1 knockdown, ChIP-seq revealed only 10% decrease in the total number of GTBS (Figure S4A). In addition, the conserved (II) and gained GTBS (III) were both strongly enhanced upon FOXA1 depletion (Figure S4B–C). There was also a clear shift of GATA2 binding from FKHD-containing sites to genomic regions that harbor the GATA motif, supporting FOXA1 being a pioneer factor of GATA2 even in the presence of androgen (Figure S4D).
To validate our observations from genomic data, we performed ChIP-qPCR analysis of several known GATA2-bound genes. Our data confirmed that, upon FOXA1 knockdown and reduced FOXA1 binding, GATA2 occupancy at PSA and TMPRSS2 enhancers was strikingly augmented, especially under androgen-depleted conditions (Figure 4F–G). Altogether, our results show that FOXA1 shifts GATA2 binding from GATA motif to FKHD motif and overall attenuates GATA2 binding events, which is very similar to the way FOXA1 regulates AR cistrome, as we have detailed in a recent paper6. Therefore, FOXA1 could act as a pioneer factor to reprogram not only AR, but also GATA2.
GATA2 is a FOXA1 collaborating factor but does not reprogram FOXA1
We next investigated how GATA2 regulates the genomic landscapes of FOXA1. By western blot analysis, we did not observe significant alterations in FOXA1 protein levels following GATA2 knockdown in both presence and absence of androgen (Figure 5A). To determine how GATA2 regulates FOXA1 cistrome, we first conducted FOXA1 ChIP-seq in control and GATA2-knockdown LNCaP cells in the absence of androgen, in order to preclude confounding effects caused by AR. Interestingly, ChIP-seq showed dramatically decreased number of FOXA1 binding sites following GATA2 depletion; over 23,000 FXBS were lost whereas only about 10,000 new sites were gained (Figure 5B). The decrease in FOXA1 binding was accompanied by reduced GATA2 ChIP-seq read intensities, suggesting that GATA2 co-occupancy might have augmented FOXA1 chromatin binding (Figure 5C). While as expected GATA motif was no longer enriched in the gained (shGATA2-only, III) FXBS, the occurrence of FKHD motif was also not significantly increased, ruling out potential FKHD-mediated reprogramming (Figure 5D). As an example, genome browser view depicted reduced FOXA1 binding at the TMPRSS2 enhancer following GATA2 knockdown and decreased GATA2 binding (Figure 5E). To validate this, ChIP-qPCR was performed and confirmed a decrease of GATA2 binding at the PSA and TMPRSS2 enhancers following GATA2 knockdown (Figure S5). Interestingly, FOXA1 ChIP-qPCR further validated the decrease in FOXA1 occupancy at these enhancers in both androgen-depleted and –replenished cells, supporting GATA2 as a collaborating transcription factor of FOXA1 (Figure 5F). Therefore, our results suggest that, unlike FOXA1 regulation of GATA2, GATA2 is not able to reprogram FOXA1.
Figure 5. GATA2 is a collaborating, but not reprogramming, factor of FOXA1.
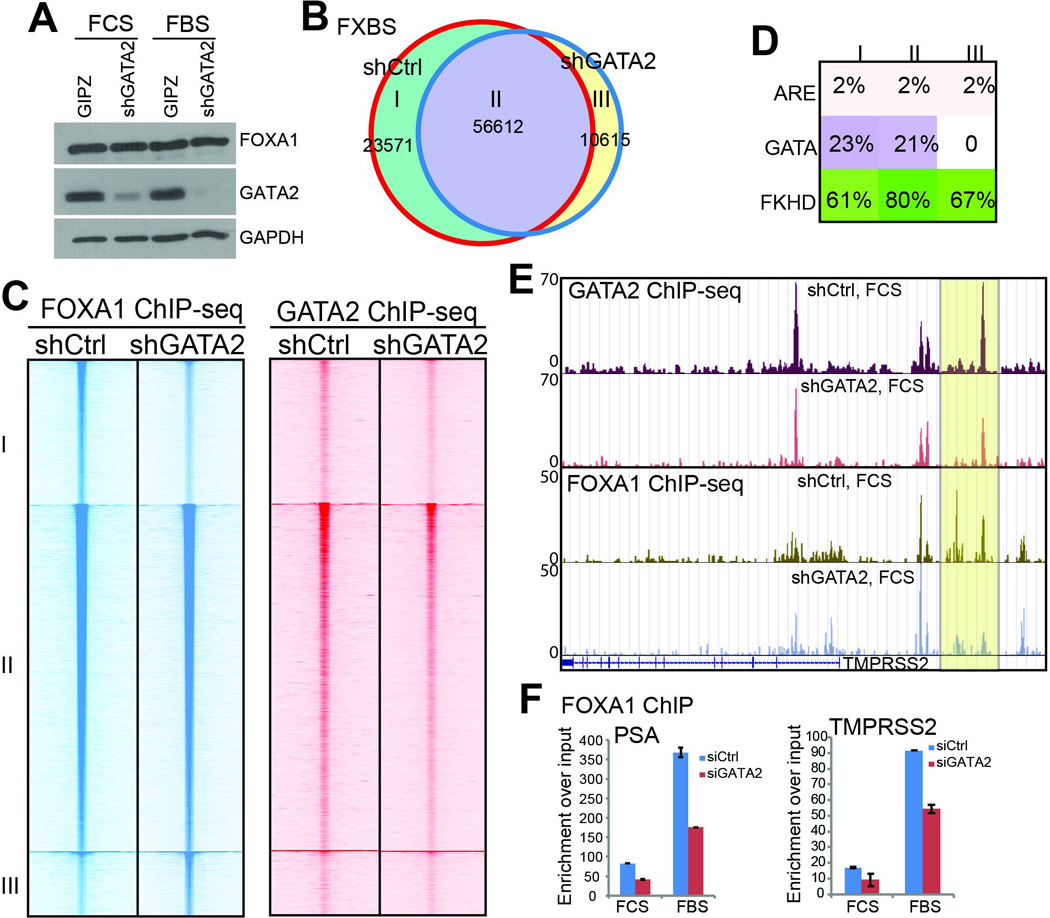
A. Western blot analysis of FOXA1 and GATA2 in control and GATA2-knockdown cells in the presence and absence of androgen.
B. Venn diagram showing overlap of FXBS identified in control and GATA2-knockdown LNCaP cells under androgen-depleted condition.
C. Heatmap view of FOXA1 and GATA2 ChIP-seq read intensity around the 3 categories of FXBS identified in B in control and GATA2-knockdown LNCaP cells.
D. Analysis of ARE, GATA, and FKHD motifs in the 3 categories of FXBS identified in B.
E. Genome browser view showing decreased GATA2 and FOXA1 binding on the TMPRSS2 enhancer following GATA2 knockdown.
F. QRT-PCR confirming decreased FOXA1 binding at the PSA and TMPRSS2 enhancers following GATA2 knockdown in LNCaP cells in the presence or absence of androgen. Data shown are mean ± SEM in triplicate qPCR and is a representative of at least two independent experiments.
Context-dependent roles of FOXA1 in regulating GATA2-mediated transcriptional program
Next, we examined the downstream transcriptional effects of GATA2 depletion on FOXA1 and AR targets, and vice versa, in order to obtain a better understanding of the regulatory hierarchy between these transcription factors. We first examined FOXA1- or GATA2-regulated genes in androgen-depleted cells. GSEA showed that FOXA1-induced genes are strongly enriched for down-regulation by GATA2 knockdown, supporting GATA2 as a collaborating transcription factor (Figure 6A). Consistent with our ChIP-seq data, GATA2 also positively regulates, although to somewhat lesser extent, FOXA1-induced genes in the presence of androgen (Figure 6B). Therefore, regardless of the androgen environment, GATA2 expression enhances FOXA1 transcriptional activities.
Figure 6. Context-dependent roles of FOXA1 in regulating GATA2 program.
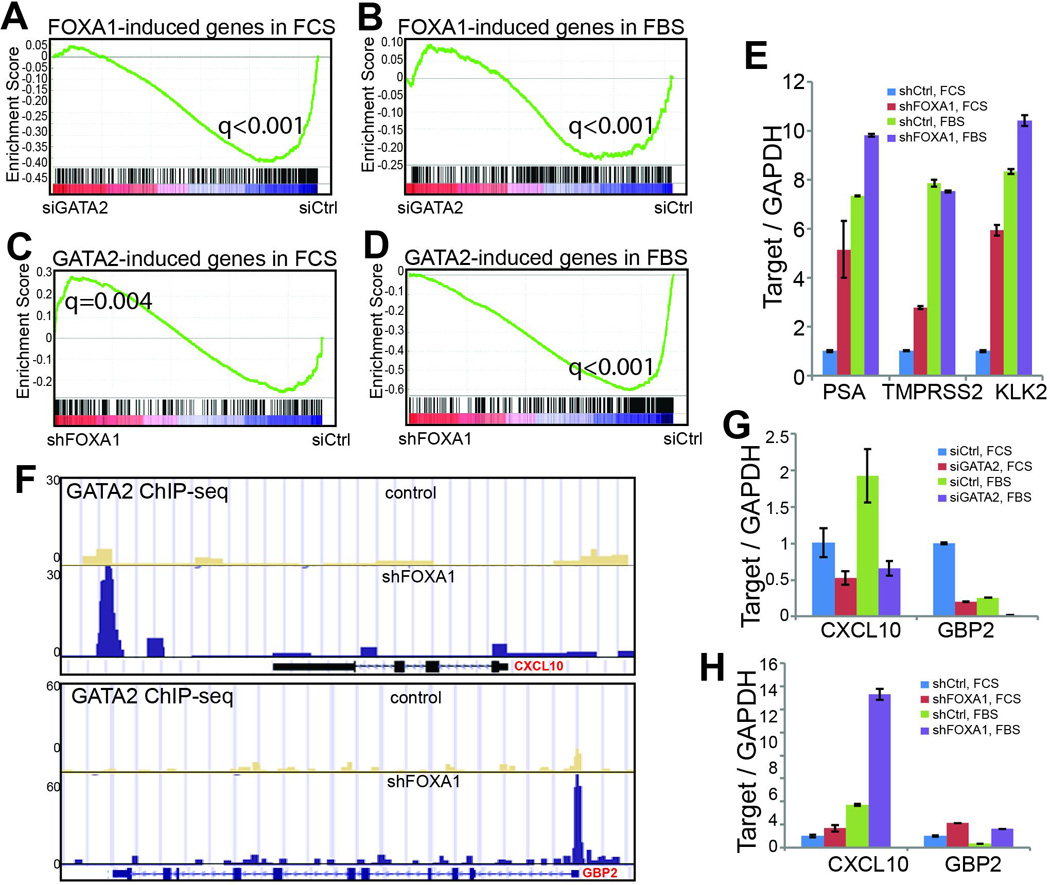
A–B. GATA2 positively regulate FOXA1-targe genes under both androgen-depleted and –replenished conditions. FOXA1-induced genes in the presence (FBS) and absence (FCS) of androgen was derived from microarray data of control and FOXA1-knockdown cells. GSEA of these gene sets was performed in microarray data of control and GATA2-knockdown cells grown under the same androgen environment.
C–D. GATA2-induced genes are significantly enriched for up-regulation upon FOXA1 knockdown in androgen-depleted cells, whereas in the presence of androgen GATA2-induced genes tend to become down-regulated following FOXA1 depletion. GATA2-induced gene sets in the presence and absence of androgen were obtained from corresponding microarray data and subjected to GSEA in microarray data of control and FOXA1-knockdown cells.
E. QRT-PCR validating that FOXA1 knockdown increased GATA2/AR-induced gene expression. Data shown are mean ± SEM in triplicate qPCR and is a representative of at least two independent experiments.
F. Genome Browser showing increased GATA2 binding at CXCL10 enhancer and GBP2 promoter following FOXA1 depletion in LNCaP cells.
G. QRT-PCR showing decreased CXCL10 and GBP2 levels following GATA2 knockdown. Androgen-deprived and –stimulated LNCaP cells were subjected to control or GATA2 knockdown through RNA interference and then qRT-PCR analysis.
H. FOXA1 depletion led to increased expression of GATA2-induced genes. QRT-PCR was performed in control and FOXA1-knockdown LNCaP cells grown in the presence or absence of androgen.
On the other hand, we investigated how FOXA1 regulates GATA2-mediated transcriptional program. Importantly, in the absence of androgen, GATA2-induced genes are enriched for further up-regulation following FOXA1 knockdown, suggesting that FOXA1 inhibits the GATA2 program (Figure 6C). This is in agreement with the remarkable increase of GTBS following FOXA1 knockdown in androgen-depleted cells as we have shown earlier (Figure 4B). Interestingly, in the presence of androgen, GATA2-induced genes were significantly down-regulated by FOXA1 knockdown, suggesting that FOXA1 is a GATA2 cofactor under this condition (Figure 6D). This result is concordant with our genome-wide location analysis showing slightly decreased GTBS following FOXA1 knockdown, potentially due to decreased total GATA2 protein level (Figure S4A). To validate the microarray findings, we conducted qRT-PCR of 3 GATA2-induced genes, PSA, TMPRSS2 and KLK2, comparing control and FOXA1-knockdown LNCaP cells. Our data demonstrated that all three GATA2-induced genes are markedly up-regulated following FOXA1 depletion in androgen-deprived cells, but only weakly up-regulated or even down-regulated in the presence of androgen (Figure 6E).
As PSA, TMPRSS2, and KLK2 are also AR-regulated genes, we sought to investigate new targets of GATA2. Examination of microarray data identified CXCL10 and GBP2 among the top GATA2-induced genes. CXCL10 belongs to the chemokines of the CXC subfamily and is the ligand for the receptor CXCR3. CXCL10 has been previously shown to promote cell motility and invasiveness in prostate cancer cells23. However, the regulation and function of CXCL10 have not been carefully investigated. GBP2 is a member of the guanine-binding protein family that is interferon-inducible to hydrolyze GTP predominantly to GDP. High expression level of GBP2 has recently been associated with better prognosis in breast cancer24. However, GBP2 has not been investigated in prostate cancer. Our ChIP-seq data demonstrated strongly increased GATA2 binding at the CXCL10 enhancer and the GBP2 promoter following FOXA1 knockdown (Figure 6F). Further, qRT-PCR showed that both CXCL10 and GBP2 were significantly down-regulated following GATA2 knockdown both in the presence and absence of androgen, supporting their being GATA2-induced genes (Figure 6G). Moreover, we found that both genes were further up-regulated following FOXA1 depletion under both androgen-depleted and –replenished conditions, in concordance with the increase in GATA2 binding (Figure 6H). We believe that this provides an independent validation of increased GATA2 regulatory activity, rather than AR activity, following FOXA1 loss, as CXCL10 appears to be induced by androgen, whereas GBP2 is strongly repressed by androgen. Taken together, like AR, GATA2 is regulated by FOXA1 in a context-dependent manner.
DISCUSSION
Pioneer factors are critical in defining the genomic actions of AR. We have recently shown that FOXA1, as a pioneer factor, assures prostate-specific AR binding events by recruiting AR from lineage-unspecific ARE sites to FXBS that concomitantly harbor a full or half ARE6. FOXA1 knockdown, therefore, results in transcriptional reprogramming of AR with AR being released to bind other ARE sites5, 7. Similar to FOXA1, GATA2 has also been suggested to behave as a pioneer factor for AR. GATA2 is known to be essential for prostate development and it cooperatively modulate gene expression with AR2, 11, 25. Notably, GATA motifs are consistently found to be enriched in AR binding sites12. The role of GATA2 in regulating AR signaling was thus thought to be comparable with that of FOXA1, but has not been explicitly delineated. In the present study, we report, for the first time, fundamental differences between the mechanisms and functions of FOXA1 and GATA2 with respect to their regulation of AR cistrome. Unlike FOXA1, which gives rise to a significant number of gained AR binding sites upon its depletion, GATA2 loss results in striking reduction in the total number of ARBS. In addition, GATA2 depletion did not enable ARE-mediated AR binding events, suggesting a lack of reprogramming effect. Moreover, at protein level, GATA2 knockdown decreases AR expression. Consistent with the respective changes in AR chromatin targeting resulting from either depletion of FOXA1 or GATA2, transcriptional changes in AR signaling also reflect the disparity between FOXA1 and GATA2. While FOXA1 knockdown led to enhanced AR signaling in the absence of androgen, GATA2 knockdown significantly impaired AR signaling regardless of the presence or absence of androgen. Taken together, these results demonstrate a clear difference distinguishing the functions of FOXA1 and GATA2 in mediating AR genomic binding and transcriptional program.
This contrast in their abilities to direct AR program may help explain, at least in part, the distinct roles they play during prostate cancer progression. Since GATA2 is a crucial AR collaborating factor, its expression is tightly linked to AR activity, therefore a potent oncogene in prostate carcinogenesis. Several reports have demonstrated the oncogenic roles for GATA2 in prostate cancer and a strong association between GATA2 and AR activities in primary specimens18, 20. Moreover, high GATA2 levels are correlated with unfavorable clinical outcome. On the other hand, FOXA1 is slightly up-regulated in primary prostate tumors but its loss was observed in more advanced disease states in metastatic castration-resistant prostate cancer (CRPC)6, 8. Recurrent mutations in the FOXA1 gene have also been consistently reported26, 27. There are also conflicting reports regarding the prognostic potential of FOXA1 in prostate cancer5, 6, 9. These context-dependent functions of FOXA1 in prostate cancer may be mediated by the regulatory mechanisms that in presence of androgen, FOXA1 loss results in abrogation of AR signaling, whereas in absence of androgen, FOXA1 loss results in enhancement of AR signaling.
In addition to illustrating the differences underlying the roles of the two transcription factors FOXA1 and GATA2, we also demonstrate that FOXA1 is not only able to pioneer AR cistrome but also that of GATA2. This is supported by the finding that FOXA1 depletion leads to a global gain in GATA2 peaks as well as a shift of GTBS from FKHD-enriched sites to GATA-containing genomic regions. This phenomenon highly resembles the effect of FOXA1 loss on AR binding, thus suggesting that FOXA1 is capable of pioneering AR as well as GATA2. Based on the molecular mechanism we previously reported underlying FOXA1-mediated pioneering effect, which involves FOXA1 increasing local chromatin accessibility and thus enhancing AR binding6, we speculate that FOXA1 may be able to pioneer and reprogram many other transcription factors in a similar fashion. On the other hand, our data show that GATA2 was not able to reprogram FOXA1. Instead, upon GATA2 knockdown, FXBS was decreased, suggesting GATA2 as a collaborating transcription factor of FOXA1. In addition, we demonstrated that AR co-occupancy can also increase genomic binding of GATA2 and FOXA1, exhibiting co-activator function.
In summary, the robust reprogramming ability shown by FOXA1, but not GATA2, suggests an interesting model wherein FOXA1 resides at the top of a hierarchical network of transcription factors that control AR cistrome (Figure 7). FOXA1 defines AR and GATA2 binding sites, which, on the other hand, further enhance FOXA1 chromatin occupancy, forming a positive feedback loop. By contrast, GATA2 is not able to define/reprogram AR cistrome but acts as a strong co-activator that positively regulate AR expression as well as potentiate AR binding on the chromatin. Consequently, GATA2 is a clear inducer of AR-mediated gene expression program, while FOXA1 exhibits context-dependent roles in its regulation of GATA2 and AR downstream pathways. Therefore, our studies provide an innovative model wherein we show for the first time the different mechanisms involved in FOXA1 and GATA2 regulation of AR cistrome and suggest that FOXA1 acts upstream of GATA2, and potential many other AR co-activators, in defining AR cistrome and determining AR-mediated transcriptional program in prostate cancer.
Figure 7. A model depicting the hierarchical regulatory network of FOXA1, GATA2, and AR.
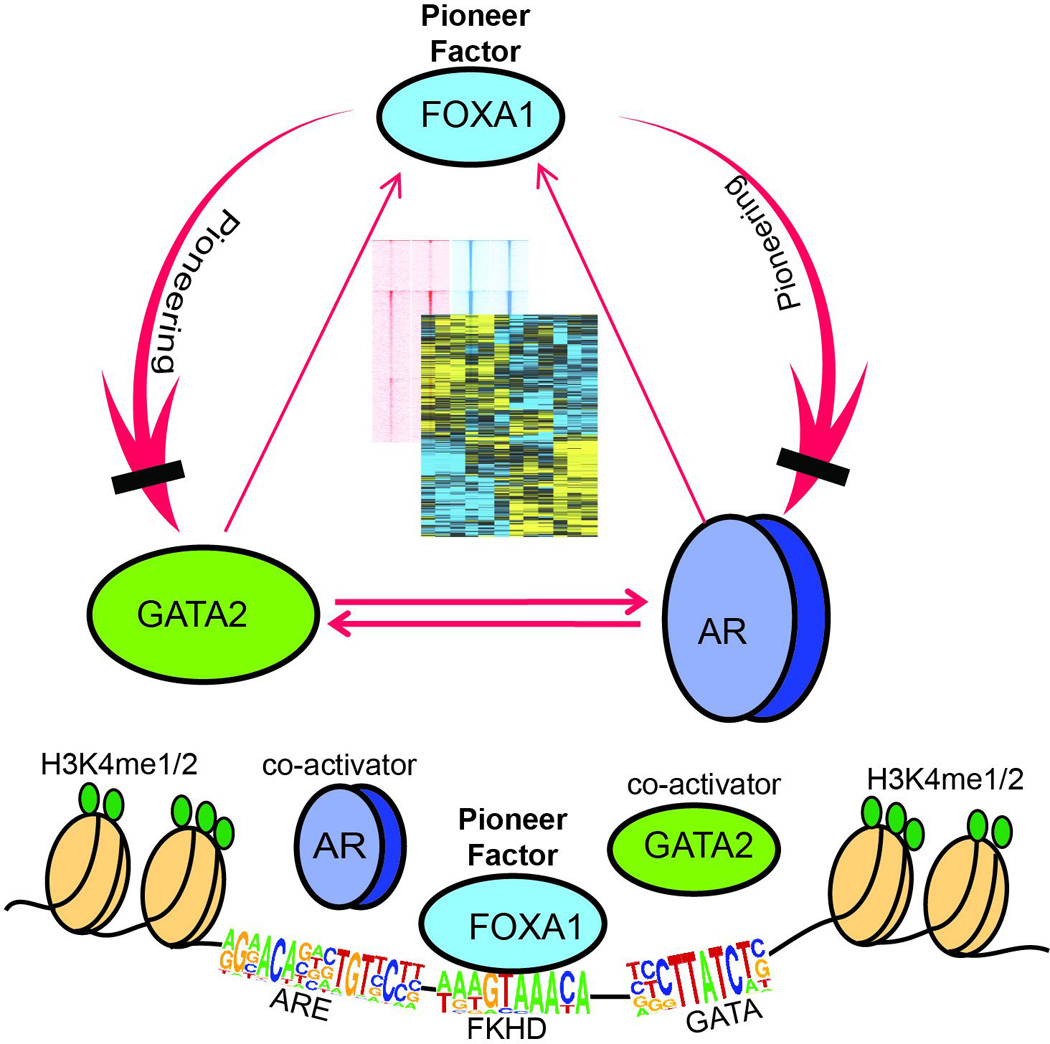
FOXA1 acts up-stream to GATA2 and AR by functioning as a pioneer factor that defines and reprograms both GATA2 and AR cistromes. By contrast, GATA2 and AR are co-activators of each other, which also potentiate, but do not reprogram, FOXA1 binding to chromatin. Our study reveal distinct molecular mechanisms by which FOXA1 and GATA2 regulate AR cistrome and suggest a unique, reprogramming, role of FOXA1 that is central to hormonal regulation of gene expression in prostate cancer.
METHODS
Cell Culture and antibodies
LNCaP cells was obtained from the American Type Culture Collection (ATCC) and grown in RPMI supplemented with 10% fetal bovine serum, 1% penicillin and streptomycin. In FCS condition, cells were cultured in phenol-red free RPMI supplement with 5% charcoal-stripped fetal bovine serum for 3 days. In FBS condition, cells were maintained in RPMI medium with fetal bovine serum.
The antibodies utilized in this study include anti-AR (06-680) from Millipore, anti-AR (39781) from active motif, anti-FOXA1 (ab23738) and anti-GAPDH (ab9385) from Abcam, anti-GATA2 (H-116) from Santa Cruz and anti-GATA2 (4595S) from Cell Signaling.
siRNA, shRNA and Plasmids
The control and pGIPZ lentiviral shRNAmir targeting FOXA1 was used in previous study, shRNA targeting GATA2 (Clone ID # V2LHS_114070 and V3LHS_354335) were obtained from Open Biosystems. A set of 4 siRNAs targeting GATA2 (ON-TARGET plus: J-009024-17, J-009024-18, J-009024-19, J-009024-20) and siRNA Luciferase GL2 Duplex (D-001100-01-20) were synthesized by Dharmacon. Primers used in this study were listed in Supplementary Table 1.
Western blot analysis
Cell lysates were mixed with 1×SDS sample buffer, boiled for 10 min at 95 °C, separated on a 10% SDS-polyacrylamide gel and transferred to an Amersham Hybond PVDF membrane. The membranes were blocked with 5% w/v BSA or milk in TBST for 1h at RT, incubated in primary antibody diluted in blocking solution overnight at 4°C, washed 3 × with TBST and incubated for 1 h in a secondary antibody (1:5000). Membranes were washed 3 × with TBST and incubated with ECL (GE Healthcare) for 2 min. Chemiluminescence was detected by film (GE Healthcare).
Chromatin Immunoprecipitation (ChIP)
ChIP were carried out as described previously28. Briefly, LNCaP cells were cross-linked with 1% formaldehyde for 10 min and the reaction is quenched by 0.125 M glycine for 5 min at RT. Cells were then rinsed with cold 1 × PBS twice, incubated with cell lysis buffer and subsequently nuclear lysis buffer. Chromatin was sonicated and fragmented to a size of 200–500bp, pre-cleared with agarose/protein A or G beads (Upstate), and incubated with 3–5 ug of antibody (anti-FOXA1 from Abcam, cat# ab23738; anti-AR from Millipore, cat#06-680; and anti-GATA2 from Santa Cruz, cat#H-116) overnight and the protein-DNA complexes were then precipitated, washed, and eluted. ChIP-seq library preparation and sequencing were performed as described previously28.
Quantitative Polymerase Chain Reaction (QPCR)
Reverse-transcribed cDNA or ChIP-DNA was mixed with corresponding primers (Supplementary Table 1) and Bullseye EvaGreen qPCR 2× Mastermix-Rox (MIDsci). QPCR reaction was run in a StepOnePlus Real-Time PCR System (Applied Biosystems). For quantitative reverse transcription-PCR (QRT–PCR) data analysis, the fold change in the target gene relative to the GAPDH (control gene) is determined by: fold change=2−Δ(ΔCt) where ΔCt=Cttarget−CtGAPDH and Δ(ΔCt)=ΔCttreatment−ΔCtcontrol. For ChIP–qPCR, enrichment analysis were performed by comparative Ct method and normalization to input: enrichment over input=2−ΔCt, where ΔCt=Ctsample−Ctinput.
ChIP-seq data analysis
ChIP-seq peak identification, overlapping, subtraction and feature annotation of enriched regions were performed using HOMER (Hypergeometric Optimization of Motif EnRichment) suite (http://homer.salk.edu/homer/). It has been suggested that equal numbers of ChIP and input reads result in best performance of peak callers. We matched the total reads of samples or input to the same size by randomly picking reads. Enriched regions of the genome were identified by comparing the ChIP samples to input samples. Weighted venn diagrams were created by R package Vennerable.
The HOMER motif discovery was used to check the enrichment of known motifs in a set of given genomic region (200 bp surrounding ChIP-seq peak center). Motif density histograms were created using HOMER for target regions. The motif percentage of occurrence was created by R packages: ggplot2, scales and gridExtra.
Gene expression microarray and data analysis
Microarray expression profiling was performed using HumanHT-12 v 4.0 Expression BeadChip (Illumina). Bead-level data were preprocessed and normalized by GenomeStudio. Differentially expressed genes were identified by Bioconductor limma package (cutoff p<0.005). Heatmap view of differentially expressed genes was created by Cluster and Java Treeview. GO terms enrichment was analyzed using DAVID and plot was drawn by R package ggplot2. GSEA was performed as described previously6.
Supplementary Material
Acknowledgments
We thank Dr. John Crispino (Northwestern University) for critical reading of the manuscript. The project described was supported by the Robert H. Lurie Comprehensive Cancer Center (P30CA060553), SPORE in Prostate Cancer (P50CA180995), and the Research Scholar Award RSG-12-085-01 (to J.Y.) from the American Cancer Society. J.K. was supported in part by the NIH Training Program in Oncogenesis and Developmental Biology (T32CA080621), and Y.A.Y. was supported in part by the NIH/NCI training grant T32CA009560. Computational analysis was supported by the computational resources and staff contributions provided for the Quest high performance computing facility at Northwestern University which is jointly supported by the Office of the Provost, the Office for Research, and Northwestern University Information Technology.
Footnotes
COMPETING INTERESTS
The authors declare that they have no competing interests.
ACCESSION NUMBERS
New high-throughput data generated in this study has been deposited in GEO database under accession numbers GSE69045.
REFERENCES
- 1.Gao N, Zhang J, Rao MA, Case TC, Mirosevich J, Wang Y, et al. The role of hepatocyte nuclear factor-3 alpha (Forkhead Box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol Endocrinol. 2003;17:1484–1507. doi: 10.1210/me.2003-0020. [DOI] [PubMed] [Google Scholar]
- 2.Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, et al. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132:958–970. doi: 10.1016/j.cell.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jozwik KM, Carroll JS. Pioneer factors in hormone-dependent cancers. Nature reviews Cancer. 2012;12:381–385. doi: 10.1038/nrc3263. [DOI] [PubMed] [Google Scholar]
- 4.Sekiya T, Muthurajan UM, Luger K, Tulin AV, Zaret KS. Nucleosome-binding affinity as a primary determinant of the nuclear mobility of the pioneer transcription factor FoxA. Genes Dev. 2009;23:804–809. doi: 10.1101/gad.1775509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–394. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jin HJ, Zhao JC, Wu L, Kim J, Yu J. Cooperativity and equilibrium with FOXA1 define the androgen receptor transcriptional program. Nat Commun. 2014;5:3972. doi: 10.1038/ncomms4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sahu B, Laakso M, Ovaska K, Mirtti T, Lundin J, Rannikko A, et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J. 2011;30:3962–3976. doi: 10.1038/emboj.2011.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jin HJ, Zhao JC, Ogden I, Bergan RC, Yu J. Androgen receptor-independent function of FoxA1 in prostate cancer metastasis. Cancer Res. 2013;73:3725–3736. doi: 10.1158/0008-5472.CAN-12-3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerhardt J, Montani M, Wild P, Beer M, Huber F, Hermanns T, et al. FOXA1 promotes tumor progression in prostate cancer and represents a novel hallmark of castration-resistant prostate cancer. Am J Pathol. 2012;180:848–861. doi: 10.1016/j.ajpath.2011.10.021. [DOI] [PubMed] [Google Scholar]
- 10.Zhang C, Wang L, Wu D, Chen H, Chen Z, Thomas-Ahner JM, et al. Definition of a FoxA1 Cistrome that is crucial for G1 to S-phase cell-cycle transit in castration-resistant prostate cancer. Cancer Res. 2011;71:6738–6748. doi: 10.1158/0008-5472.CAN-11-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Q, Li W, Liu XS, Carroll JS, Janne OA, Keeton EK, et al. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell. 2007;27:380–392. doi: 10.1016/j.molcel.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Y, Chi P, Rockowitz S, Iaquinta PJ, Shamu T, Shukla S, et al. ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nature medicine. 2013;19:1023–1029. doi: 10.1038/nm.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pevny L, Simon MC, Robertson E, Klein WH, Tsai SF, D'Agati V, et al. Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA-1. Nature. 1991;349:257–260. doi: 10.1038/349257a0. [DOI] [PubMed] [Google Scholar]
- 14.Shivdasani RA, Fujiwara Y, McDevitt MA, Orkin SH. A lineage-selective knockout establishes the critical role of transcription factor GATA-1 in megakaryocyte growth and platelet development. The EMBO journal. 1997;16:3965–3973. doi: 10.1093/emboj/16.13.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu M, Riva L, Xie H, Schindler Y, Moran TB, Cheng Y, et al. Insights into GATA-1-mediated gene activation versus repression via genome-wide chromatin occupancy analysis. Molecular cell. 2009;36:682–695. doi: 10.1016/j.molcel.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boyes J, Omichinski J, Clark D, Pikaart M, Felsenfeld G. Perturbation of nucleosome structure by the erythroid transcription factor GATA-1. Journal of molecular biology. 1998;279:529–544. doi: 10.1006/jmbi.1998.1783. [DOI] [PubMed] [Google Scholar]
- 17.Bossard P, Zaret KS. GATA transcription factors as potentiators of gut endoderm differentiation. Development. 1998;125:4909–4917. doi: 10.1242/dev.125.24.4909. [DOI] [PubMed] [Google Scholar]
- 18.He B, Lanz RB, Fiskus W, Geng C, Yi P, Hartig SM, et al. GATA2 facilitates steroid receptor coactivator recruitment to the androgen receptor complex. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:18261–18266. doi: 10.1073/pnas.1421415111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu D, Sunkel B, Chen Z, Liu X, Ye Z, Li Q, et al. Three-tiered role of the pioneer factor GATA2 in promoting androgen-dependent gene expression in prostate cancer. Nucleic Acids Res. 2014;42:3607–3622. doi: 10.1093/nar/gkt1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vidal SJ, Rodriguez-Bravo V, Quinn SA, Rodriguez-Barrueco R, Lujambio A, Williams E, et al. A targetable GATA2-IGF2 axis confers aggressiveness in lethal prostate cancer. Cancer cell. 2015;27:223–239. doi: 10.1016/j.ccell.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell. 2011;20:457–471. doi: 10.1016/j.ccr.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao JC, Yu J, Runkle C, Wu L, Hu M, Wu D, et al. Cooperation between Polycomb and androgen receptor during oncogenic transformation. Genome Res. 2012;22:322–331. doi: 10.1101/gr.131508.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu Q, Dhir R, Wells A. Altered CXCR3 isoform expression regulates prostate cancer cell migration and invasion. Mol Cancer. 2012;11:3. doi: 10.1186/1476-4598-11-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Godoy P, Cadenas C, Hellwig B, Marchan R, Stewart J, Reif R, et al. Interferon-inducible guanylate binding protein (GBP2) is associated with better prognosis in breast cancer and indicates an efficient T cell response. Breast Cancer. 2014;21:491–499. doi: 10.1007/s12282-012-0404-8. [DOI] [PubMed] [Google Scholar]
- 25.Andreu-Vieyra C, Lai J, Berman BP, Frenkel B, Jia L, Jones PA, et al. Dynamic nucleosome-depleted regions at androgen receptor enhancers in the absence of ligand in prostate cancer cells. Mol Cell Biol. 2011;31:4648–4662. doi: 10.1128/MCB.05934-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012 doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–689. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu J, Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010;17:443–454. doi: 10.1016/j.ccr.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu L, Runkle C, Jin HJ, Yu J, Li J, Yang X, et al. CCN3/NOV gene expression in human prostate cancer is directly suppressed by the androgen receptor. Oncogene. 2013 doi: 10.1038/onc.2012.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.