

Abstract
Background and Purpose
δ Opioid receptor agonists are being developed as potential treatments for depression and alcohol use disorders. This is particularly interesting as depression is frequently co‐morbid with alcohol use disorders. Yet we have previously shown that δ receptor agonists range widely in their ability to modulate alcohol intake; certain δ receptor agonists actually increase alcohol consumption in mice. We propose that variations in β‐arrestin 2 recruitment contribute to the differential behavioural profile of δ receptor agonists.
Experimental Approach
We used three diarylmethylpiperazine‐based non‐peptidic δ receptor selective agonists (SNC80, SNC162 and ARM390) and three structurally diverse δ receptor agonists (TAN‐67, KNT127 and NIH11082). We tested these agonists in cAMP and β‐arrestin 2 recruitment assays and a behavioural assay of alcohol intake in male C57BL/6 mice. We used β‐arrestin 2 knockout mice and a model of depression‐like behaviour to further study the role of β‐arrestin 2 in δ receptor pharmacology.
Key Results
All six tested δ receptor agonists were full agonists in the cAMP assay but displayed distinct β‐arrestin 2 recruitment efficacy. The efficacy of δ receptor agonists to recruit β‐arrestin 2 positively correlated with their ability to increase alcohol intake (P < 0.01). The effects of the very efficacious recruiter SNC80 on alcohol intake, alcohol place preference and depression‐like behaviour were β‐arrestin 2‐dependent.
Conclusions and Implications
Our finding that δ receptor agonists that strongly recruit β‐arrestin 2 can increase alcohol intake carries important ramifications for drug development of δ receptor agonists for treatment of alcohol use disorders and depressive disorders. © 2015 The British Pharmacological Society
Abbreviations
- (−)NIH11082
(−)‐(1R,5R,9R)‐5,9‐dimethyl‐2′‐hydroxy‐2‐(6‐hydroxyhexyl)‐6,7‐benzomorphan
- ADL5859
N,N‐diethyl‐4‐(5‐hydroxyspiro[chromene‐2,4′‐piperidine]‐4‐yl)benzamide
- ARM390
N,N‐diethyl‐4‐(phenylpiperidin‐4‐ylidene‐methyl)‐benzamide
- AZD2327
4‐[(R)‐(3‐aminophenyl)[4‐(4‐fluorobenzyl)‐piperazin‐1‐yl]methyl]‐N,N‐diethylbenzamide
- KNT‐127
1,2,3,4,4a,5,12,12a‐octahydro‐2‐methyl‐4aβ,1β‐([1,2]benzenomethano)‐2,6‐diazanaphthacene‐12aβ,17‐diol
- KO
knockout
- SNC80
(+)‐4‐[(alphaR)‐α‐[(2S,5R)‐2,5‐dimethyl‐4‐(2‐propenyl)‐1‐piperazinyl]‐(3‐methoxyphenyl)methyl‐N,N‐diethylbenzamide]
- SNC162
(+)‐4‐[(alphaR)‐α‐[(2S,5R)‐2,5‐dimethyl‐4‐(2‐propenyl)‐1‐piperazinyl]‐(3‐phenyl)methyl]‐N,N‐diethylbenzamide
- TAN‐67
(R*,S*)‐(±)‐2‐methyl‐4aa‐(3‐hydroxyphenyl)‐1,2,3,4,4a,5,12,12aa‐octahydroquinolino[2,3,3‐g]isoquinoline dihydrobromide
Tables of Links
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Drugs, like morphine, oxycodone and fentanyl, targeting μ opioid receptors are broadly known for their therapeutic use in providing analgesic relief. Yet, the opioid receptor family consists of three other members that may be of therapeutic use as well. The δ opioid receptor is widely expressed throughout the body and as such is involved in many different physiological responses including pain transmission, ischaemic protection, depression and drug abuse (Pradhan et al., 2011; Chu Sin Chung and Kieffer, 2013; van Rijn et al., 2013a). Over the last couple of decades, medicinal chemists have synthesized a wide array of structurally diverse compounds that selectively target δ receptors. Several δ receptor agonists are in clinical trials for treatment of acute and chronic pain, and anxious major depressive disorder (Chu Sin Chung and Kieffer, 2013; van Rijn et al., 2013a). Still, no δ receptor‐selective drugs have made it past clinical trials, let alone entered phase III clinical trials, suggesting perhaps that the pharmacology of δ receptor drugs is not fully understood. It is apparent from recent reports that δ receptors have the ability to engage in heteromeric receptor–receptor interactions (Gomes et al., 2000; Gomes et al., 2004; van Rijn and Whistler, 2009; Gupta et al., 2010; He et al., 2011; Gomes et al., 2013) and that their expression and trafficking are more dynamic than those of the μ and κ opioid receptors (Cahill et al., 2001; Whistler et al., 2002; Bao et al., 2003; Pradhan et al., 2010; He et al., 2011; Margolis et al., 2011; van Rijn et al., 2012a). Thus, δ receptor pharmacology is more complex than initially assumed (van Rijn et al., 2013a).
Transgenic mice lacking the δ receptor show enhanced depressive‐like behaviour (Filliol et al., 2000) and many δ receptor agonists including SNC80 (Jutkiewicz et al., 2004; Saitoh et al., 2004), SNC162 (Jutkiewicz et al., 2004), AZD2327 (Hudzik et al., 2011), ADL5859 (Le Bourdonnec et al., 2008), (−)‐NIH11082 (Naidu et al., 2007) and KNT‐127 (Saitoh et al., 2011) are able to reduce depression‐like behaviour. However, the pharmacology of δ receptor agonists with regard to alcohol use is more complex. Recently, we showed that two structurally distinct δ receptor agonists, SNC80 and TAN‐67, have opposite effects on alcohol intake and alcohol‐induced place preference (van Rijn et al., 2009; van Rijn et al., 2010; van Rijn et al., 2012b). Microinjection of the δ receptor agonist DPDPE in the ventral tegmental area also decreases alcohol intake in rats (Margolis et al., 2008). Pharmaceutical companies have used the diarylmethylpiperazine scaffold of the highly selective δ receptor agonist SNC80 (Knapp et al., 1996) as a template for the development of drugs currently undergoing clinical trials [e.g. AZD2327 (Hudzik et al., 2011) and ADL5859 (Le Bourdonnec et al., 2008; Nozaki et al., 2012)] (Figure 1). Considering that SNC80 can increase alcohol consumption (Nielsen et al., 2012b; van Rijn et al., 2012b), it is important to understand the pharmacology underlying these behavioural effects when compared with a δ receptor agonist like TAN‐67 that decreases alcohol intake.
Figure 1.

Structural similarities and dissimilarities between δ receptor selective agonists. Chemical structures of the δ receptor selective agonists TAN‐67 and SNC80, SNC162, ARM390, KNT‐127 and NIH11082 and the SNC80‐like δ receptor agonists AZD2327 and ADL5859 that have been in clinical trials.
We have previously proposed that TAN‐67 may differ from SNC80 in that it interacts with δ –μ receptor heteromers (van Rijn et al., 2009; van Rijn et al., 2012a; van Rijn et al., 2013b). Yet, another possible hypothesis is that the two agonists have different signalling bias and may show a preference to signal through G‐proteins or β‐arrestins (DeWire et al., 2007). This is an intriguing hypothesis to explore as δ receptor agonists have been shown to differentially interact with β‐arrestins. Due to a difference in β‐arrestin 2 recruitment, certain δ receptor agonists cause the receptor to internalize rapidly but recycle poorly, while other δ receptor agonists are good internalizers and recyclers, whereas yet other agonists are poor receptor internalizers (Pradhan et al., 2010; Audet et al., 2012). Here, we examined four other δ receptor agonists, together with SNC80 and TAN‐67, for their efficacy to recruit β‐arrestin 2 and modulate alcohol intake. We used two SNC80‐like agonists, ARM390 and SNC162, and two δ receptor agonists that are structurally dissimilar to SNC80 (KNT‐127 and NIH11082, Figure 1). Our results indicate that δ receptor agonists that are super‐recruiters of β‐arrestin 2 increase alcohol intake whereas δ receptor agonists with low β‐arrestin 2 efficacy decrease alcohol intake in mice.
Methods
Interpretation
Power analyses were performed to determine the optimal group size to determine a behavioural effect with sufficient statistical power. The strong correlation between in vitro data and in vivo effect on alcohol intake will allow for replacement and reduction of animal use in future studies, by testing new compounds first in the cellular β‐arrestin 2 recruitment assay.
Animals
Wild‐type male C57BL/6 mice were obtained from Taconic Biosciences, Inc (Cambridge City, IN, USA). Mice arrived aged 4–5 weeks and were habituated to the new housing. Male β‐arrestin 2 knockout (KO) C57BL/6 mice, originally obtained from Dr. Lefkowitz at Duke University, originated from breeding colonies in house. The β‐arrestin 2 KO C57BL/6 mice were originally delivered to Dr. Jennifer Whistler at the University of California San Francisco (UCSF) and, while there, had been backcrossed to C57BL/6 mice for >10 generations. Every two to three generations, the homozygous KO mice would be crossed with C57BL/6 wild‐type mice to reduce effects from inbreeding. Mice were shipped to Purdue University, and due to the presence of norovirus in the UCSF strain, the mice were rederived and bred to a homozygous β‐arrestin 2 KO genotype again. Genotype was confirmed by PCR analysis from tissue of mice tails collected 3 weeks after birth. Due to the particular breeding history of our colony of β‐arrestin 2 KO C57BL/6 mice, they may genetically differ from other β‐arrestin 2 KO C57BL/6 that are used elsewhere. For alcohol experiments, mice started drinking alcohol at an age of 5–6 weeks (weighing 18–23 g). Drug injections occurred when mice were between 9–12 weeks of age. For conditioned place preference, locomotor and depression‐like behaviour studies, mice were tested during the ages of 6–9 weeks. For alcohol metabolism studies, wild‐type mice with an age range of 9–16 weeks were used, whereas β‐arrestin 2 KO mice ranged in age from 5 to 15 weeks.
Validity of animal species or model selection
Mice, in particular C57BL/6 mice, are willing to consume unsweetened alcohol. Drugs used clinically to treat alcohol use disorders are able to reduce alcohol intake in these mice. The forced swim test is commonly used to determine depression‐like behaviour in rodents and clinically used anti‐depressants are able to reduce immobility time in this assay. Thus, our mouse models have face and construct validity. Additionally, the rationale for using mice was driven by previous results obtained by us in these mouse models and the availability of transgenic β‐arrestin 2 KO mice, necessary for further interpretation of the acquired data.
Ethical statement
All animal procedures were pre‐approved by our Institutional Animal Care and Use Committee and were in accordance with National Institutes of Health Guide for the Care and Use of Laboratory Animals. Mice were not deprived of food or water at any time. Efforts were made to minimize animal suffering and to reduce the number of animals used, and all in vivo experiments have been performed in accordance with ARRIVE Guidelines (Kilkenny et al., 2010).
Housing and husbandry
For alcohol intake studies, mice were individually housed in double‐grommet ventilated Plexiglas cages. For all other studies, mice were grouped housed (three to five mice per cage) in single grommet cages at ambient temperature (22–24°C, 40–60% humidity) in a room maintained on a reversed 12L : 12D cycle (lights off at 10.00 h, lights on at 22.00 h) in our animal research facility that has Association for Assessment and Accreditation of Laboratory Animal Care approval. Mice are house in specific pathogen‐free zones, with sentinel mice for monitoring health of the colonies. Cages were lined with Harlan 7092 corncob bedding material with nestlets provided for enrichment purposes. Food (Harlan 2018–18% protein dry food pellets) and reverse osmosis water were provided ad libitum. The mice were given ~10–14 days to acclimatize to the individual housing conditions and reverse light cycle as well as habituate to handling before the start of the experiments. Mice were weighed weekly. Cages of individually housed mice were changed every 2 weeks. Cages of group housed mice were changed weekly. Mice were monitored by the investigating team and veterinary staff for abnormal behaviour and symptoms that would be reasons for removing the animal from the experiment prematurely and killing the affected animal. Signs that were monitored included the following: rapid or progressive weight loss of more than 20% of the body weight, unrelenting/unresolved diarrhoea, dehydration determined by an increase in skin tenting, abdominal swelling and/or ascites, progressive dermatitis and/or self‐induced trauma, rough hair coat/poor grooming, hunched posture, lethargy, an inability to stand or loss of righting reflex and respiratory symptoms. To reduce stress and suffering, mice were only tested once in the forced swim test.
Group sizes
The exact group size for each experimental group/condition is provided in the figure and figure legends and provided as range in the Methods section describing the entire experiment. When used, the term ‘n’ refers to independent values, not replicates. Group sizes at the start were identical, but experimental loss (accidental spillage of bottles, clogged sipper tubes, accidental death, lack of responsiveness in pre‐tests and inability to swim) changed final group size on occasions. No mice for which end data recorded were excluded. On many occasions, one to two mice per group drank less alcohol compared with the other mice in the group; as we did not exclude these mice, there is a decent amount of variability. We have empirically found that starting with a group size of 12 provides us with enough statistical power to observe significant differences in drug‐induced modulation of alcohol intake compared with saline‐treated mice. Additionally, we measured the effect of TAN‐67 on alcohol intake on two separate occasions as a positive control. The data for these experiments were combined.
Randomization
Within an experiment, animals were randomized by arbitrarily placing them in treatment groups. For alcohol drinking studies, mice were trained to consume alcohol until a steady state was reached. Mice were then subdivided in groups such that each group showed identical levels of alcohol intake. These ‘identical’ groups were then randomized to their treatment. Similarly for place preference studies, mice were subdivided in groups that showed equal and identical preference for the drug‐paired and vehicle‐paired chambers. These ‘identical’ groups were then randomized to their treatment.
Experimental procedures – behavioural assays
Voluntary alcohol consumption
Wild‐type and β‐arrestin 2 KO C57BL/6 mice (n = 8–18) age 4–5 weeks were individually housed in double‐grommet cages and presented with a two‐bottle choice of water and 10% alcohol for a 4 h period each day during the animal's dark cycle, starting 1 h into the dark as previously described (van Rijn et al., 2009). From Monday through to Friday, mice were presented with a two‐bottle choice (water and 10% alcohol) for a 4 h period (11.00–15.00 h) while in the dark cycle. Outside the two‐bottle choice period, all mice had unlimited access to water. All fluids were presented in 50 mL graduated falcon tubes fitted with stainless‐steel drinking spouts inserted through two grommets in front of the cage. Bottles were weighed to the nearest decigram (0.1 g) at the start and end of the two‐bottle choice period. Loss due to spillage and evaporation was accounted for by measuring the weights of a pair of bottles on an empty double‐grommet cage. The positions of the tubes containing water and alcohol were reversed daily to limit the effects of positional preference. Mice were trained for 3 weeks, during which their alcohol intake and preference stabilized. During this time, mice were habituated to handling and injections. After the training period, mice (n = 9–12) were injected s.c. or for ARM390 by oral gavage on Friday, 30 min before the start of the two‐bottle choice period. Alcohol and water consumption was measured and compared with the average of the consumption on Tuesday, Wednesday and Thursday (baseline value). The first injection was with saline, and the following weeks, each individual group received increasing doses of a δ receptor agonist (Supporting Information Fig. S1). Mice did not receive drug or alcohol on Saturday or Sunday. Mice that were unable to consume alcohol or showed unpredictable fluctuations in their alcohol drinking pattern during the week (Monday–Thursday) were excluded.
Alcohol place preference
Alcohol place preference was performed as previously described (van Rijn et al., 2012b). In short, wild‐type or β‐arrestin 2 KO C57BL/6 mice (n = 11–12) age 6–7 weeks were conditioned to saline and 1.5 mg⋅kg−1 ethyl alcohol (15%) in a two‐chamber place preference paradigm (Med. Associates, St. Albans, VA, USA). One chamber was outfitted with a rod floor, whereas the second chamber had a wire mesh floor. To further enhance contextual difference, the chambers were outfitted with either horizontal or vertical black and white stripes. On days 2–9, mice were conditioned for 5 min, with mice receiving either saline or alcohol on alternating days. Mice were placed in the conditioned place preference (CPP) box directly after i.p. injection. Mice were randomly assigned to receive alcohol or saline on a particular chamber. An unbiased approach was used to measure place preference. On days 1 and 10, mice were free to explore both chambers for 30 min. On these days, all mice still received an i.p. saline injection immediately before the preference assessment. Movement and time spent were tracked by infrared beams.
Alcohol metabolism measurement
Wild‐type and β‐arrestin 2 KO C57BL/6 mice aged 5–16 weeks (n = 8) were injected with saline or 20 mg ⋅kg−1 SNC80 30 min before an i.p. injection with 2.5 g⋅kg−1 ethanol. Blood was drawn after 30 and 150 min. Blood alcohol content was determined as mentioned previously. Blood samples were precipitated in 3% perchloric acid (200 μL final volume) and stored at 4°C until analysis. Ethanol concentration was quantified by the alcohol dehydrogenase assay. Samples (20 μL) were incubated in duplicate in 1 mL of 0.5 M Tris–HCl buffer (pH 8.8) containing 5.5 μg⋅mL−1 of alcohol dehydrogenase and 1.5 mM β‐nicotinamide adenine dinucleotide (β‐NAD) for 40 min at room temperature. Accumulation of β‐NADH was measured by reading sample absorbance at 340 nm. The ethanol concentration in the samples was determined using a standard calibration curve, which was linear between 0.1 and 50 mM ethanol (r 2 = 0.98).
Locomotor activity
Wild‐type or β‐arrestin 2 KO C57BL/6 mice aged 6–7 weeks (n = 8–10), housed in a standard light–dark cycle, were habituated to s.c. injection handling. To reduce the effect of novelty, we habituated mice to the locomotor box for 60 min the day before the test. On the test day, mice were injected with saline or drug and placed in the Med. Associated locomotor activity box for 90 min. Movement was monitored for 60 min by infrared beams. For the comparison of locomotor effects induced by ARM390, SNC80 and SNC162, 10 week old wild‐type C57BL/6 mice (n = 6) that were housed in a reversed light cycle were used.
Depression‐like behaviour – forced swim test
One swim session was performed per mouse, as described in Enquist et al. (2012). Wild‐type or β‐arrestin 2 KO C57BL/6 mice aged 6–7 weeks (n = 10–13) were injected with SNC80 (20 mg⋅kg−1, s.c.) or TAN‐67 (25 mg⋅kg−1 s.c. in saline) 30 min before the test. Swim sessions lasted 10 min (in ambient light of 580–680 lux) and were conducted in individual clear plastic cylinders (35.5 cm tall × 25 cm diameter) filled with water (26.5°C, ±0.7°C) to a depth of 20 ± 1.5 cm. A camera positioned at an angle to the cylinders recorded the sessions. An experimenter blind to treatment scored the animals. Manual scoring was for total immobile time during the last 6 min of the session. Immobility was defined as the absence of movement, except that necessary to keep afloat.
cAMP inhibition assay
HEK 293FT (Life Technologies, Grand Island, NY, USA) cells were transfected with pcDNA3.1‐FLAG‐DOR and pGloSensor22F‐cAMP plasmids (Promega, Madison, WI, USA) in a 3:7 ratio using X‐tremeGENE9 (Roche, Indianapolis, IN, USA) according to manufacturer's protocol. On day 2, cells were dislodged and counted, and 7.5 μL of cell suspension was seeded (25 000 cells/well) in a low volume, round bottom white 384‐well CulturPlate‐384 (Perkin Elmer, Waltham, MA, USA). Four hours later, cells were stimulated with 7.5 μL 4% GloSensor reagent (Promega) in HBSS/HEPES (Life Technologies) and incubated for 90 min at room temperature. Cells were stimulated for 20 min with a dilution series of δreceptor agonists (5 μL per well). Each dilution was performed in triplicate. Following stimulation, cells were incubated for 15 min with 5 μL 31.6 μM forskolin (Sigma, St. Louis, MO, USA), and luminescence was measured on a Flexstation3 (Molecular Devices, Sunnyvale, CA, USA). Data were plotted using graphpad prism 5 software (GraphPad Software, La Jolla, CA, USA). Each experiment was performed a minimum of three independent times.
β‐Arrestin 2 recruitment assay
CHO‐OPRD PathHunter β‐arrestin 2 cells (DiscoverX, Fremont, CA, USA) were seeded (2500 cells per well) in a low volume, round bottom 384‐well plate. The next day, cells were stimulated for 90 min with a dilution series of δ receptor agonists at 37°C/5% CO2. Each dilution was performed in triplicate. β‐Arrestin 2 recruitment was detected following a 60 min incubation period with PathHunter reagent according to the manufacturer's guidelines. Luminescence was measured using a Flexstation3. Data were plotted using graphpad prism 5 software. Each experiment was performed a minimum of three independent times.
Drugs
SNC80, SB205607 (TAN‐67), SNC162 and ARM390 were purchased from R&D systems (Minneapolis, MN, USA). DPDPE, leu‐enkephalin and ethyl alcohol (200 % proof) were obtained from Sigma. KNT‐127 and (−)‐NIH11082 were custom synthesized by ChemPartner (Shanghai, China). For cellular assays, compounds were dissolved in water, whereas for behavioural assays, compounds were dissolved in saline. SNC80 and SNC162 were dissolved in saline with 2 μL of 1 M hydrochloric acid mg‐1 drug. All drugs were administered by s.c. injection with the exception of ARM390, which was administered by oral gavage using a flexible plastic needle. The final pH of the solution ranged between 5 and 6, as measured with MColorpHast pH indicator test strips (Millipore, Temecula, CA, USA).
Blinding
Data for locomotor activity and place preference were electronically monitored and recorded without human input. The operator was blind to the injected drugs. Additionally, data were frequently not analysed by the operator of the experiments.
Normalization
Results from the β‐arrestin 2 recruitment assay were normalized against DPDPE/leucine‐enkephalin as the ‘endogenous’ full agonist response. Normalization was employed to account for differences in luminescence caused by differences in cell density between experiments and to compare the exogenous agonist response relative to the endogenous agonist response. To determine how δ receptor agonists affected alcohol intake and depression‐like behaviour, a baseline removal was performed using the saline control data.
Statistical comparison
All data are presented as means ± SEM. Pharmacokinetics of drug were analysed using repeated‐measures anova followed by Dunnett's post hoc analysis to determine difference versus vehicle. The analysis of pharmacological drug effects between wild‐type and β‐arrestin 2 KO was performed using two‐way anova for drug effect and genotype, followed by a Bonferroni post hoc test, to determine statistically significant differences. A level of probability of P < 0.05 was deemed to constitute the threshold for statistical significance and marked with an asterisk. For transparency, results with a level of probability of P < 0.01 were marked with **, P < 0.001 with *** and P < 0.0001 with ****.
Results
The δ receptor agonist TAN‐67 is a very weak recruiter of β‐arrestin 2
Previous results have shown that TAN‐67 and SNC80 (Figure 1) modulate alcohol consumption in an opposite direction (van Rijn et al., 2009; van Rijn et al., 2012b). To understand the molecular basis for this paradoxical effect, we studied the ability of these drugs to activate their cognate Gi/o pathway and thereby inhibit cAMP production. For reference compound, we used the cyclized enkephalin analogue DPDPE. We found that both TAN‐67 and SNC80 act as full agonists in their ability to inhibit forskolin‐mediated cAMP release in HEK 293FT cells and had similar potency (Figure 2 and Table 1). All agonists inhibited cAMP production by roughly 40%, in accordance with previously published work (Lecoq et al., 2004). Next, we investigated the ability of these compounds to recruit β‐arrestin 2 using the DiscoverX PathHunter assay. Using this assay, we found that in comparison with DPDPE, SNC80 was a β‐arrestin 2 ‘super‐recruiter’ (α =142 ± 9), whereas TAN‐67 was a very weak recruiter (α =41 ± 3, Table 1, Figure 2).
Figure 2.
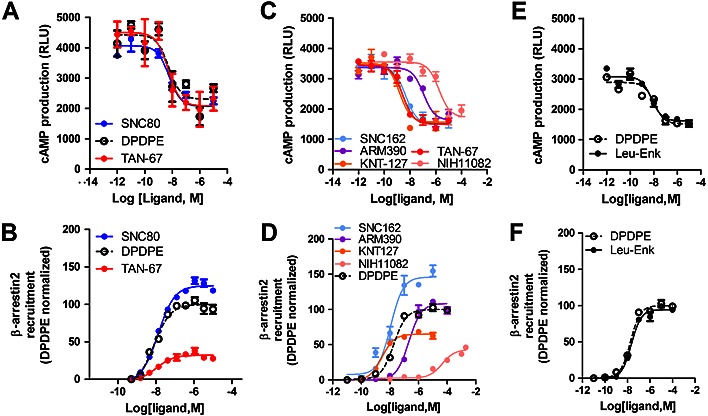
The δ receptor agonists exhibit distinct differences in β‐arrestin 2 recruitment efficacy. (A) The δ receptor selective agonists TAN‐67, SNC80 and DPDPE inhibit forskolin‐induced cAMP production in HEK293 cells expressing δ receptors. (B) Compared with DPDPE, TAN‐67 is a weak recruiter of β‐arrestin 2, whereas SNC80 is a ‘super‐recruiter’ as measured in CHO PathHunter cells expressing δ receptors and β‐arrestin 2‐galactosidase. (C) The δ receptor selective agonists SNC162, ARM390, KNT‐127 and NIH11082 fully inhibit forskolin‐induced cAMP production in HEK293 cells expressing δ receptors. TAN‐67 was used for comparison. (D) Compared with DPDPE, the δ receptor agonist NIH1108 is a weak recruiter, KNT‐127 a moderate recruiter of β‐arrestin 2, ARM390 is a regular full agonist and SNC162 is a ‘super‐recruiter’ as measured in CHO PathHunter cells expressing δ receptors and β‐arrestin 2‐galactosidase. (E and F) DPDPE and the endogenous leu‐enkephalin have identical pharmacology in inhibiting cAMP production and recruiting β‐arrestin 2. Representative curves are depicted.
Table 1.
δ Receptor agonist potencies for inhibiting cAMP production and recruiting β‐arrestin 2 at the δ receptor expressed in HEK cells and CHO cells respectively
| NIH11082 | TAN‐67 | KNT‐127 | Leu‐Enk | DPDPE | ARM390 | SNC162 | SNC80 | |
|---|---|---|---|---|---|---|---|---|
| pIC50 (cAMP) | 5.2 ± 0.2 (5) | 8.6 ± 0.2 (6) | 8.7 ± 0.1 (4) | 8.5 ± 0.1 (4) | 8.2 ± 0.1 (5) | 6.9 ± 0.1 (5) | 8.4 ± 0.1 (22) | 8.2 ± 0.2 (7) |
| pEC50 (βARR2) | 4.9 ± 0.2 (4) | 7.9 ± 0.1 (4) | 8.5 ± 0.1 (5) | 7.6 ± 0.1 (5) | 7.6 ± 0.1 (4) | 6.5 ± 0.1 (6) | 7.9 ± 0.1 (4) | 8.2 ± 0.1 (4) |
| Efficacy (%) | 36 ± 4 | 41 ± 3 | 71 ± 2 | 100 ± 4 | 100 ± 1 | 103 ± 7 | 137 ± 7 | 142 ± 9 |
Potency of δ receptor agonists to inhibit cAMP production is depicted as concentration of 50% inhibition (pIC50) and the SEM. Potency (pEC50) and efficacy (normalized to DPDPE and leu‐enkephalin) of δ receptor agonists to recruit β‐arrestin 2 are depicted with SEM. The number of repetitions for each drug is indicated in parentheses.
β‐arrestin efficacy of δ receptor agonists is correlated with increased alcohol consumption
We next wanted to determine if δ receptor agonists, which are structurally similar to SNC80, are also β‐arrestin super‐recruiters and would, like SNC80, increase alcohol intake, as this could be a clear adverse effect for δ receptor agonists currently in clinical trials. We tested two SNC80‐like δ receptor agonists (SNC162 and ARM390) and compared it with two δ receptor agonists (KNT‐127 and NIH11082) that are less structurally related to SNC80 (Figure 1). All four δ receptor agonists were full agonists in their ability to inhibit forskolin‐induced cAMP production (Figure 2, Table 1). Interestingly, these four compounds had distinct β‐arrestin 2 recruitment profiles, with the SNC80‐like agonist being super‐recruiters compared with DPDPE and the endogenous opioid leu‐enkephalin and KNT‐127 and NIH11082 being moderate to weak recruiters of β‐arrestins, respectively (Figure 2, Table 1). As it is more appropriate to normalize responses to an endogenous ligand, we compared the DPDPE responses in the cAMP and β‐arrestin 2 recruitment assays against the endogenous δ receptor agonist leu‐enkephalin. We found that the DPDPE and leu‐enkephalin both acted as full agonist and recruited β‐arrestin 2 with identical efficacy (Figure 2, Table 1).
We next determined the efficacy of these compounds to modulate alcohol intake in wild‐type mice at two doses. We also repeated our previous findings for TAN‐67 and SNC80. For each agonist, we used doses that have previously been reported to produce significant behavioural effects (Jutkiewicz et al., 2004; Saitoh et al., 2004; Naidu et al., 2007; Pradhan et al., 2010; Saitoh et al., 2011). We found that at the highest dose, the strong β‐arrestin 2 recruiters SNC162 (F 2, 30 = 7.089, P < 0.01) and ARM390 (F 2, 24 = 3.723, P < 0.05) significantly increased alcohol intake, the intermediate β‐arrestin 2 recruiter KNT‐127 was unable (F 3, 48 = 2.180, P = 0.1) to significantly modulate alcohol intake at any of the three doses tested and the weak β‐arrestin 2 recruiter NIH11082 decreased alcohol intake (F 2, 25 = 2.553, P = 0.10) at the highest dose (Figure 3). We also tested higher doses of ARM390 (20 mg⋅kg−1), SNC162 (10 mg⋅kg−1) SNC80 (30 mg⋅kg−1) and NIH11082 (32 mg⋅kg−1); however, it appears that the functionality of these drugs reach either a plateau or are bell shaped (Supporting Information Fig. S2). This is in accordance with previous reports; for example, the optimal dose for NIH11082 to reduce depressant‐like behaviour is 16 and not 32 mg⋅kg−1 (Naidu et al., 2007).
Figure 3.
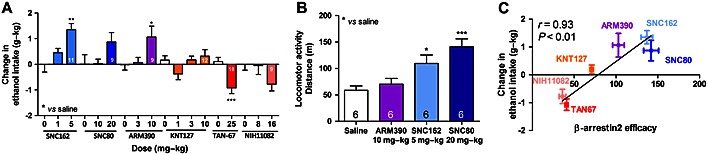
δ Receptor agonist‐induced alcohol intake is positively correlated with β‐arrestin 2 recruitment efficacy, (A) Modulation of alcohol intake in wild‐type C57BL/6 mice trained to consume 10% alcohol in a two‐bottle choice paradigm limited access by different doses of δ receptor agonists with distinct difference in β‐arrestin 2 recruitment efficacy. The group size for each drug is depicted inside the column of that drug. (B) Locomotor activity of wild‐type C57BL/6 mice (n = 6) induced by 10 mg⋅kg−1 ARM390, 5 mg⋅kg−1 SNC162, 20 mg⋅kg−1 SNC80 or saline. (C) Positive correlation between the β‐arrestin 2 recruitment efficacy of the tested compounds and their modulation of alcohol intake. Significance was determined by paired one‐way anova followed by Dunnet's post hoc analysis versus saline. *P < 0.05, **P < 0.01, ***P < 0.001.
δ Receptor agonists did not significantly change water intake at any dose; changes in alcohol intake caused concomitant changes in alcohol preference, but these were not significant (Supporting Information Fig. S3). As SNC80 is well known to increase locomotor activity, we measured locomotor activity of all the drugs (SNC80, SNC162 and ARM390) that increased alcohol consumption in wild‐type mice. We found that SNC80 (20 mg⋅kg−1) and to a lesser extent SNC162 (5 mg⋅kg−1), but not ARM390 (10 mg⋅kg−1), significantly increased locomotor activity (F 3, 20 = 8.573, P < 0.001) over saline injection (Figure 3). When we plotted the β‐arrestin 2 efficacy of the tested drugs against their maximum effect in modulating alcohol intake, we found that the ability of these DOR agonists to recruit β‐arrestin 2 showed a clear positive correlation (Pearson r = 0.93, P < 0.01) and had a significant goodness of fit (r 2 = 0.47, P < 0.0001) with alcohol intake in mice (Figure 3). While, depending on dose, KNT‐127's effect on alcohol intake was bi‐directional, we used the 3 mg⋅kg−1 dose of KNT‐127 for this correlation, as this dose was shown to be effective in decreasing anxiety and depression‐like behaviour (Saitoh et al., 2011; Saitoh et al., 2013). In comparison, alcohol intake is not significantly correlated with DOR agonist potency in the cAMP assay or the β‐arrestin 2 recruitment assay (Supporting Information Fig. S4).
The δ receptor agonist SNC80 increases alcohol consumption in a β‐arrestin 2‐dependent manner
To establish if the observed correlation between β‐arrestin 2 recruitment efficacy and alcohol intake has a causal base, we next investigated if the ability of the super‐recruiter SNC80 to increase alcohol intake (Figure 2) would be affected by β‐arrestin 2 KO. We also injected mice with TAN‐67 based on our hypothesis that the behavioural effects of this poor β‐arrestin 2 recruiter may be less reliant on β‐arrestin 2. For this, we trained β‐arrestin 2 KO mice to consume 10% alcohol in a two‐bottle choice paradigm (refer to Methods). We found that TAN‐67 (25 mg⋅kg−1, s.c.) was able to reduce alcohol consumption in β‐arrestin 2 KO mice (F 2, 23 = 7.487, P < 0.01). In contrast, SNC80 (20 mg⋅kg−1, s.c.) was unable to significantly increase alcohol intake in the KO mice (Figure 4). We further confirmed SNC80's functional dependence on β‐arrestin 2 using an alcohol place preference paradigm. In this model, alcohol produces a significant conditioned place preference (F 1, 21 = 58.42, P < 0.0001). We previously showed in wild‐type mice that SNC80 (20 mg⋅kg−1) blocks the acquisition of alcohol place preference (van Rijn et al., 2012b); yet in β‐arrestin 2 KO mice, we found no significant difference between saline‐treated and SNC80‐treated mice in alcohol place preference (F 1, 21 = 1.22, P = 0.28). There was no significant interaction between drug effect and conditioning state (F 1,21 = 0.504, P = 0.49) on alcohol place preference in β‐arrestin 2 KO mice (Figure 4).
Figure 4.
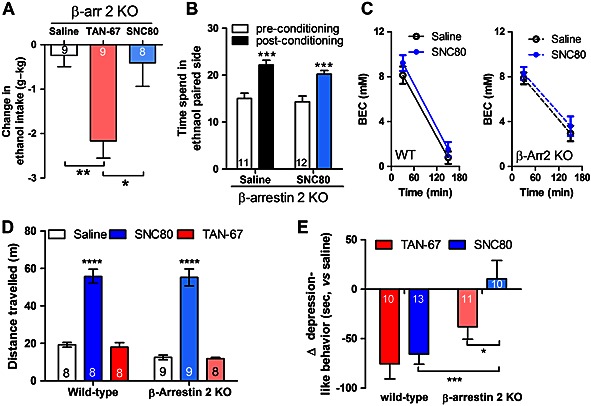
Behavioural effects of β‐arrestin 2 super‐recruiters like SNC80 are β‐arrestin 2 dependent. (A) The ability of SNC80 (20 mg⋅kg−1, s.c.) to increase alcohol intake is abolished in transgenic β‐arrestin 2 KO mice. In contrast, TAN‐67 (25 mg⋅kg−1, s.c.) still decreased alcohol intake in β‐arrestin 2 KO mice. (B) SNC80 (20 mg⋅kg−1, s.c.) did not alter the expression of alcohol place preference in β‐arrestin 2 KO mice. (C) SNC80 (20 mg⋅kg−1, s.c.) did not affect alcohol metabolism in wild‐type and β‐arrestin 2 KO mice. (D) β‐Arrestin 2 KO did not change SNC80 (20 mg⋅kg−1, s.c.) induced hyperlocomotion, or induce locomotor activity to TAN‐67 (25 mg⋅kg−1, s.c.). (E) The ability of SNC80 (20 mg⋅kg−1 s.c.) and to a lesser extent TAN‐67 (25 mg⋅kg−1 s.c.) to reduce depression‐like behaviour was attenuated in β‐arrestin 2 KO mice. The group size for each drug is depicted inside the column of that drug. Significance was determined by two‐way anova followed by a Bonferonni post hoc test. *P < 0.05, ***P < 0.001, ****P < 0.0001.
To investigate if SNC80 affects alcohol metabolism, we injected wild‐type and β‐arrestin 2 KO mice with saline or 20 mg⋅kg−1 SNC80 (s.c.) prior to a bolus of 2.5 g⋅kg−1 alcohol and tracked blood alcohol concentrations. In line with previous results published for TAN‐67 (van Rijn et al., 2009), we found no significant difference between saline and SNC80 injection in either wild‐type or β‐arrestin 2 KO mice (F 3, 28 = 1.471, P = 0.25, Figure 4). Two‐way anova revealed a small significance effect for genotype × time interaction (F 3, 28 = 3.124, P = 0.42), which could indicate that alcohol metabolism between wild‐type and β‐arrestin 2 KO mice differs slightly.
SNC80 is well known to increase locomotor activity (Jutkiewicz et al., 2004; Pradhan et al., 2010), and at a dose of 20 mg⋅kg−1 (s.c.), it significantly increases locomotion (F 2, 45 = 75.70, P < 0.0001) in our mice as well. We did not find significant differences (F 1, 45 = 1.31, P = 0.26) in drug‐induced locomotor effects between wild‐type and in β‐arrestin 2 KO mice (Figure 4). Two‐way anova did not reveal a significant interaction (F 2, 45 = 0.141, P = 0.87) between drug treatment and genotype (Figure 4). TAN‐67 (25 mg⋅kg−1, s.c.) did not alter locomotion in wild‐type mice, and disruption of β‐arrestin 2 does not change the locomotor activity of mice injected with TAN‐67 (Figure 4).
Both SNC80 and TAN‐67 decrease depression‐like behaviour
Next, we investigated the role of β‐arrestin 2 in the anti‐depressant‐like effects of δ receptor agonists with different β‐arrestin 2 recruitment efficacy. For this, we injected wild‐type and β‐arrestin 2 KO mice with SNC80 (20 mg⋅kg−1, s.c.) and TAN‐67 (25 mg⋅kg−1, s.c.) measured anti‐depressant‐like behaviour using a forced swim paradigm. We found that both TAN‐67 and SNC80 reduced depressant‐like behaviours (Figure 4). To assess the role of β‐arrestin 2 in the anti‐depressant‐like effects of SNC80 and TAN‐67, we injected β‐arrestin 2 KO mice with SNC80 (20 mg⋅kg−1, s.c.) and TAN‐67 (25 mg⋅kg−1, s.c.). We found significant differences in drug effect (F 1, 41 = 4.33, P < 0.05) and genotype (F 1, 41 = 16.33, P < 0.001) in the ability of SNC80 to reduce depression‐like behaviour in wild‐type and β‐arrestin 2 KO mice compared with TAN‐67 (Figure 4). Two‐way anova did not reveal a significant interaction between drug effect and genotype (F 1, 41 = 1.93, P = 0.17). Transgenic KO of β‐arrestin 2 appears to attenuate TAN‐67‐mediated anti‐depression‐like behaviour; however, this was not significant.
Discussion
Here, we tested G‐protein signalling and β‐arrestin 2 recruitment of six δ receptor selective agonists and compared their in vitro activity with their ability to modulate alcohol intake in mice. We found a strong correlation between β‐arrestin 2 recruitment efficacy and alcohol intake. Using transgenic β‐arrestin 2 KO mice, we further confirmed this correlation. The δ receptor agonists that we used in this study, with the exception of NIH11082 for which we could not find relative efficacy data, have previously been reported to act as full agonists in GTPyS binding assays (Quock et al., 1997; Wei et al., 2000; Jutkiewicz et al., 2004; Nemoto et al., 2013). We confirmed the full agonism of these δ receptor agonists in the G‐protein coupled cAMP inhibition assay, although the artificial overexpressed nature of our cell system may preclude us from observing partial agonism in this assay. Interestingly, the six agonists differed in their efficacy to recruit β‐arrestin 2. We next tested the ability of the six δ receptor agonists to modulate alcohol intake. With the exception of TAN‐67, for which we previously tested multiple doses in the same assay (van Rijn et al., 2009), we tested three doses of each drug (Figure 3 and Supporting Information Fig. S2). Previous studies using δ receptor KO mice or δ receptor antagonists show that at the tested dose, the functional effect of the agonists is through their selective action on δ receptors (Jutkiewicz et al., 2004; Naidu et al., 2007; van Rijn and Whistler, 2009; Saitoh et al., 2011; Nozaki et al., 2014; Chu Sin Chung et al., 2015). The tested doses were based on therapeutically effective doses as previously published, and we found that the most effective dose of each δ receptor agonist corresponded well with the published reports (Jutkiewicz et al., 2004; Naidu et al., 2007; Pradhan et al., 2010; Saitoh et al., 2011; Saitoh et al., 2013; Nozaki et al., 2014). With the exception of KNT‐127, all agonists displayed a one‐directional dose–response curve, either increasing or decreasing alcohol intake. Our β‐arrestin 2 recruitment versus alcohol intake correlation intersects at a β‐arrestin 2 recruitment efficacy of 78% (Figure 3). One would predict that near the intersection, the dose of a δ receptor agonist is crucial in determining whether that agonist will increase or decrease alcohol intake. Indeed, we found that KNT‐127 (β‐arrestin 2 efficacy = 71%, Table 1) exhibits a bi‐directional dose–response: decreasing alcohol consumption, albeit not significant, at a low dose, but increasing alcohol intake at a high dose.
Thus, we showed that δ receptor agonists that are super‐recruiters of β‐arrestin 2 may display a clear unwanted side effect, in that those agonists increased alcohol intake in mice. We report here for the first time as well that TAN‐67 is a weak recruiter of β‐arrestin 2 that decreases alcohol intake and depression‐like behaviour in a β‐arrestin 2‐independent manner. Our results are highly significant as the two δ receptor agonists AZD2327 (Hudzik et al., 2011) and ADL5859 (Le Bourdonnec et al., 2008; Nozaki et al., 2012) that have been in clinical trials for treatment of major depressive disorder, and acute and chronic pain, respectively, are structurally similar to SNC80 and may be expected to increase alcohol intake.
δ Receptors play a role in mood disorders (Filliol et al., 2000; Lutz and Kieffer, 2013), and both academic and industrial laboratories have developed δ receptor agonists that reduce depression‐like behaviour (Saitoh and Yamada, 2012). Several research groups have also investigated δ receptors as potential target for treatment of alcohol use disorders (Froehlich et al., 1998; Margolis et al., 2008; van Rijn et al., 2010; Nielsen and Bartlett, 2012a). Considering that depression is co‐morbid with alcohol use disorders (Petrakis et al., 2002), δ receptors could be a particular interesting target for the developing drugs that simultaneously reduce depression and alcohol use. Yet our data on δ receptor agonist modulation of alcohol intake indicate that strong β‐arrestin 2 recruiters such as SNC80, while effective as anti‐depressants, will carry a potential risk for increasing alcohol use in patients. On the other hand, a drug like TAN‐67 could therapeutically be very efficacious in treating patients suffering from alcohol use disorders co‐morbid with depression or anxiety (van Rijn et al., 2010).
Are there correlations between β‐arrestin 2 recruitment and other behavioural effects of δ receptor agonists?
Depression
Previous studies have shown that SNC80, KNT‐127 and NIH 11082 can reduce depression‐like behaviour (Jutkiewicz et al., 2005a; Naidu et al., 2007; Saitoh et al., 2011). Here, we demonstrated for the first time that SNC80 and TAN‐67 also decrease depression‐like behaviour in mice. Considering the wide range in β‐arrestin 2 efficacy between these δ receptor agonists, it appears that β‐arrestin 2 recruitment is not important in mediating depression‐like behaviour. We found that the anti‐depressant‐like effects of SNC80 were abolished in β‐arrestin 2 KO mice; however, it is important to note that the β‐arrestin 2 KO mice displayed altered basal depression levels that confound proper interpretation of our results (Supporting Information Fig. S5). Yet, despite the altered basal depression, we still observed differences between SNC80 and TAN‐67.
Hyperlocomotion
SNC80, SNC162 and AZD2327 are known to induce hyperlocomotion (Jutkiewicz et al., 2004; Hudzik et al., 2011). Here, we showed that the SNC80‐induced hyperlocomotion is not dependent on β‐arrestin 2. As has been previously suggested (Pradhan et al., 2010; Hudzik et al., 2011), we confirmed that acute ARM390 does not induce hyperlocomotion. The finding that in our assay ARM390 has strong β‐arrestin 2 recruitment efficacy would agree with our hypothesis that δ receptor agonist‐induced locomotion is independent from β‐arrestin 2. For additional support, we also measured TAN‐67‐induced locomotion in wild‐type and β‐arrestin 2 KO mice and did not find any changes in locomotion compared with saline in either strain of mice. These results are also important, as they indicate that locomotor activity does not confound the behavioural responses observed for δ receptor agonists in alcohol intake and depression‐like behaviour. Considering the long breeding history of our β‐arrestin 2 KO mice, it would be valuable to have some of our findings be confirmed in β‐arrestin 2 KO mice bred in other labs to ensure that the obtained data are truly due to the disruption of β‐arrestin 2 and not due to other genotype differences introduced in maintaining the colony.
Convulsions
A potentially severe side effect commonly associated with δ receptor activation are convulsions. It is known that SNC80 and SNC162 induce convulsions at high doses (Jutkiewicz et al., 2004; Jutkiewicz et al., 2005a, 2005b) as does AZD2327 in some species (Hudzik et al., 2011; Hudzik et al., 2014). Many other δ receptor agonists, like ADL5859, ARM390 and KNT‐127, are reportedly devoid of convulsion activity (Le Bourdonnec et al., 2008; Saitoh et al., 2011; Chu Sin Chung et al., 2015). Considering that SNC80 and SNC162 were the most efficacious recruiters, it would be worthy to follow up on this study by investigating the correlation between β‐arrestin 2 recruitment and convulsive properties of δ receptor agonists and observe if selective knockdown of β‐arrestin 2 in GABAergic forebrain neurons eliminates SNC80‐induced seizures (Chu Sin Chung et al., 2015).
Drug tolerance
δ Receptors, in contrast with μ receptors, are more frequently targeted to lysozomes for degradation (Whistler et al., 2002; Pradhan et al., 2009). Another issue with SNC80‐like compounds is that due to their strong ability to recruit and interact with β‐arrestin 2, they are more likely to direct internalized δ receptors to a path of degradation (Audet et al., 2012) in comparison with agonists that are moderate recruiters like DPDPE that interact with β‐arrestin 2 more transiently and promote δ receptor recycling (Audet et al., 2012) and ARM390 and KNT‐127 that are a poor internalizers of δ receptors compared with the more efficacious β‐arrestin 2 recruiter SNC80 (Pradhan et al., 2009; Nozaki et al., 2014). When compared with δ receptor agonists that are poor internalizers of the δ receptor, SNC80 may cause receptor tolerance (Pradhan et al., 2010). Thus, strong β‐arrestin 2 recruiting δ receptor agonists may be less therapeutically interesting not only because they can increase alcohol intake but also because chronic use can more readily lead to tolerance development.
Conclusion
GPCRs remain a major drug target, and new insights into their pharmacology have revealed opportunities to produce efficacious drugs that may be less hampered by side effects. In particular, development of drugs that prefer activating either G‐protein‐mediated signal transduction pathways or β‐arrestin 2‐mediated signalling are of therapeutic interest (Whalen et al., 2011). Based on our current data, previously reported work by us and others, we would recommend for this moment to avoid developing δ receptor agonists that have a high efficacy for β‐arrestin 2 recruitment. Our data suggest that these super‐recruiters can increase alcohol consumption in mice, whereas other reports have suggest that drug tolerance is also a bigger concern for these agonists (Pradhan et al., 2010) as are convulsions (Jutkiewicz et al., 2004; Hudzik et al., 2014). The δ receptor crystal structure was resolved (Granier et al., 2012), and amino acids in the δ receptor structure that are important for β‐arrestin 2 recruitment were identified in another study (Fenalti et al., 2014). Recently, structural data have also emerged for β2‐adrenoceptors recruiting β‐arrestin 1 (Shukla et al., 2014) and rhodopsin bound to arrestin (Kang et al., 2015). Together, it may be possible to build a model that will help us better understand potential differences in δ receptor conformation induced by SNC80‐like β‐arrestin 2 super‐recruiters compared with weak β‐arrestin 2 recruiters like TAN‐67 and NIH11082. Such modelling studies would aid future drug development of δ receptor agonists with a lower risk of severe adverse effects.
Author contributions
R.M.v.R conceived and designed the experiments T.C., K.C. and R.M.v.R. conducted experiments and analyzed data T.C., K.C. and R.M.v.R. wrote the manuscript.
Conflict of interest
The authors declare to have no potential conflict of interest in this study.
Supporting information
Figure S1 S.c, and gavage injections of saline do not affect water and alcohol intake. Wild‐type and β‐arrestin 2 knockout (B‐Arr2 KO) C57BL/6 male mice were trained to consume 10% alcohol. Mice were habituated to handling and injection during training. The s.c. or p.o. saline injections did not change baseline water (A) or alcohol (B) intake.
Figure S2 Lack of linear dose–response effect for δ receptor agonists with regard to modulation of alcohol intake. Modulation of alcohol intake in wild‐type C57BL/6 male mice trained to consume 10% alcohol in a two‐bottle choice paradigm limited access by different doses of δ receptor agonists with distinct difference in β‐arrestin 2 recruitment efficacy. The group size for each drug is depicted inside the column of that drug.
Figure S3 Impact of water intake and alcohol preference of δ receptor agonists. Modulation of water intake (A) and alcohol preference (B) in wild‐type C57BL/6 male mice trained to consume 10% alcohol in a two‐bottle choice paradigm limited access by different doses of δ receptor agonists.
Figure S4 Weak correlation between potency of δ receptor agonists and their modulation of alcohol intake. δ receptor agonists show a weak correlation when their behaviour modulation alcohol intake is plotted against their potency in inhibiting cAMP or their potency to recruit β‐arrestin 2.
Figure S5 β‐Arrestin 2 knockout mice display reduced basal depression‐like behaviour. Depression‐like behaviour in wild‐type and β‐arrestin 2 knockout (B‐Arr2 KO) male C57Bl/6 mice (n = 10) was tested by measuring time spent immobile in a 6 min forced swim test.
Supporting Info item
Acknowledgements
We thank Dr. Lefkowitz for providing us with the β‐arrestin 2 KO mice. This work was financially supported by a National Institutes of Health K99/R00 grant through the National Institute on Alcohol Abuse and Alcoholism (AA20539).
Chiang, T. , Sansuk, K. , and van Rijn, R. M. (2016) β‐Arrestin 2 dependence of δ opioid receptor agonists is correlated with alcohol intake. British Journal of Pharmacology, 173: 332–343. doi: 10.1111/bph.13374.
References
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013). The concise guide to pharmacology 2013/14: G protein‐coupled receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audet N, Charfi I, Mnie‐Filali O, Amraei M, Chabot‐Dore AJ, Millecamps M et al (2012). Differential association of receptor‐Gbetagamma complexes with beta‐arrestin2 determines recycling bias and potential for tolerance of delta opioid receptor agonists. J Neurosci 32: 4827–4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L, Jin S‐X, Zhang C, Wang L‐H, Xu Z‐Z, Zhang F‐X et al (2003). Activation of delta opioid receptors induces receptor insertion and neuropeptide secretion. Neuron 37: 121–133. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Morinville A, Lee MC, Vincent JP, Collier B, Beaudet A (2001). Prolonged morphine treatment targets delta opioid receptors to neuronal plasma membranes and enhances delta‐mediated antinociception. J Neurosci 21: 7598–7607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Sin Chung P, Kieffer BL (2013). Delta opioid receptors in brain function and diseases. Pharmacol Ther 140: 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Sin Chung P, Boehrer A, Stephan A, Matifas A, Scherrer G, Darcq E et al (2015). Delta opioid receptors expressed in forebrain GABAergic neurons are responsible for SNC80‐induced seizures. Behav Brain Res 278: 429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK (2007). β‐Arrestins and cell signaling. Annu Rev Physiol 69: 483–510. [DOI] [PubMed] [Google Scholar]
- Enquist J, Ferwerda M, Madhavan A, Hok D, Whistler JL (2012). Chronic ethanol potentiates the effect of neuropeptides in the basolateral amygdala and shows increased anxiolytic and anti‐depressive effects. Neuropsychopharmacology 37: 2436–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenalti G, Giguere PM, Katritch V, Huang XP, Thompson AA, Cherezov V et al (2014). Molecular control of delta‐opioid receptor signalling. Nature 506: 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filliol D, Ghozland S, Chluba J, Martin M, Matthes HW, Simonin F et al (2000). Mice deficient for delta‐ and mu‐opioid receptors exhibit opposing alterations of emotional responses. Nat Genet 25: 195–200. [DOI] [PubMed] [Google Scholar]
- Froehlich JC, Badia‐Elder NE, Zink RW, McCullough DE, Portoghese PS (1998). Contribution of the opioid system to alcohol aversion and alcohol drinking behavior. J Pharmacol Exp Ther 287: 284–292. [PubMed] [Google Scholar]
- Gomes I, Jordan BA, Gupta A, Trapaidze N, Nagy V, Devi LA (2000). Heterodimerization of mu and delta opioid receptors: A role in opiate synergy. J Neurosci 20: RC110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Gupta A, Filipovska J, Szeto HH, Pintar JE, Devi LA (2004). A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia. Proc Natl Acad Sci U S A 101: 5135–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Fujita W, Gupta A, Saldanha AS, Negri A, Pinello CE et al (2013). Identification of a mu‐delta opioid receptor heteromer‐biased agonist with antinociceptive activity. Proc Natl Acad Sci U S A 110: 12072–12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI et al (2012). Structure of the delta‐opioid receptor bound to naltrindole. Nature 485: 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Mulder J, Gomes I, Rozenfeld R, Bushlin I, Ong E et al (2010). Increased abundance of opioid receptor heteromers after chronic morphine administration. Sci Signal 3: ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He SQ, Zhang ZN, Guan JS, Liu HR, Zhao B, Wang HB et al (2011). Facilitation of mu‐opioid receptor activity by preventing delta‐opioid receptor‐mediated codegradation. Neuron 69: 120–131. [DOI] [PubMed] [Google Scholar]
- Hudzik TJ, Pietras MR, Caccese R, Bui KH, Yocca F, Paronis CA et al (2014). Effects of the delta opioid agonist AZD2327 upon operant behaviors and assessment of its potential for abuse. Pharmacol Biochem Behav 124C: 48–57. [DOI] [PubMed] [Google Scholar]
- Hudzik TJ, Maciag C, Smith MA, Caccese R, Pietras MR, Bui KH et al (2011). Preclinical pharmacology of AZD2327: a highly selective agonist of the delta‐opioid receptor. J Pharmacol Exp Ther 338: 195–204. [DOI] [PubMed] [Google Scholar]
- Jutkiewicz EM, Rice KC, Traynor JR, Woods JH (2005a). Separation of the convulsions and antidepressant‐like effects produced by the delta‐opioid agonist SNC80 in rats. Psychopharmacology (Berl) 182: 588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutkiewicz EM, Kaminsky ST, Rice KC, Traynor JR, Woods JH (2005b). Differential behavioral tolerance to the delta‐opioid agonist SNC80 ([(+)‐4‐[(alphaR)‐alpha‐[(2S,5R)‐2,5‐dimethyl‐4‐(2‐propenyl)‐1‐piperazinyl]‐(3‐methoxyphenyl)methyl]‐N,N‐diethylbenzamide) in Sprague–Dawley rats. J Pharmacol Exp Ther 315: 414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutkiewicz EM, Eller EB, Folk JE, Rice KC, Traynor JR, Woods JH (2004). Delta‐opioid agonists: differential efficacy and potency of SNC80, its 3‐OH (SNC86) and 3‐desoxy (SNC162) derivatives in Sprague–Dawley rats. J Pharmacol Exp Ther 309: 173–181. [DOI] [PubMed] [Google Scholar]
- Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A et al (2015). Crystal structure of rhodopsin bound to arrestin by femtosecond X‐ray laser. Nature 523: 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW (2010). Animal research: reporting in vivo experiments: the ARRIVE Guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp RJ, Santoro G, de Leon IA, Lee KB, Edsall SA, Waite S et al (1996). Structure–activity relationships for SNC80 and related compounds at cloned human delta and mu opioid receptors. J Pharmacol Exp Ther 277: 1284–1291. [PubMed] [Google Scholar]
- le Bourdonnec B, Windh RT, Ajello CW, Leister LK, Gu M, Chu GH et al (2008). Potent, orally bioavailable delta opioid receptor agonists for the treatment of pain: discovery of N,N‐diethyl‐4‐(5‐hydroxyspiro[chromene‐2,4′‐piperidine]‐4‐yl)benzamide (ADL5859). J Med Chem 51: 5893–5896. [DOI] [PubMed] [Google Scholar]
- Lecoq I, Marie N, Jauzac P, Allouche S (2004). Different regulation of human delta‐opioid receptors by SNC‐80 [(+)‐4‐[(alphaR)‐alpha‐((2S,5R)‐4‐allyl‐2,5‐dimethyl‐1‐piperazinyl)‐3‐meth oxybenzyl]‐N,N‐diethylbenzamide] and endogenous enkephalins. J Pharmacol Exp Ther 310: 666–677. [DOI] [PubMed] [Google Scholar]
- Lutz PE, Kieffer BL (2013). Opioid receptors: distinct roles in mood disorders. Trends Neurosci 36: 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Fields HL, Hjelmstad GO, Mitchell JM (2008). Delta‐opioid receptor expression in the ventral tegmental area protects against elevated alcohol consumption. J Neurosci 28: 12672–12681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Mitchell JM, Hjelmstad GO, Fields HL (2011). A novel opioid receptor‐mediated enhancement of GABAA receptor function induced by stress in ventral tegmental area neurons. J Physiol 589: 4229–4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidu PS, Lichtman AH, Archer CC, May EL, Harris LS, Aceto MD (2007). NIH 11082 produces anti‐depressant‐like activity in the mouse tail‐suspension test through a delta‐opioid receptor mechanism of action. Eur J Pharmacol 566: 132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto T, Ida Y, Iihara Y, Nakajima R, Hirayama S, Iwai T et al (2013). The most effective influence of 17‐(3‐ethoxypropyl) substituent on the binding affinity and the agonistic activity in KNT‐127 derivatives, delta opioid receptor agonists. Bioorg Med Chem 21: 7628–7647. [DOI] [PubMed] [Google Scholar]
- Nielsen CK, Bartlett SE (2012a). The role of delta opioid receptors in ethanol consumption and seeking: implications for new treatments for alcohol use disorders In: {0} (ed)Contreras CM. Neuroscience – Dealing with frontiers. InTech.: Reijeka, pp. 205–240. [Google Scholar]
- Nielsen CK, Simms JA, Li R, Mill D, Yi H, Feduccia AA et al (2012b). Delta‐opioid receptor function in the dorsal striatum plays a role in high levels of ethanol consumption in rats. J Neurosci 32: 4540–4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki C, Nagase H, Nemoto T, Matifas A, Kieffer BL, Gaveriaux‐Ruff C (2014). In vivo properties of KNT‐127, a novel delta opioid receptor agonist: receptor internalization, antihyperalgesia and antidepressant effects in mice. Br J Pharmacol 171: 5376–5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki C, le Bourdonnec B, Reiss D, Windh RT, Little PJ, Dolle RE et al (2012). Delta‐opioid mechanisms for ADL5747 and ADL5859 effects in mice: analgesia, locomotion, and receptor internalization. J Pharmacol Exp Ther 342: 799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, Davenport AP, McGrath JC, Peters JA, Southan C, Spedding M, Yu W, Harmar AJ; NC‐IUPHAR . (2014) The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 42 (Database Issue): D1098‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrakis I, Gonzales G, Rosenheck R, Krystal JH (2002). Comorbidity of alcoholism and psychiatric disorders – an overview. Alcohol Res Health J Natl Inst Alcohol Abuse Alcoholism 26: 81–89. [Google Scholar]
- Pradhan AA, Befort K, Nozaki C, Gaveriaux‐Ruff C, Kieffer BL (2011). The delta opioid receptor: an evolving target for the treatment of brain disorders. Trends Pharmacol Sci 32: 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Walwyn W, Nozaki C, Filliol D, Erbs E, Matifas A et al (2010). Ligand‐directed trafficking of the delta‐opioid receptor in vivo: two paths toward analgesic tolerance. J Neurosci 30: 16459–16468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Becker JA, Scherrer G, Tryoen‐Toth P, Filliol D, Matifas A et al (2009). In vivo delta opioid receptor internalization controls behavioral effects of agonists. PLoS One 4: e5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quock RM, Hosohata Y, Knapp RJ, Burkey TH, Hosohata K, Zhang X et al (1997). Relative efficacies of delta‐opioid receptor agonists at the cloned human delta‐opioid receptor. Eur J Pharmacol 326: 101–104. [DOI] [PubMed] [Google Scholar]
- Saitoh A, Yamada M (2012). Antidepressant‐like effects of delta opioid receptor agonists in animal models. Curr Neuropharmacol 10: 231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh A, Kimura Y, Suzuki T, Kawai K, Nagase H, Kamei J (2004). Potential anxiolytic and antidepressant‐like activities of SNC80, a selective delta‐opioid agonist, in behavioral models in rodents. J Pharmacol Sci 95: 374–380. [DOI] [PubMed] [Google Scholar]
- Saitoh A, Sugiyama A, Yamada M, Inagaki M, Oka J, Nagase H et al (2013). The novel delta opioid receptor agonist KNT‐127 produces distinct anxiolytic‐like effects in rats without producing the adverse effects associated with benzodiazepines. Neuropharmacology 67: 485–493. [DOI] [PubMed] [Google Scholar]
- Saitoh A, Sugiyama A, Nemoto T, Fujii H, Wada K, Oka J et al (2011). The novel delta opioid receptor agonist KNT‐127 produces antidepressant‐like and antinociceptive effects in mice without producing convulsions. Behav Brain Res 223: 271–279. [DOI] [PubMed] [Google Scholar]
- Shukla AK, Westfield GH, Xiao K, Reis RI, Huang LY, Tripathi‐Shukla P et al (2014). Visualization of arrestin recruitment by a G‐protein‐coupled receptor. Nature 512: 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rijn RM, Whistler JL (2009). The delta(1) opioid receptor is a heterodimer that opposes the actions of the delta(2) receptor on alcohol intake. Biol Psychiatry 66: 777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rijn RM, Brissett DI, Whistler JL (2010). Dual efficacy of delta opioid receptor selective ligands for ethanol drinking and anxiety. J Pharmacol Exp Ther 335: 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rijn RM, Brissett DI, Whistler JL (2012a). Emergence of functional spinal delta opioid receptors after chronic ethanol exposure. Biol Psychiatry 71: 232–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rijn RM, Brissett DI, Whistler JL (2012b). Distinctive modulation of ethanol place preference by delta opioid receptor‐selective agonists. Drug Alcohol Depend 122: 156–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rijn RM, Defriel JN, Whistler JL (2013a). Pharmacological traits of delta opioid receptors: pitfalls or opportunities? Psychopharmacology (Berl) 228: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rijn RM, Harvey JH, Brissett DI, DeFriel JN, Whistler JL (2013b). Novel screening assay for the selective detection of G‐protein‐coupled receptor heteromer signaling. J Pharmacol Exp Ther 344: 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei ZY, Brown W, Takasaki B, Plobeck N, Delorme D, Zhou F et al (2000). N,N‐Diethyl‐4‐(phenylpiperidin‐4‐ylidenemethyl)benzamide: a novel, exceptionally selective, potent delta opioid receptor agonist with oral bioavailability and its analogues. J Med Chem 43: 3895–3905. [DOI] [PubMed] [Google Scholar]
- Whalen EJ, Rajagopal S, Lefkowitz RJ (2011). Therapeutic potential of beta‐arrestin‐ and G protein‐biased agonists. Trends Mol Med 17: 126–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whistler JL, Enquist J, Marley A, Fong J, Gladher F, Tsuruda P et al (2002). Modulation of postendocytic sorting of G protein‐coupled receptors. Science 297: 615–620. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 S.c, and gavage injections of saline do not affect water and alcohol intake. Wild‐type and β‐arrestin 2 knockout (B‐Arr2 KO) C57BL/6 male mice were trained to consume 10% alcohol. Mice were habituated to handling and injection during training. The s.c. or p.o. saline injections did not change baseline water (A) or alcohol (B) intake.
Figure S2 Lack of linear dose–response effect for δ receptor agonists with regard to modulation of alcohol intake. Modulation of alcohol intake in wild‐type C57BL/6 male mice trained to consume 10% alcohol in a two‐bottle choice paradigm limited access by different doses of δ receptor agonists with distinct difference in β‐arrestin 2 recruitment efficacy. The group size for each drug is depicted inside the column of that drug.
Figure S3 Impact of water intake and alcohol preference of δ receptor agonists. Modulation of water intake (A) and alcohol preference (B) in wild‐type C57BL/6 male mice trained to consume 10% alcohol in a two‐bottle choice paradigm limited access by different doses of δ receptor agonists.
Figure S4 Weak correlation between potency of δ receptor agonists and their modulation of alcohol intake. δ receptor agonists show a weak correlation when their behaviour modulation alcohol intake is plotted against their potency in inhibiting cAMP or their potency to recruit β‐arrestin 2.
Figure S5 β‐Arrestin 2 knockout mice display reduced basal depression‐like behaviour. Depression‐like behaviour in wild‐type and β‐arrestin 2 knockout (B‐Arr2 KO) male C57Bl/6 mice (n = 10) was tested by measuring time spent immobile in a 6 min forced swim test.
Supporting Info item