

Abstract
Background and Purpose
Endothelin‐1 (ET‐1) is increased in patients with sickle cell disease and may contribute to the development of sickle cell nephropathy. The current study was designed to determine whether ET‐1 acting via the ETA receptor contributes to renal injury in a mouse model of sickle cell disease.
Experimental Approach
Adult, humanized HbSS (homozygous for sickle Hb) mice had increased ET‐1 mRNA expression in both the cortex and the glomeruli compared with mice heterozygous for sickle and Hb A (HbAS controls). In the renal cortex, ETA receptor mRNA expression was also elevated in HbSS (sickle) mice although ETB receptor mRNA expression was unchanged. Ligand binding assays confirmed that sickle mice had increased ETA receptors in the renal vascular tissue when compared with control mice.
Key Results
In response to PKC stimulation, reactive oxygen species production by isolated glomeruli from HbSS sickle mice was increased compared with that from HbSA controls, an effect that was prevented by 1 week in vivo treatment with the selective ETA antagonist, ABT‐627. Protein and nephrin excretion were both elevated in sickle mice, effects that were also significantly attenuated by ABT‐627. Finally, ETA receptor antagonism caused a significant reduction in mRNA expression of NADPH oxidase subunits, which may contribute to nephropathy in sickle cell disease.
Conclusions and Implications
These data support a novel role for ET‐1 in the progression of sickle nephropathy, specifically via the ETA receptor, and suggest a potential role for ETA receptor antagonism in a treatment strategy.
Abbreviations
- ET‐1
endothelin‐1
- HbAS
heterozygous for sickle and Hb A
- HbSS
homozygous for sickle Hb
- ROS
reactive oxygen species
- SAD
HbS Antillies and D‐Punjab
- SCD
sickle cell disease
- SCN
sickle cell nephropathy
Tables of Links
| TARGETS |
| ETA receptor |
| ETB receptor |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Despite significant gains in life expectancy over the past three decades, adult patients with sickle cell disease (SCD) continue to suffer from progressive organ damage. Although hydroxyurea typically confers significant long‐term treatment benefits, most patients still suffer chronic effects of the disease. Sickle cell nephropathy (SCN) has emerged as one of the major causes of morbidity and mortality in SCD (Abbott et al., 2002; Yanni et al., 2009). Nearly 70% of adults with SCD will develop either micro or macroalbuminuria with about 30% developing overt proteinuria, an early indicator for chronic kidney disease (Guasch et al., 2006). Currently, it is estimated that nearly one in five of all SCD patients will progress to end‐stage renal disease requiring long‐term dialysis (Powars et al., 1991).
Renal pathology in SCD is thought to begin in childhood and progress through adulthood. Early stages are characterized by ischaemic medullary damage, urine concentrating deficiency and increased GFR (Statius van Eps et al., 1970). This is followed by progressive proteinuria, glomerulosclerosis and eventual decline in GFR (Falk et al., 1992; Falk and Jennette, 1994). Despite the growing prevalence of this chronic manifestation, the mechanisms behind SCN are largely unknown and commonly believed to be due to recurring vaso‐occlusive crises and intermittent hypoxia. Currently, there are no effective treatment modalities to adequately address the long‐term effects of this progressive renal disease.
The endothelium‐derived peptide, endothelin‐1 (ET‐1), has strong vasoconstrictor, proliferative and pro‐inflammatory effects that are mediated primarily through endothelin type A (ETA) receptors (Kohan et al., 2011). ET‐1 acts in either a paracrine or autocrine fashion and is released in response to shear stress, hypoxia, hemin, angiotensin II, thrombin activation and inflammatory cytokines – all are prevalent in SCD. Indeed, ET‐1 is elevated in the plasma and urine of patients with SCD (Rybicki and Benjamin, 1998; Tharaux et al., 2005). Furthermore, ET‐1 was recently shown to have potent systemic adverse effects in one of the first mouse models of SCD, potentially exacerbating hypoxia/reoxygenation injury and stimulating the progression of SCN (Sabaa et al., 2008).
ET‐1 also increases the production of reactive oxygen species (ROS). ROS are chemically active molecules containing oxygen and persist physiologically as a product of metabolism and as a defence mechanism utilized by the immune system to neutralize bacterial threats. However, when ROS are produced in amounts that overwhelm endogenous antioxidants, significant cellular damage can result due to oxidative stress. ET‐1 stimulates the production of ROS in arterial smooth muscle cells, endothelial cells and isolated arteries, demonstrating a putative mechanism for the pathological actions of ET‐1 (Dong et al., 2005; Laplante et al., 2005; Loomis et al., 2005).
ROS are implicated in a wide range of disease processes such as hypertension and chronic kidney disease and also SCD (Wolin, 2000; Wilcox, 2012). SCD creates a constant state of increased oxidative stress, owing to increased ROS and chemically active nitrogen metabolites, the effects of which are thought to contribute significantly to the long‐term pathology of SCD (Kupesiz et al., 2012). Previous studies demonstrated that neutrophils, erythrocytes and platelets from patients with SCD produce significantly higher levels of ROS compared with controls (Fibach and Rachmilewitz, 2008; Marcal et al., 2008). Furthermore, SCD patients have decreased antioxidant capacity, disabling protective endogenous mechanisms to prevent oxidative stress‐induced injury (Almeida et al., 2010).
While there are multiple enzymatic sources of ROS, one of the most important and perhaps ubiquitous is NADPH oxidase. NADPH oxidase is a multi‐subunit enzyme that produces superoxide when activated. Plasma from SCD patients stimulates the production of ROS by leukocytes from unaffected controls via NADPH oxidase activation, and another recent report observed mRNA expression of NADPH oxidase subunits was elevated in neutrophils from SCD patients (Marcal et al., 2008; Almeida et al., 2010). NADPH oxidase participates in the later phase of hypoxia/reoxygenation injury through the overproduction of superoxide, further implicating NADPH oxidase and subsequent ROS over production in SCD (Wood et al., 2005).
All of these studies suggest that ET‐1 is involved in the initiation of SCN and contributes to the progression of this form of chronic kidney disease. However, pre‐clinical support for the use of selective ETA blockade as a viable treatment for SCN is lacking. Therefore, we sought to test the hypothesis that ET‐1 via the ETA receptor is responsible for elevated glomerular ROS production and other markers of renal injury seen in a mouse model of SCD.
Methods
Animals
All mice were maintained and studied in accordance with the US National Research Council's Guide for the Care and Use of Laboratory Animals and the experiments were approved and monitored by the Georgia Regents University or the University of Alabama at Birmingham Institutional Animal Care and Use Committees. All animal care and experimental procedures complied with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010). Studies utilized humanized homozygous for sickle Hb (HbSS)‐only transgenic knock‐in mice with notation: B6;129‐Hba tm1(HBA)Tow Hbb tm2(HBG1,HBB*)Tow/Hbb tm3(HBG1,HBB)Tow/J, originally developed by Townes et al. (Wu et al., 2006; Belcher et al., 2014; Rivers et al., 2015). These animals will be referred to as ‘humanized sickle mice’ or simply ‘sickle mice’. These animals are bred on a mixed C57BL/6 J and 129 backgrounds. Control animals used in the current experiments were heterozygotes [heterozygous for sickle and Hb A (HbAS)] from the same colony. Animals in all groups ranged in age from 12 to 16 weeks‐old at the time of experimentation and were age and sex matched with the exception of the binding studies, which were carried out with age‐matched mice of mixed sexes. Group numbers varied depending on availability of mice. Mice were housed under conditions of constant temperature and humidity and exposed to a 12 h light/dark cycle. All mice were given free access to mouse chow (Harlan Teklad) and tap water. Separate groups of mice were placed in standard metabolic cages and allowed to adapt for 2 days prior to obtaining a 24 h urine sample.
Glomerular isolation
Glomeruli were isolated as previously described (Savin et al., 1992; Saleh et al., 2010). Renal cortices were isolated and passed through a series of cellulose filters to separate the tubular and vascular elements from the glomeruli. Final cellulose filter pore size was 70 μm to ensure uniform collection of glomeruli. The resulting suspension was purified via centrifugation twice at 1000× g, and the pellet was resuspended in HBSS (5.33 mM potassium chloride, 0.44 mM potassium phosphate monobasic, 138 mM sodium chloride, 4 mM sodium bicarbonate, 0.3 mM sodium phosphate dibasic and 5.6 mM glucose) to produce a suspension of >95% glomeruli. Isolations were snap‐frozen in liquid nitrogen and stored at −80°C for mRNA analysis or used immediately for ROS measurements.
Renal pre‐glomerular vessel isolation
Arterioles, interlobular and arcuate arteries were isolated as previously reported (Schneider et al., 2010). Mice were anaesthetized by isoflurane induction and then given an injection of sodium pentobarbital (65 mg·kg−1 i.p.). Kidneys were removed, decapsulated and placed in ice‐cold HBSS. One kidney at a time was gently pressed against a 70 μm micro‐cellulose filter while submerged in HBSS to separate vascular from tubular and microvascular renal tissue using a flat, rounded spatula. Care was taken to minimize cellular damage; rather, tissue was gently massaged until the tubular structures were separated from the vasculature. Once separated, kidney vessels were placed in a 1.5 mL tube, re‐suspended in HBSS and snap‐frozen with liquid nitrogen for later use in the binding assay.
Drug treatments
Where indicated, the ETA antagonist, ABT‐627 (also known as atrasentan; AbbVie, Inc., Abbott Park, IL, USA), was administered via drinking water and the concentration adjusted daily according to water intake to deliver a consistent 5 mg·kg−1·day−1 for 7 days as previously reported (Honing et al., 2000; Elmarakby et al., 2004; Sasser et al., 2007; Boesen et al., 2011; Saleh et al., 2011b). ABT‐627 is an orally bioavailable ETA receptor antagonist considered highly specific for the ETA receptor (K i of 69 pM) (Opgenorth et al., 1996; Winn et al., 1996; Wessale et al., 2002).
Glomerular ROS assay
Glomerular ROS production was quantified using a luminescence assay (Heimlich et al., 2015). Isolated glomeruli were plated on a 96‐well clear‐bottom plate and incubated with 1 mM L012, a luminol derivative. Baseline luminescence was obtained for 5 min prior to exposure to 1 mM PMA. ROS production was calculated by the change in luminescence in response to PMA stimulation. Data were collected at 1 min intervals for 25 min. AUC based on an average of the baseline period was calculated with Graph Pad Prism Software (San Diego, CA, USA).
Assays
Urinary protein concentration was measured by Bradford Assay (BioRad, Hercules, CA, USA) and multiplied by 24 h urine volume to attain excretion rate. Absorbance was measured at 595 nm and compared with a known quantity of protein. The glomerular barrier protein, nephrin, was measured using an indirect competitive ELISA according to the manufacturer's instructions (Exocell, Philadelphia, PA, USA).
To determine the quantity of ETA and ETB receptors present in the renal vasculature, tissue was harvested and frozen at −80°C. Tissues were thawed and homogenized in homogenization buffer (250 mM sucrose, 50 mM Tris–HCl, pH 7.4, 5 mM EDTA, 15 μM PMSF). The homogenate was centrifuged at 1000× g for 30 min at 4°C. The supernatant was collected and centrifuged at 30 000× g for 45 min at 4°C and the pellet resuspended in homogenization buffer to obtain the membrane fraction. Protein concentrations of membrane fractions were determined by Comassie blue Bradford assay. Binding assays were conducted as previously reported (Pollock et al., 2000; Taylor et al., 2002; Kittikulsuth et al., 2011; Kittikulsuth et al., 2012). In brief, [125I]‐ET‐3 binding was used to determine the maximum binding (Bmax) values for ETB receptor expression. To determine ETA receptor number, the value for [125I]‐ET‐3 binding was subtracted from the Bmax value for [125I]‐ET‐1 binding.
mRNA isolation and measurement
mRNA was isolated using the Pure Link Mini RNA extraction kit (Ambion, Austin, TX, USA) as previously described (Boesen et al., 2008). Real‐time PCR was carried out using iTaq Universal Probes Mastermix (BioRad) and TaqMan primer gene expression assays (Applied Biosystems, Foster City, CA, USA; ET‐1: Mm00438656_m1, ETA: Mm01243722_m1, ETB: Mm00432989_m1, NADP oxidase 2 (NOX2): Mm01287743_m1, NADP oxidase 4 (NOX4): Mm00479246_m1, p47PHOX: Mm00447921_m1). Results were quantified using 2ΔΔCT method and calculated as a fold change from the appropriate control values (Livak and Schmittgen, 2001).
Statistical analysis
All groups of data were assessed for normality using the Shapiro–Wilk test. Where appropriate, Student's t test was used to compare normally distributed means of two groups, whereas the non‐parametric Kruskal–Wallis test was used to compare non‐normally distributed groups. For two‐factor analysis, a two‐way ANOVA was used with Bonferroni's post hoc correction to compare individual means. Results are expressed as means ± SEM, with P < 0.05 being considered statistically significant.
Results
Physical data
Mice were assessed for physical characteristics and results displayed in Table 1. Sickle mice and heterozygous controls showed no difference in overall body mass or kidney‐to‐body mass ratio. The spleens of sickle mice were significantly larger when compared with body weight than the spleens of controls (P < 0.05). Sickle mice also had lower haematocrit than their heterozygous counterparts (P < 0.05).
Table 1.
Physical data for sickle and homozygous control mice
| Parameter | HbAS | HbSS | Units | P value |
|---|---|---|---|---|
| Haematocrit | 39.5 ± 0.6 | 31.2 ± 1.3 | % | <0.05 |
| Body mass | 26.4 ± 0.7 | 28.8 ± 1.2 | g | 0.14 |
| Spleen/body mass | 9.6E−3 ± 8.2E−4 | 6.7E−2 ± 6.1E−3 | g | <0.05 |
| Kidney/body mass | 1.5E−2 ± 8.5E−4 | 1.6E−2 ± 5.6E−4 | g | 0.23 |
| Urine output | 1.1 ± 0.3 | 2.2 ± 0.2 | mL | <0.05 |
| Water consumption | 4.5 ± 0.4 | 5.1 ± 0.7 | mL | 0.46 |
ET‐1, ETA and ETB receptor mRNA expression
Renal cortical tissue and isolated glomeruli from HbAS and HbSS mice were collected for determination of ET‐1 mRNA expression. HbSS mice had significantly higher levels of ET‐1 mRNA at baseline in both cortex (Figure 1) and glomeruli (Figure 1), confirming the presence of elevated tissue ET‐1 mRNA in the humanized mouse similar to previous models of SCD. ETA and ETB receptor expression was also determined in the cortex of HbAS and HbSS mice. HbSS mice had increased expression of ETA receptor but not ETB receptor compared with controls (Figure 1).
Figure 1.
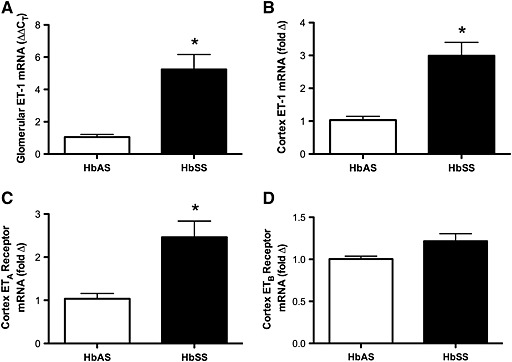
ET‐1 mRNA expression in heterozygous control and SCD mice in isolated glomeruli (A) and renal cortex (B). ETA (C) and ETB (D) receptor mRNA expression was measured in the renal cortex. Data are mean ± SEM. *P < 0.05; n = 5, 6 or 7.
ET receptor binding in renal vasculature
To further characterize the renal endothelin system in the Townes humanized mouse model, we isolated renal vascular tissue of HbAS and HbSS mice to determine ET receptor binding activity. Specific binding of [125I]‐ET‐1 in the renal vessel preparation was higher in HbSS animals than in the HbAS controls (Figure 2; Bmax = 181 ± 19 vs. 117 ± 11 fmol·mg−1 respectively); however, [125I]‐ET‐3‐specific binding was similar between the groups (Figure 2; Bmax = 116 ± 14 vs. 94 ± 18 fmol mg−1 for HbAS vs. HbSS respectively). ET‐1 has equal affinity for both ETA and ETB receptors, while ET‐3 only binds to ETB receptors at low concentrations. Therefore, Bmax values for ET‐1 represent the total binding to both receptors, while Bmax values for ET‐3 binding at the concentrations used represent ETB receptor‐specific binding. ETA receptor‐specific binding was then determined by subtracting ET‐1 Bmax from ET‐3 Bmax. HbSS cell mice had significantly increased ETA receptor‐specific binding in the renal vasculature but had comparable ETB‐specific binding (Figure 2). The K d for ET‐1 and ET‐3 was determined from the Scatchard analysis. ET‐1 K d in HbSS mice was significantly increased compared with that in HbAS controls (0.11 ± 0.02 nM vs. 0.05 ± 0.01 nM, n = 9–11, P < 0.05), while there were no significant differences in the K d for ET‐3 binding between genotypes (0.032 ± 0.004 nM and 0.034 ± 0.005 nM).
Figure 2.
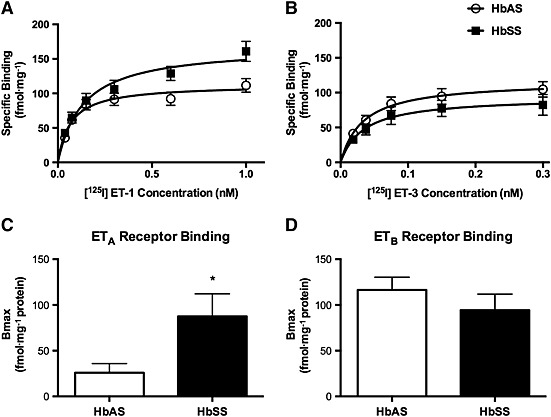
Specific ET‐1 (A) and ET‐3 (B) binding in membrane preparations from dissected renal vasculature. Bmax for the ETA receptor was calculated as the difference between Bmax for ET‐1 and ET‐3 binding (C), while ETB receptor binding was equated to maximum ET‐3‐specific binding (D). Data are mean ± SEM. *P < 0.05; n = 9, 10 or 11.
Stimulation of glomerular ROS production via ETA receptor activation
To determine the level of ETA receptor contribution to glomerular ROS production, HbSS mice were treated with vehicle or ABT‐627 for 1 week. ROS production was measured by PMA stimulation of isolated mouse glomeruli, and representative data from each treatment group are displayed in Figure 3A. First, ROS production in glomeruli from untreated HbSS mice was significantly greater than that from HbAS control mice (Figure 3). After in vivo ETA receptor blockade with ABT‐627, the average response to PMA for glomeruli from HbSS animals was significantly reduced compared with that from untreated HbSS mice (Figure 3). To test the etiology of the ROS in glomeruli, glomeruli from HbSS mice were treated in vitro with the ROS inhibitor apocynin (100 μM) before stimulation with PMA. Apocynin completely abolished the PMA‐stimulated ROS production, suggesting that NADPH oxidase could be a potential source of glomerular ROS (0.82 ± 0.02 × 103 ALU·mg−1 protein vs. 26.3 ± 4.8 103 ALU·mg−1 protein, n = 6–9, P < 0.05).
Figure 3.
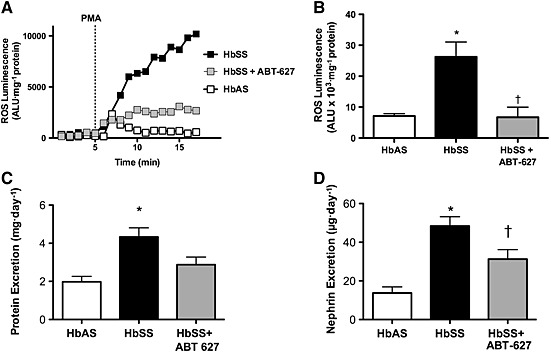
Representative luminescence reading in glomeruli isolated from control, SCD and SCD mice treated with the ETA receptor antagonist, ABT‐627. (A) Total ROS luminescence was calculated from the area under the luminescence curve and normalized to mg protein (B). Panels C and D depict the average protein and nephrin excretion, respectively, for the three groups of mice. Data are mean ± SEM. *P < 0.05 versus control; † P < 0.05 versus untreated sickle mice; n = 6 or 8.
Effects of ETA receptors on renal injury markers
To test the hypothesis that ETA receptors contribute to proteinuria in SCD, HbSS mice were treated with or without ABT‐627. Urine was collected over a 24 h period following 1 week of treatment. In untreated mice, HbSS animals had significantly higher levels of proteinuria compared with heterozygous HbAS mice (Figure 3). As a result of treatment with ABT‐627, HbSS mice exhibited significantly decreased 24 h protein excretion when compared with untreated HbSS mice (Figure 4). HbSS mice also had significantly reduced nephrin excretion when treated with the ETA antagonist. The untreated HbSS animals had significantly elevated nephrinuria compared with heterozygous HbAS controls, an effect that was significantly attenuated by ABT‐627 (Figure 3).
Figure 4.
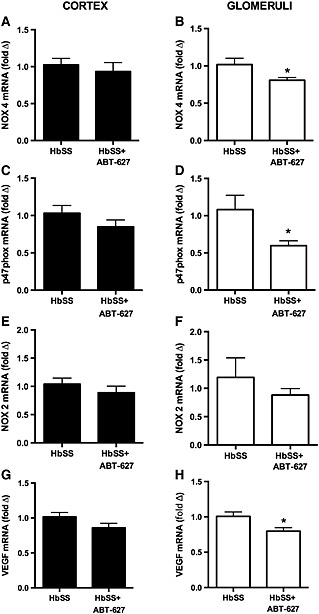
Expression of NADPH oxidase subunits, NOX4 and p47phox, in renal cortex and glomeruli of SCD mice treated with or without the ETA receptor antagonist, ABT‐627. Data are mean ± SEM. *P < 0.05; n = 6 or 7.
ETA receptor antagonism with ABT‐627 significantly decreased mRNA expression of oxidative stress markers in glomeruli of HbSS mice. Both NOX4 and the subunit p47phox were significantly decreased compared with glomeruli from untreated HbSS (Figure 4). In contrast, there was no discernable difference in either NOX4 or p47phox between HbAS and HbSS within whole cortex homogenates (Figure 4). NOX2 was comparable between treatment groups in both the cortex and the glomeruli (Figure 4).
Discussion
These data support a novel role for ET‐1 via the ETA receptor in the progression of SCN in a humanized mouse model of SCD. ET‐1 as well as ETA receptor mRNA was elevated in renal tissue from sickle mice. Additionally, sickle mice exhibited enhanced ET‐1 binding and, specifically, increased ETA receptor binding in the renal vasculature. Because oxidative stress is well‐established in SCD, our findings also support the hypothesis that ET‐1 contributes to oxidative stress and glomerular injury in this model. In vivo and in vitro ETA receptor antagonism decreased glomerular ROS production, proteinuria and nephrinuria in addition to down‐regulating mRNA expression for NADPH oxidase subunits.
The sickle mice utilized in these studies expressed similar overall body mass and kidney‐to‐body mass ratio between genotypes. Spleen‐to‐body mass ratio was expectedly elevated in sickle mice. This aspect was recorded in the first publication of this mouse model, by noting marked expansion of red pulp and intrasplenic pooling of erythrocytes (Levasseur et al., 2003). Haematocrit is a rough estimate of the percentage erythrocyte volume in whole blood and can indicate anaemia. Haematocrit was also significantly decreased but not to the extent that is typically seen in humans, usually around 0.25 (Kaul et al., 1983). Sickle cell mice as well as humans are anaemic by definition, but the degree of anaemia is variable. Given the current data, our sickle mice might experience less anaemia than a typical human SCD patient. However, haematocrit does not necessarily indicate disease severity except in the more extreme values such as those in the teens and lower, which might indicate aplastic crisis (Mallouh and Qudah, 1993). Therefore, we believe this model to represent the typical anaemia status found in SCD.
Multiple studies have reported the presence of increased plasma and urine ET‐1 in humans with SCD (Rybicki and Benjamin, 1998; Tharaux et al., 2005; Hatzipantelis et al., 2013). The SAD mouse was one of the first models developed that used a combination of human Hb mutations (HbS, Antillies and D‐Punjab, SAD) in the mouse genome to increase the propensity for RBCs to sickle and mirror human pathophysiology (Trudel et al., 1991). However, the SAD differs in certain aspects of the human disease course such as the lack of chronic anaemia and a more severe response to hypoxia/reoxygenation. Our current studies were motivated, in part, by observations that SAD mice have increased ET‐1 mRNA expression as determined in whole kidney homogenates, which is exacerbated by hypoxia–reoxygenation (Sabaa et al., 2008). To our knowledge, tissue levels of ET‐1 have not been previously evaluated for the humanized knock‐out, knock‐in mouse model of SCD developed by the Townes lab where mouse genes for α, β and γ Hb have been replaced by corresponding human genes that include the mutation for SCD. Using this model, we observed increased ET‐1 mRNA both in the cortex and to a greater extent in the glomeruli of the humanized sickle mouse. Based on previous experiments in our laboratory, the endothelium is the most likely source of elevated ET‐1 following hypoxia (Heimlich et al., 2015). Vascular endothelial ET‐1 knock‐out animals exhibited no change in ET‐1 mRNA expression after acute hypoxia, whereas animals with intact endothelial ET‐1 showed significant elevations in ET‐1 mRNA after hypoxia (Heimlich et al., 2015). Further supporting this theory is the finding that cultured endothelial cells incubated with sickled RBCs produce increases in ET‐1 (Phelan et al., 1995; Shiu et al., 2002; Ergul et al., 2004). Nonetheless, it remains possible that cell types other than the endothelium, such as macrophages, may be important sources of ET‐1 production in SCD.
ET‐1 at physiological levels is necessary for proper renal function, especially in the collecting duct system to stimulate natriuresis via the ETB receptor (Kohan et al., 2011). However, transgenically‐derived mice constitutively overexpressing ET‐1 mRNA have a glomerulosclerosis phenotype without increased BP lending support to the notion that ET‐1 has powerful trophic effects, independent of BP (Hocher et al., 1997; Amiri et al., 2004). In the Townes humanized sickle mice, increased ET‐1 mRNA expression in the glomeruli combined with enhanced ETA receptor expression within the renal cortex provides the circumstances requisite for extensive renal injury induced by overactivation of ETA receptors.
Because SCD creates a chronic state of oxidative stress, we aimed to evaluate ROS production in the glomeruli with the understanding that increased levels of ROS can cause cellular damage and appear to contribute to glomerular injury in other disorders such as diabetic nephropathy (Sharma et al., 2005). ROS production was elevated upon stimulation with PMA in the HbSS mouse, a phenomenon that was preventable by in vivo treatment with ABT‐627 and in vitro incubation with apocynin. PMA stimulation gives little insight as to the cellular origin of the ROS production; however, we would suspect the source to be endothelial. Given our findings that ETA receptor blockade reduces the expression of NADPH oxidase subunits, these data indicate that glomerular ROS are most likely enzymatically derived from NADPH oxidase and that the ETA receptor perhaps promotes the activity of this ROS‐producing enzyme, especially in the setting of chronic exposure to elevated ET‐1. Previous studies also discovered that ET‐1 increases oxidative stress via up‐regulation of NADPH oxidase through ETA receptor activation, but the mechanism for this effect is not completely clear (Touyz et al., 2004; Loomis et al., 2005). These findings do not exclude the possibility of additional enzymatic sources of ROS. Nonetheless, our findings provide mechanistic insight into the pathophysiology of SCN with particular emphasis on the role of ETA receptor activation.
NOX4, a gp91phox homologue, and p47phox were decreased in response to ETA receptor blockade only in the glomeruli, while NOX2 showed no change in response to ETA antagonism in either tissue. NOX4 is the predominant isoform found in the kidney but is also expressed throughout the vasculature (Geiszt et al., 2000; Ellmark et al., 2005; Hu et al., 2005). Previous studies have shown that increased superoxide production was associated with increased mRNA expression of gp91phox and p47phox (Kitiyakara et al., 2003). Our data indicate that the ETA receptor has a glomerular‐specific modulatory effect upon important NADPH oxidase subunits that contribute to oxidative stress in SCD.
The humanized SCD mice exhibited proteinuria at baseline that was reduced by ETA receptor blockade. Albuminuria and proteinuria represent an early sign of glomerular injury, and their presence predicts not only an elevated risk for nephropathy but also cardiovascular disease (Raptis and Viberti, 2001). ETA receptor activation has been reported in an array of proteinuric renal diseases including both diabetic and non‐diabetic forms of nephropathy. ET‐1 and ETA receptor involvement in chronic kidney disease is well‐established in a large variety of animal models as well (Benigni et al., 1993; Kohan and Pollock, 2013). Previous studies attempting to address proteinuria in SCD have revealed ACE inhibitors can reduce proteinuria and microalbuminuria in humans (Falk et al., 1992). ET‐1 production is stimulated by angiotensin II, thus providing a potential explanation for how both ACE inhibition and ETA antagonism would be beneficial in SCD‐associated proteinuria (Resink et al., 1990; Emori et al., 1991).
Chronic administration of sub‐pressor doses of ET‐1 is also known to directly cause nephrin shedding in vivo and can be reversed by ETA receptor blockade (Saleh et al., 2010). Similarly, ETA antagonism reduces urinary nephrin excretion in a model of diabetic nephropathy (Saleh et al., 2011b; Saleh et al., 2011a). Our current data showing increased urinary nephrin excretion in the humanized SCD mouse provide a novel biomarker for glomerular injury in SCN. Similar to other models of kidney disease, nephrinuria in sickle mice is corrected through ETA receptor antagonism, further validating the paradigm of renal injury via ETA receptor activation. Further studies examining the structural and histological changes associated with ETA receptor activation are needed to confirm the role of ET‐1 in the progression of renal disease in SCD.
Conclusions
Overall, our results point to a mechanism describing the initiation and progression of nephropathy in a humanized mouse model of SCD. Our experiments offer a potential pathophysiological explanation for elevated ROS production in the setting of SCD within the kidney. There are many aspects of SCD that trigger elevated activity of the ET‐1 system such as hypoxia, inflammation, reduced NO bioavailability, increased angiotensin II and a potentially vicious cycle of ROS production. In any event, our findings offer convincing evidence to support ETA receptor blockade as a potential treatment strategy for approaching the increasingly prevalent and deadly manifestation of kidney injury in human SCD.
Author contributions
J. B. H. and J. S. S. performed the research. J. B. H., S. E. M., J. S. P., A. K. and D. M. P. designed the research study. T. M. T., S. E. M. and J. S. P. contributed essential reagents or tools. J. B. H., P. M. O. and D. M. P. analysed the data. J. B. H. and D. M. P. wrote the paper.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by National Heart, Lung, and Blood Institute grants P01 HL69999 (D. M. P., J. S. P.), P01 HL95499 (D. M. P., J. S. P.), U01HL117684 (D. M. P., J. S. P.), T32 HL76146 (J. B. H) and an American Heart Association Post‐doctoral Fellowship (J. S. S.).
Brett Heimlich, J. , Speed, J. S. , O'Connor, P. M. , Pollock, J. S. , Townes, T. M. , Meiler, S. E. , Kutlar, A. , and Pollock, D. M. (2016) Endothelin‐1 contributes to the progression of renal injury in sickle cell disease via reactive oxygen species. British Journal of Pharmacology, 173: 386–395. doi: 10.1111/bph.13380.
References
- Abbott KC, Hypolite IO, Agodoa LY (2002). Sickle cell nephropathy at end‐stage renal disease in the United States: patient characteristics and survival. Clin Nephrol 58: 9–15. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al (2013). The Concise Guide to PHARMACOLOGY 2013/14: G protein‐coupled receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida CB, Franco‐Penteado C, Saad ST, Costa FF, Conran N (2010). Sickle cell disease serum induces NADPH enzyme subunit expression and oxidant production in leukocytes. Hematology 15: 422–429. [DOI] [PubMed] [Google Scholar]
- Amiri F, Virdis A, Neves MF, Iglarz M, Seidah NG, Touyz RM, et al (2004). Endothelium‐restricted overexpression of human endothelin‐1 causes vascular remodeling and endothelial dysfunction. Circulation 110: 2233–2240. [DOI] [PubMed] [Google Scholar]
- Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, et al (2014). Heme triggers TLR4 signaling leading to endothelial cell activation and vaso‐occlusion in murine sickle cell disease. Blood 123: 377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benigni A, Zoja C, Corna D, Orisio S, Longaretti L, Bertani T, et al (1993). A specific endothelin subtype A receptor antagonist protects against injury in renal disease progression. Kidney Int 44: 440–444. [DOI] [PubMed] [Google Scholar]
- Boesen EI, Krishnan KR, Pollock JS, Pollock DM (2011). ETA activation mediates angiotensin II‐induced infiltration of renal cortical T cells. J Am Soc Nephrol 22: 2187–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boesen EI, Sasser JM, Saleh MA, Potter WA, Woods M, Warner TD, et al (2008). Interleukin‐1beta, but not interleukin‐6, enhances renal and systemic endothelin production in vivo. Am J Physiol Renal Physiol 295: F446–F453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong F, Zhang X, Wold LE, Ren Q, Zhang Z, Ren J (2005). Endothelin‐1 enhances oxidative stress, cell proliferation and reduces apoptosis in human umbilical vein endothelial cells: role of ETB receptor, NADPH oxidase and caveolin‐1. Br J Pharmacol 145: 323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellmark SH, Dusting GJ, Fui MN, Guzzo‐Pernell N, Drummond GR (2005). The contribution of Nox4 to NADPH oxidase activity in mouse vascular smooth muscle. Cardiovasc Res 65: 495–504. [DOI] [PubMed] [Google Scholar]
- Elmarakby AA, Dabbs Loomis E, Pollock JS, Pollock DM (2004). ETA receptor blockade attenuates hypertension and decreases reactive oxygen species in ETB receptor‐deficient rats. J Cardiovasc Pharmacol 44 (Suppl 1): S7–10. [DOI] [PubMed] [Google Scholar]
- Emori T, Hirata Y, Ohta K, Kanno K, Eguchi S, Imai T, et al (1991). Cellular mechanism of endothelin‐1 release by angiotensin and vasopressin. Hypertension 18: 165–170. [DOI] [PubMed] [Google Scholar]
- Ergul S, Brunson CY, Hutchinson J, Tawfik A, Kutlar A, Webb RC, et al (2004). Vasoactive factors in sickle cell disease: in vitro evidence for endothelin‐1‐mediated vasoconstriction. Am J Hematol 76: 245–251. [DOI] [PubMed] [Google Scholar]
- Falk RJ, Jennette JC (1994). Sickle cell nephropathy. Adv Nephrol Necker Hosp 23: 133–147. [PubMed] [Google Scholar]
- Falk RJ, Scheinman J, Phillips G, Orringer E, Johnson A, Jennette JC (1992). Prevalence and pathologic features of sickle cell nephropathy and response to inhibition of angiotensin‐converting enzyme. N Engl J Med 326: 910–915. [DOI] [PubMed] [Google Scholar]
- Fibach E, Rachmilewitz E (2008). The role of oxidative stress in hemolytic anemia. Curr . Mol Med 8: 609–619. [DOI] [PubMed] [Google Scholar]
- Geiszt M, Kopp JB, Varnai P, Leto TL (2000). Identification of renox, an NAD(P)H oxidase in kidney. Proc Natl Acad Sci U S A 97: 8010–8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guasch A, Navarrete J, Nass K, Zayas CF (2006). Glomerular involvement in adults with sickle cell hemoglobinopathies: prevalence and clinical correlates of progressive renal failure. J Am Soc Nephrol 17: 2228–2235. [DOI] [PubMed] [Google Scholar]
- Hatzipantelis ES, Pana ZD, Gombakis N, Taparkou A, Tzimouli V, Kleta D, et al (2013). Endothelial activation and inflammation biomarkers in children and adolescents with sickle cell disease. Int J Hematol 98: 158–163. [DOI] [PubMed] [Google Scholar]
- Heimlich JB, Speed JS, Bloom CJ, O'Connor PM, Pollock JS, Pollock DM (2015). ET‐1 increases reactive oxygen species following hypoxia and high‐salt diet in the mouse glomerulus. Acta Physiol (Oxf) 213: 722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocher B, Thone‐Reineke C, Rohmeiss P, Schmager F, Slowinski T, Burst V, et al (1997). Endothelin‐1 transgenic mice develop glomerulosclerosis, interstitial fibrosis, and renal cysts but not hypertension. J Clin Invest 99: 1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honing ML, Hijmering ML, Ballard DE, Yang YP, Padley RJ, Morrison PJ, et al (2000). Selective ET(A) receptor antagonism with ABT‐627 attenuates all renal effects of endothelin in humans. J Am Soc Nephrol 11: 1498–1504. [DOI] [PubMed] [Google Scholar]
- Hu T, Ramachandrarao SP, Siva S, Valancius C, Zhu Y, Mahadev K, et al (2005). Reactive oxygen species production via NADPH oxidase mediates TGF‐beta‐induced cytoskeletal alterations in endothelial cells. Am J Physiol Renal Physiol 289: F816–F825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul DK, Fabry ME, Windisch P, Baez S, Nagel RL (1983). Erythrocytes in sickle cell anemia are heterogeneous in their rheological and hemodynamic characteristics. J Clin Invest 72: 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). NC3Rs Reporting Guidelines Working Group. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitiyakara C, Chabrashvili T, Chen Y, Blau J, Karber A, Aslam S, et al (2003). Salt intake, oxidative stress, and renal expression of NADPH oxidase and superoxide dismutase. J Am Soc Nephrol 14: 2775–2782. [DOI] [PubMed] [Google Scholar]
- Kittikulsuth W, Pollock JS, Pollock DM (2011). Sex differences in renal medullary endothelin receptor function in angiotensin II hypertensive rats. Hypertension 58: 212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittikulsuth W, Pollock JS, Pollock DM (2012). Loss of renal medullary endothelin B receptor function during salt deprivation is regulated by angiotensin II. Am J Physiol Renal Physiol 303: F659–F666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohan DE, Pollock DM (2013). Endothelin antagonists for diabetic and non‐diabetic chronic kidney disease. Br J Clin Pharmacol 76: 573–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohan DE, Inscho EW, Wesson D, Pollock DM (2011). Physiology of endothelin and the kidney. Compr Physiol 1: 883–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupesiz A, Celmeli G, Dogan S, Antmen B, Aslan M (2012). The effect of hemolysis on plasma oxidation and nitration in patients with sickle cell disease. Free Radic Res 46: 883–890. [DOI] [PubMed] [Google Scholar]
- Laplante MA, Wu R, Moreau P, de Champlain J (2005). Endothelin mediates superoxide production in angiotensin II‐induced hypertension in rats. Free Radic Biol Med 38: 589–596. [DOI] [PubMed] [Google Scholar]
- Levasseur DN, Ryan TM, Pawlik KM, Townes TM (2003). Correction of a mouse model of sickle cell disease: lentiviral/antisickling beta‐globin gene transduction of unmobilized, purified hematopoietic stem cells. Blood 102: 4312–4319. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001). Analysis of relative gene expression data using real‐time quantitative PCR and the 2(−delta delta C(T)) method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- Loomis ED, Sullivan JC, Osmond DA, Pollock DM, Pollock JS (2005). Endothelin mediates superoxide production and vasoconstriction through activation of NADPH oxidase and uncoupled nitric‐oxide synthase in the rat aorta. J Pharmacol Exp Ther 315: 1058–1064. [DOI] [PubMed] [Google Scholar]
- Mallouh AA, Qudah A (1993). Acute splenic sequestration together with aplastic crisis caused by human parvovirus B19 in patients with sickle cell disease. J Pediatr 122: 593–595. [DOI] [PubMed] [Google Scholar]
- Marcal LE, Dias‐da‐Motta PM, Rehder J, Mamoni RL, Blotta MH, Whitney CB, et al (2008). Up‐regulation of NADPH oxidase components and increased production of interferon‐gamma by leukocytes from sickle cell disease patients. Am J Hematol 83: 41–45. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C. Wainwright, C. (2010) Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol, 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opgenorth TJ, Adler AL, Calzadilla SV, Chiou WJ, Dayton BD, Dixon DB, et al (1996). Pharmacological characterization of A‐127722: an orally active and highly potent ETA‐selective receptor antagonist. J Pharmacol Exp Ther 276: 473–481. [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, Davenport AP, McGrath JC, Peters JA, Southan C, Spedding M, Yu W, Harmar AJ; NC‐IUPHAR. (2014) The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 42 (Database Issue): D1098–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan M, Perrine SP, Brauer M, Faller DV (1995). Sickle erythrocytes, after sickling, regulate the expression of the endothelin‐1 gene and protein in human endothelial cells in culture. J Clin Invest 96: 1145–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock DM, Allcock GH, Krishnan A, Dayton BD, Pollock JS (2000). Upregulation of endothelin B receptors in kidneys of DOCA‐salt hypertensive rats. Am J Physiol Renal Physiol 278: F279–F286. [DOI] [PubMed] [Google Scholar]
- Powars DR, Elliott‐Mills DD, Chan L, Niland J, Hiti AL, Opas LM, et al (1991). Chronic renal failure in sickle cell disease: risk factors, clinical course, and mortality. Ann Intern Med 115: 614–620. [DOI] [PubMed] [Google Scholar]
- Raptis AE, Viberti G (2001). Pathogenesis of diabetic nephropathy. Exp Clin Endocrinol Diabetes 109 (Suppl 2): S424–S437. [DOI] [PubMed] [Google Scholar]
- Resink TJ, Hahn AW, Scott‐Burden T, Powell J, Weber E, Buhler FR (1990). Inducible endothelin mRNA expression and peptide secretion in cultured human vascular smooth muscle cells. Biochem Biophys Res Commun 168: 1303–1310. [DOI] [PubMed] [Google Scholar]
- Rivers A, Vaitkus K, Ruiz MA, Ibanez V, Jagadeeswaran R, Kouznetsova T, et al (2015). RN‐1, a potent and selective lysine‐specific demethylase 1 inhibitor, increases gamma‐globin expression, F reticulocytes, and F cells in a sickle cell disease mouse model. Exp Hematol 43 (546–553): e543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybicki AC, Benjamin LJ (1998). Increased levels of endothelin‐1 in plasma of sickle cell anemia patients. Blood 92: 2594–2596. [PubMed] [Google Scholar]
- Sabaa N, de Franceschi L, Bonnin P, Castier Y, Malpeli G, Debbabi H, et al (2008). Endothelin receptor antagonism prevents hypoxia‐induced mortality and morbidity in a mouse model of sickle‐cell disease. J Clin Invest 118: 1924–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh MA, Pollock JS, Pollock DM (2011a). Distinct actions of endothelin A‐selective versus combined endothelin A/B receptor antagonists in early diabetic kidney disease. J Pharmacol Exp Ther 338: 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh MA, Boesen EI, Pollock JS, Savin VJ, Pollock DM (2010). Endothelin‐1 increases glomerular permeability and inflammation independent of blood pressure in the rat. Hypertension 56: 942–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh MA, Boesen EI, Pollock JS, Savin VJ, Pollock DM (2011b). Endothelin receptor A‐specific stimulation of glomerular inflammation and injury in a streptozotocin‐induced rat model of diabetes. Diabetologia 54: 979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasser JM, Sullivan JC, Hobbs JL, Yamamoto T, Pollock DM, Carmines PK, et al (2007). Endothelin A receptor blockade reduces diabetic renal injury via an anti‐inflammatory mechanism. J Am Soc Nephrol 18: 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savin VJ, Sharma R, Lovell HB, Welling DJ (1992). Measurement of albumin reflection coefficient with isolated rat glomeruli. J Am Soc Nephrol 3: 1260–1269. [DOI] [PubMed] [Google Scholar]
- Schneider MP, Wach PF, Durley MK, Pollock JS, Pollock DM (2010). Sex differences in acute ANG II‐mediated hemodynamic responses in mice. Am J Physiol Regul Integr Comp Physiol 299: R899–R906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, McCarthy ET, Savin VJ, Lianos EA (2005). Nitric oxide preserves the glomerular protein permeability barrier by antagonizing superoxide. Kidney Int 68: 2735–2744. [DOI] [PubMed] [Google Scholar]
- Shiu YT, McIntire LV, Udden MM (2002). Sickle erythrocytes increase prostacyclin and endothelin‐1 production by cultured human endothelial cells under flow conditions. Eur J Haematol 68: 163–169. [DOI] [PubMed] [Google Scholar]
- Statius van Eps LW, Pinedo‐Veels C, de Vries GH, de Koning J (1970). Nature of concentrating defect in sickle‐cell nephropathy. Microradioangiographic studies. Lancet 1: 450–452. [DOI] [PubMed] [Google Scholar]
- Taylor TA, Gariepy CE, Pollock DM, Pollock JS (2002). Unique endothelin receptor binding in kidneys of ETB receptor deficient rats. Am J Physiol Regul Integr Comp Physiol 7: 7. [DOI] [PubMed] [Google Scholar]
- Tharaux PL, Hagege I, Placier S, Vayssairat M, Kanfer A, Girot R, et al (2005). Urinary endothelin‐1 as a marker of renal damage in sickle cell disease. Nephrol Dial Transplant 20: 2408–2413. [DOI] [PubMed] [Google Scholar]
- Touyz RM, Yao G, Viel E, Amiri F, Schiffrin EL (2004). Angiotensin II and endothelin‐1 regulate MAP kinases through different redox‐dependent mechanisms in human vascular smooth muscle cells. J Hypertens 22: 1141–1149. [DOI] [PubMed] [Google Scholar]
- Trudel M, Saadane N, Garel MC, Bardakdjian‐Michau J, Blouquit Y, Guerquin‐Kern JL, et al (1991). Towards a transgenic mouse model of sickle cell disease: hemoglobin SAD. EMBO J 10: 3157–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessale JL, Adler AL, Novosad EI, Calzadilla SV, Dayton BD, Marsh KC, et al (2002). Pharmacology of endothelin receptor antagonists ABT‐627, ABT‐546, A‐182086 and A‐192621: ex vivo and in vivo studies. Clin Sci (Lond) 103 (Suppl 48): 112S–117S. [DOI] [PubMed] [Google Scholar]
- Wilcox CS (2012). Asymmetric dimethylarginine and reactive oxygen species: unwelcome twin visitors to the cardiovascular and kidney disease tables. Hypertension 59: 375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M, von Geldern TW, Opgenorth TJ, Jae HS, Tasker AS, Boyd SA, et al (1996). 2,4‐Diarylpyrrolidine‐3‐carboxylic acids–potent ETA selective endothelin receptor antagonists. 1. Discovery of A‐127722. J Med Chem 39: 1039–1048. [DOI] [PubMed] [Google Scholar]
- Wolin MS (2000). Interactions of oxidants with vascular signaling systems. Arterioscler Thromb Vasc Biol 20: 1430–1442. [DOI] [PubMed] [Google Scholar]
- Wood KC, Hebbel RP, Granger DN (2005). Endothelial cell NADPH oxidase mediates the cerebral microvascular dysfunction in sickle cell transgenic mice. FASEB J 19: 989–991. [DOI] [PubMed] [Google Scholar]
- Wu LC, Sun CW, Ryan TM, Pawlik KM, Ren J, Townes TM (2006). Correction of sickle cell disease by homologous recombination in embryonic stem cells. Blood 108: 1183–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanni E, Grosse SD, Yang Q, Olney RS (2009). Trends in pediatric sickle cell disease‐related mortality in the United States, 1983–2002. J Pediatr 154: 541–545. [DOI] [PubMed] [Google Scholar]