

Abstract
The transcriptional activation mechanisms that regulate tissue‐specific expression of cardiac muscle genes have been extensively investigated, but little is known of the regulatory events involved in repression of cardiac‐specific genes in non‐cardiac cells. We have previously reported that Nished, a ubiquitous transcription factor, interacts with a positive sequence element, the Intron Regulatory Element (IRE) as well as a negatively acting element, the Cardiac‐Specific Sequence (CSS), in myosin light chain‐2 (MLC2v) gene to promote activation and repression of the gene in cardiac and skeletal muscle cells respectively. Here, we show that the negative regulation of cardiac MLC2v gene in skeletal muscle cells is mediated via the interaction of Nished with histone deacetylase (HDAC) co‐repressor. Treatment of cells with the HDAC inhibitor, Trichostatin A (TSA), alleviates the repressor activity of Nished in a dose‐dependent manner. Co‐transfection studies in primary muscle cells in culture and in Nished expressing stable skeletal muscle cell line demonstrate that Nished down‐regulates the cardiac MLC2 gene expression when its association is restricted to CSS alone. Chromatin immunoprecipitation data suggest that the CSS‐mediated repression of cardiac MLC2v gene in skeletal muscle cells excludes the participation of the positive element IRE despite the presence of an identical Nished binding site. Taken together, it appears that the negative control of MLC2v transcription is based on a dual mode of regulations, one that affords inaccessibility of IRE to Nished and second that promotes the formation of the transcription repression complex at the inhibitory CSS site to silence the cardiac gene in skeletal muscle cell.
Keywords: Nished, myosin light chain‐2v, transcription repression, histone deacetylases
Introduction
Repression of gene transcription is obligatory for establishment of cell‐specific gene expression and cellular differentiation. Eukaryotic gene transcription is regulated in part by the enzymatic activity of histone acetylases (HATs) and deacetylases (HDACs) (see ref. [1 and 2] for review). Analysis of genetically manipulated mice has documented the functions of these chromatin‐modifying enzymes as central to control of gene expression in an animal context. HDACs promote chromatin compaction and repress transcription by limiting the access of transcription factors to DNA. Hypoacetylated histones, therefore, are generally found in transcriptonally silent genes. Several recent studies have implicated HDACs in control of muscle development and differentiation through their association with myogenic transcription factors MEF‐2 and MyoD to their cognate binding sites [3, 4, 5]. The class II HDACs (HDACs 4,5,7) associate with MEF‐2 and inhibit the MEF‐2‐dependent activation of target genes. The class I HDACs 1,2,3, on the other hand, do not interact with MEF‐2, but associate with MyoD and inhibit the MyoD‐mediated gene transcription that involves recruitment of co‐repressors [6]. During skeletal myogenesis, MyoD‐dependent expression of muscle gene requires the association of MyoD with members of the MEF‐2 family. Thus, the events that target the chromatin remodelling enzymes for interactions with myogenic transcription factors are important and orchestrate the positive and negative regulation of muscle genes during myogenic cell development and differentiation.
The cardiac muscle cell‐restricted expression of myosin light‐chain 2 (MLC2v) gene offers an attractive experimental paradigm for uncovering the role of specific transcription factors and their co‐regulators in modulation of gene activity in response to different muscle cell type signalling [7, 8, 9]. The expression of chicken cardiac MLC2v is regulated positively in cardiac muscle and negatively in skeletal muscle cells, even though the basal promoter architecture of both cardiac and skeletal muscle MLC2 genes is almost identical. In order to understand the underlying mechanism(s), we have identified and characterized several cis‐elements in MLC2v gene and their cognate DNA binding proteins [10, 11, 12, 13, 14, 15]. The activation of chicken MLC2v gene transcription is orchestrated by the combinational interaction of MEF‐2 [7], SRF [11] and Nished [15] with the proximal promoter elements in concert with the co‐activators NFAT‐c4 and p300 [15], whereas its inhibition in skeletal muscle cell requires an intact upstream element (CSS) [10] and a downstream modulator element [14]. We have recently reported that a ubiquitous transcription factor, Nished, recognizes the palindrome sequence present in both the negative CSS and the positive IRE elements to facilitate the negative and positive transcription activity of MLC2v gene, respectively [15].
In this report, we provide evidence that Nished promotes repression of the cardiac MLC2v in skeletal muscle cells via its association with histone deacetylases co‐repressor complex at the CSS site. The HDAC inhibitor, TSA, releases the Nished‐mediated repression of MLC2v gene transcription in a dose‐dependent manner. Chromatin immunoprecipitation data suggest that IRE is inaccessible to Nished in skeletal muscle cells affording thereby the preferential binding of Nished to CSS and the concurrent repression of MLC2v gene transcription. Taken together, our data define the mechanism in which Nished plays a key role via its interaction with HDAC. In addition, the inaccessibility of the activator IRE site influences the formation of a functional complex promoting the repression of the cardiac MLC2v gene in skeletal muscle cell.
Materials and methods
Construction of mutant CSS and IRE reporter plasmids
A 2.1 kb Sma I/Stu I blunt‐ended fragment of MLC‐2v gene derived from plLC5.2 [16] was cloned into the Sma I site of the promoterless vector, pGL2Basic, that carries the coding region for firefly (Photinus pyralis) luciferase (Promega, Madison, WI, USA) to generate the pMLC2.1Luc reporter plasmid. GeneEditor in vitro Site‐Directed Mutagenesis System (Promega) was used to introduce mutations within CSS and IRE sequences of pMLC2.1Luc as per the manufacturer’s instructions. Mutagenic oligonucleotides for CSS and IRE were synthesized (see below). The underlined sequence denotes the mutation within the core motif. CSSMutB: 5′‐CGAGGAGGTAGTACTACCCTGAAGCAAAAG‐3′ MutIRE: 5′‐GCAGAGAGCAAGGGTACCCCGGGGGTCTGATGGC‐3′. CSS oligonucleotides corresponding to −355 to –323 bp as shown below were used as probes for GMSA. CSS 5: 5′‐GACGAGGAGGTACTTCTACCCTGAAGCAAAAGG‐′3 and CSS 3: 5′‐CCTTTTGCTTCAGGGTAGAAGTACCTCCTCGTC‐3′.
Cloning of Nished cDNA sequence in mammalian expression vector pcDNA6 V5/HisB
The coding region of Nished was amplified by PCR using the primer pair Nished‐V5 5′Hind III: 5′‐CCCAAGCTTGCCACCATGTGCAGGAATTCCCGCCA‐3′ and Nished‐V5 3′Not 1: 5′‐ATAAGAATGCGGCCGCCCCCGGGAGGTGACAGAAGTGA‐3.’ The amplified PCR DNA fragment was cloned into pcDNA6‐V5‐HisB vector DNA and transformed into DH5a competent cells. Plasmid DNA isolated from the clones was sequenced to ascertain that no errors were introduced into Nished cDNA during PCR and that it was in frame with the C‐terminal V5 epitope.
Generation of N‐terminal and C‐terminal mutants of Nished
The primer pair used to generate the N‐terminal mutants, (pNΔ1) are NΔ1 5′ Hind III: 5′‐ACCAAGCTTGCCGCCACCATGAACAAGGGAGGA‐3′ where the underlined sequence corresponds to 177–188 bp of Nished cDNA and NΔ1 3′ Not I: 5‐′ ATAAGAATGCGGCCGCCCCCGGGAGGTGACAGAAGTGA‐3′ where the underlined sequence corresponds to 468–488 bp of Nished cDNA. The primer pair for pNΔ2 corresponding to 345–356 bp and 468–488bp of Nished cDNA are NΔ2 5′ Hind III: 5′ACCAAGCTTGCCGCCACCATGATGCAGCAGCCA‐3′ and NΔ2 3′ Not I: 5′‐ATAAGAATGCGGCCGCCCCCGGGAGGTGACAGAAGTGA‐3′. To generate the C‐terminal mutants, pCΔ1 primer pairs corresponding to 78–97 bp and 252–269 bp are shown below: CΔ1 5′ Hind III: 5′‐CCCAAGCTTGCCACCATGTGCAGGAATTCCCGCCA‐3′ and CΔ1 3′ Not I: 5′‐ATAAGAATGCGGCCGCCTGCCCAGACAAAGCCGAA‐3′. The primer for pCΔ2 corresponds to 78–97 bp and 405–422 bp are CΔ2 5′ Hind III: 5′‐CCCAAGCTTGCCACCATGTGCAGGAATTCCCGCCA‐3′ and Cdel2 3′ Not I: 5′‐ATAAGAATGCGGCCGCCGCACGCAAACCAGAACCC‐3′. N‐and C‐terminal mutants were cloned into the mammalian expression vector, pcDNA6‐V5/HisB. 35S‐methionine was used to synthesize radiolabelled proteins and the gel was subjected to fluorography using EN3HANCE (NEN). After electrophoresis, the gel was fixed in 10% glacial acetic acid, 30% methanol for 1 hr after which it was impregnated with ENHANCE, washed, dried and exposed to X‐ray film.
Cell culture
Hearts of the 11‐day‐old White Leghorn chicken embryos (Charles River Laboratories, CT, USA) were collected for primary cardiac skeletal muscle cell cultures as described previously [10, 15]. C2C12 mouse myoblast cells were grown in DMEM containing 20% heat‐inactivated foetal bovine serum. Primary cells were transfected using FuGENE 6 transfection reagent according to the manufacturer’s conditions (Roche Applied Science) with 1 μg of luciferase vectors mixed with 100 ng of pcDNA6 vector, Nished‐V5 or its N‐and C‐terminal mutants. C2C12 cells were transfected as above with 1 μg Nished‐V5 or pcDNA6 vector. After Blasticidin S selection (10 μg/ml), individual clones were selected based on the existence of Nished and/or Blasticidin S resistant gene (BGH) transcripts.
Gel mobility shift assay (GMSA)
Tissues from either fresh or frozen embryonic hearts and skeletal soleus muscles were minced finely and the dissociated cells were lysed in lysis buffer as described previously [10, 15]. Nuclei were lysed in the nuclei lysis buffer (20 mM HEPES, pH 7.6, 20% glycerol, 500 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.1% Triton X‐100) with 1 mM DTT, 1 mM PMSF, 2.5 mM Na‐vanadate, 10 mM NaF and protease inhibitor cocktail (Sigma, P8340; Sigma, St Louis, MO, USA) and extracts were used for binding reactions [7, 10, 15]. Protein ‐DNA complexes were separated by electrophoresis on 6–8% non‐denaturing polyacrylamide gels as previously described [7, 13]. When Nished antibodies were used, 5 ml of 1:2.5 diluted antibodies in PBS were pre‐incubated on ice with nuclear extracts for 20–30 min. followed by incubation with radiolabelled probe. Similar conditions were used for pre‐immune serum incubations. NIH Image (version 1.57) software was used to determine the relative density of the DNA‐protein complexes.
Immunoprecipitation and Western blot analysis
For co‐immunoprecipitations, 10 μg of antibody against HDAC 3 (ab47237) and 5 (ab1439) (Abcam) was incubated overnight with 2.5 mg of pre‐cleared cell or tissue lysate. Western blotting was carried out as described earlier with anti‐Nished antibody [15].
Microaffinity purification
Microaffinity purification of CSS or IRE‐binding proteins was performed using chemically synthesized CSS or IRE oligonucleotides containing 5′ biotin (Bt) on a flexible linker (Invitrogen). Forty pmol of duplex Bt‐IRE was incubated with 500 μg of cardiac and skeletal muscle nuclear extracts from embryos or with 35S methione labelled in vitro translated proteins in the presence of 5 μg of poly (dI/dC) in a 1000‐μl volume of a buffer containing 4.5% (v/v) glycerol, 5 mM MgCl2, 10 mM KCl, 0.42 mM EDTA, 0.8 mM DTT, 4 mM HEPES (pH 7.5) for 1 hr at 4°C. Proteins bound to Bt‐CSS or ‐IRE were captured by addition of 50 μl of a 50% (v/v) slurry of streptavidin‐agarose beads (Thermo Scientific, Pierce, Rockford, IL, USA) and washed twice in binding buffer. Bt‐CSS or IRE‐binding proteins were then eluted by SDS‐PAGE loading buffer for Western immunoblot analysis.
ChIP assays
The ChIP assay was perfromed following manufacturer’s protocol (ChIP Assay Kit Upstate 17‐295). The frozen tissue (−100–500 mg) was cut into small pieces with a razor blade and cross‐link with 1% formaldehyde in 1× PBS plus protease inhibitor Sigma (P8340) for 15 min. at room temperature. Then, the formaldehyde was quenched with glycine at room temperature for 5 min. Tissue was homogenized on ice in 1× PBS plus protease inhibitors. The cell pellet was washed twice with cold 1× PBS with protease inhibitors and lysed in SDS‐lysis buffer (50 mM Tris‐HCl, ph = 8.1, 1% SDS, 10 mM EDTA) with protease inhibitors. Then, the extracts were sonicated with 30 sec. pulses (30 times) and resuspended in Chip Dilution Buffer (16.7 mM Tris‐HCl, pH 8.1, 167 mM NaCl, 0.01% SDS, 1% Triton X‐100, 2 mM EDTA). The immunoprecipitation was performed by pre‐clearing with Salmon Sperm DNA/Protein A Agarose‐50% slurry for 30 min. Then, 5 ug of antibodies H3K4M3, and H3K9M1 (Abcam) were incubated with 10–15 mg DNA/sonicated extract overnight with rotation at 4°C followed by addition of 60 ml protein A, per IP and incubated 1 hr at 4°C. The immunoprecipitates were washed as recommended by the manufacturer (Upstate ChIP assay kit) for 5 min., 1 wash low salt immune complex wash buffer, 1 wash high salt immune complex wash buffer, 1 wash LiCl immune complex wash buffer, 2 washes 1× TE buffer. The elution was obtained with 1% SDS, 0.1 M NaHCO3. Rotate 15 min. 250 ml 2 times. Add 20 ml of 5 M NaCl and reverse cross‐linked at 65°C overnight. Add 10 ml of 0.5 M EDTA, 20 ml 1 M Tris‐HCL, pH 6.5, 1 ml 20 mg/ml proteinase K, 45°C 1 hr. The DNA was recovered using the Qiagen QIAquick kit followed by PCR. The primer pairs used for the PCR reactions are; IRE‐F: 5′‐CCTGTGGCACATGCGTTCTCATT‐3′, IRE‐R: 5′‐TGCCCCCAAATGACCTGTGG‐3′, GAPDH‐F: 5′‐AGAGAGCTCGATGGGGATG‐3′, GAPDH‐R:5′‐CCGTTGACTCCGACTTTCAC‐3′, and ApoB‐F: 5‐′AAAACCAACCCAACAACTGG‐3′, ApoB‐R: 5′‐CCACCTCAGAGGGAGAATGA‐3′.
Statistical analysis
Data are expressed as mean ± S.E. Differences between experimental groups were evaluated for statistical significance using either one sample t‐test with Bonferroni correction or one‐way ANOVA test. Tamhane’s test were performed post hoc to test for significant differences; P‐values <0.05 were considered statistically significant.
Results
Role of CSS and IRE in tissue‐specific expression of MLC2v gene
We have previously identified a negative regulatory cis‐element (CSS) in the MLC2v promoter that contains a palindrome sequence 5′GAAGCTTC3′[10]. We observed subsequently that an identical sequence motif resides in the downstream activator element IRE located in the first intron of the MLC2v gene [15]. To examine the possibility of functional interaction between CSS and IRE in transcription regulation of the cardiac MLC2v gene, transient transfection was done with gene constructs that contain mutations in either IRE or in both IRE and CSS sequences. Specifically, the GAAGCTTC palindrome in IRE was mutated in the 2.1 kb SmaI‐StuI genomic fragment that extends from −1293 to +815 bp and encompasses the promoter and the first intron of the gene. The DNA fragment was linked to the luciferase reporter (pMutIRLuc) (see Materials and Methods). The same domain was also mutated in CSS as above to generate a double mutant pMutCSS+pMutIRELuc (Fig. 1A). The activity of these constructs was examined in transient transfection assay. In the wild‐type construct, the IRE positive activity apparently overrides the inhibitory activity of CSS. Mutation in IRE in pMutIRELuc resulted in a significant loss (70%) of the promoter function. However, when the same mutation was introduced in CSS as well as in IRE in the double mutant pMutIRE+MutCSSLuc, there was a loss of inhibition (Fig. 1B), suggesting that CSS and IRE interact with common DNA binding protein(s) that recognizes the conserved GAAG/CTTC motif present in both regulatory elements. Indeed, we have previously identified a transcription factor, Nished, that binds with sequence specificity to the palindrome in CSS [14] as well as in IRE where the IRE/Nished complex mediates the activation function of MLC2v gene promoter in cardiac cells [15].
Figure 1.
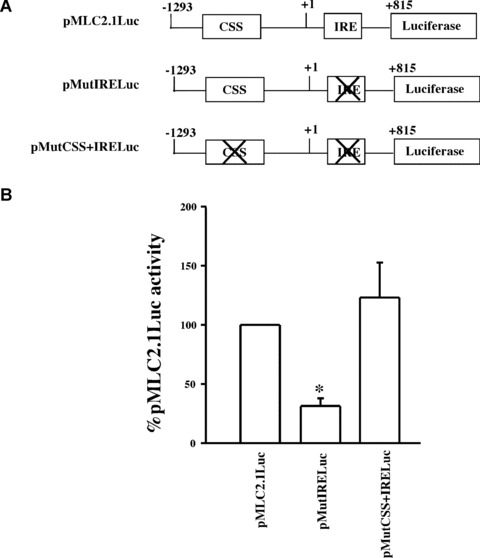
(A) Schematic representation of the chicken cardiac MLC2 promoter constructs with either the wild‐type (pMLC2.1Luc), IRE mutant, (pMutIRELuc) or mutations in both CSS and IRE, (pMutCSSIRELuc). (B) Transient transfection and MLC2 promoter activity. Chicken primary skeletal muscle cells in culture were transfected with plasmids pMLC2.1Luc, pMutIRELuc or pMutCSSIRELuc and activity of luciferase was measured as described in ‘Material and Methods’. Data are shown as percentage of wild‐type promoter activity. (n= 4 in triplicate; □ one sample t‐test with Bonferroni correction, P < 0.0167).
In order to test whether the CSS/Nished complex is formed in skeletal muscle cells, we performed the gel shift assay with CSS DNA as probe using the skeletal muscle nuclear extract (Fig. 2). We observed that one major (CSSBPA) and two minor (CSSBP1 & 2) complexes are formed and that preincubation of the extracts with anti‐Nished antibody disrupts the major complex, CSSBPA, and one of the minor complexes, CSSBP1. Thus, Nished appears to be the common CSS and IRE DNA binding protein that is likely to be involved in both activation and repression mechanism of transcription of cardiac MLC2v gene in the distinct cellular environments, i.e. cardiac and skeletal muscle.
Figure 2.
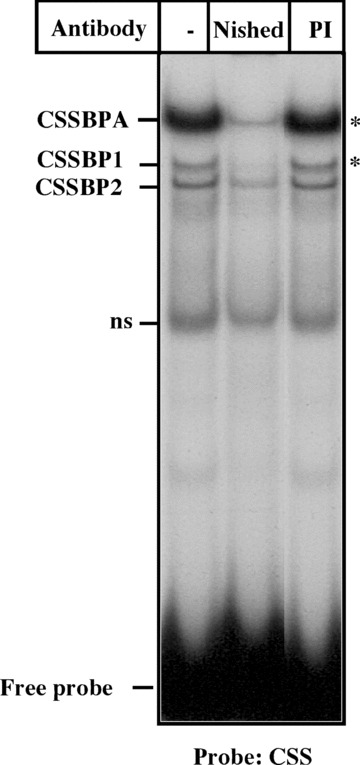
Gel mobility shift assay. Nuclear extracts of Chicken skeletal muscle cells pre‐incubated with anti‐Nished antibody (Nished) or pre‐immune serum (PI) were incubated with radiolabelled CSS oligonucleotide and subjected to gel mobility shift assay as described in ‘Materials and Methods’. □ denotes the DNA‐protein complexes disrupted by anti‐Nished antibody.
Nished represses cardiac MLC2v gene in skeletal muscle cells
In previous studies, Arnold and co‐workers [16] have noted the existence of a DNase‐I hypersensitive region (HR1) in the first intron of the cardiac MLC2v gene. Since IRE appeared to be the only functional sequence element in that region [15], we speculated that IRE is in an open conformation in cardiac cells, and perhaps not in skeletal muscle cells where the gene is repressed. To test this possibility, we performed the ChIP assay (see Materials and Methods) to read the histone code in the vicinity of IRE in cardiac and skeletal muscle cells. Euchromatin is characterized by the presence of histone modifications such as acetylated H3K9 and dimethylated H3K4. The heterochromatin is enriched by the presence of mono‐, di‐ and trimethylated H3K9. We used anti‐K4 methyl antibody to immunoprecipate chromatin from both heart and skeletal muscle tissues. Anti‐K4 methyl antibody was raised against the methylated lysine residue 4 and serves as an indicator of transcriptionally active DNA. PCR was performed with DNA primers flanking the IRE sequence. Results in Fig. 3 confirmed that the IRE containing DNA is in open conformation and hence accessible in cardiac cells, but under identical conditions it was not accessible in skeletal muscle cells. The use of monomethylated H3K9 antibody revealed the closed conformation in skeletal muscle. The expression of GAPDH gene was measured as a control reaction for evaluating the transcriptionally active selection of Anti‐K4 methyl antibody. Conversely, inactive chromatin activity was determined by evaluating the close chromatin status of the ApoB gene.
Figure 3.
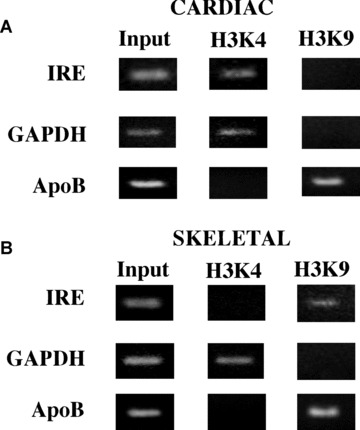
Chip assay with extracts from Chicken cardiac and skeletal muscle tissues. (A) Upper panel shows the ChIP assay performed in cardiac tissue for detection of active chromatin antiH3K4 (trimethylated) and inactive chromatin anti‐H3K9 (monomethylated) antibodies. We used primer flanking the IRE region in the cMLC2 gene, and as positive control the GAPDH primer, and as negative control primers that recognize the ApoB gene. (B) Lower panel shows a representative PCR using the same antibodies and target primers described in A but using skeletal muscle tissue.
We then examined the functional activity of Nished/IRE complex in transient cotransfection assay using the wild‐type promoter, pMLC2.1Luc, and mutant IRE‐ containing plasmid, pMutIRELuc, as reporters along with the Nished expression vector pNished‐V5 in primary cardiac cells (see Materials and Methods). We reasoned that in pMutIRE plasmid the presence of non‐functional IRE will simulate the transcriptional state of the gene in skeletal muscle cells where IRE is not accessible to DNA binding protein(s). We observed (Fig. 4) that the ectopic expression of Nished does not repress the activity of MLC2v promoter with wild‐type IRE, supporting our argument (see Fig. 1) that IRE/Nished interaction overrides the repressor activity of the CSS/Nished complex. Repression of promoter activity occurs when IRE is mutated. Apparently, the absence of wild‐type IRE and loss of IRE/Nished interaction would drive Nished to CSS to cause inhibition of transcription.
Figure 4.
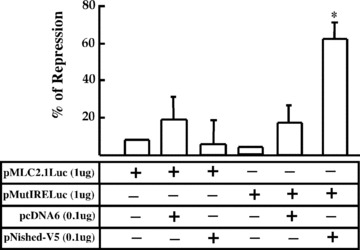
Primary chicken skeletal cells were co‐transfected with the DNA constructs containing wild‐type IRE, (pMLC2.1Luc) or IRE mutant (pMutIRELuc) along with Nished expression vector, pNished‐V5, or the control mammalian expression vector (pcDNA6V5/HisB). Data are plotted as percentage increase or decrease of promoter activity relative to that of pMLC2.1LucDNA. Experiments were performed in triplicate (n= 7). □ signifies differences with one sample t‐test with Bonferroni correction, P < 0.008.
To further investigate the role of Nished as repressor, we used the mouse skeletal muscle cell line C2C12 to produce Nished expressing stable transfectants, N1, (see ‘Materials and Methods’). Immunoprecipitation and Western blotting of cell extracts with anti‐Nished antibody showed an enhanced level of Nished expression in N1 cells relative to the parent cells C2C12. GHAPDH levels were detected as loading control (Fig. 5A). Upon transient transfection of mutant MLC2 promoter (pMutIRELuc) in N1 cells, there was repression of the reporter plasmid compared to the level of expression in the parent cell line C2C12 (Fig. 5B). Taken together, these results suggest that Nished effectively down‐regulates the MLC2v promoter activity when its binding is restricted to CSS alone, an environment mimicking that of the skeletal muscle cell.
Figure 5.
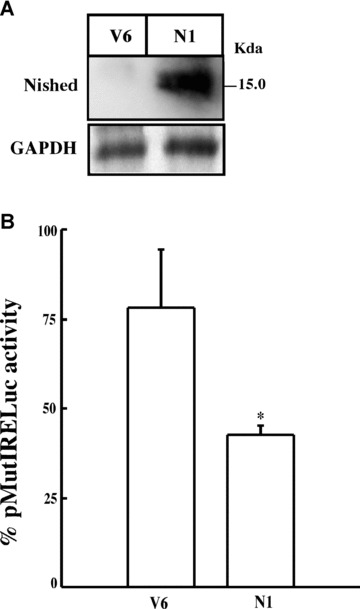
Western blot of total cell lysates from C2C12 cells stably transfected with pcDNA6 vector (V6) or with plasmid expressing Nished (N1). (A) cell lysates were immunoprecipitated and blotted with anti‐Nished antibody or pre‐immune serum. As loading control we used equal amount of proteins from each cell line, and by Western blot determined the GAPDH levels. (B) Transient transfection of plasmid pMutIRELuc in C2C12 cells stably transfected with pNished (N1) or vector alone (V5). Data are expressed as percentage activity of pMutIRELuc transfected in wild‐type C2C12 cells. (n= 4 in triplicate; □, one sample t‐test; P < 0.05).
DNA binding domain in Nished
In an attempt to localize the DNA binding domain in Nished, we created two N‐terminal, NΔ1 (34–137) and NΔ2 (90–137) and two C‐terminal CΔ1 (1–64) and CΔ2 (1–115) mutants (Fig. 6A) (see Materials & Methods). The wild‐type Nished and the mutants in lanes NΔ1, CΔ1, NΔ2 and CΔ2 were tested by micro‐affinity isolation assay (see Materials & Methods) where the in vitro translated and S35‐labelled products were subjected to binding to the biotinylated oligonucleotide containing CSS. Results in Fig. 6B show that mutants CΔ1 (1–64) and NΔ2 (90–137) that lack the segment do not bind CSS suggesting that the DNA binding activity resides within this region. The weak signals might be due to the proteolysis activity during transcription/translation assay. The results were nonetheless reproducible. The input of the reaction mixtures used in the assay, where the oligonucleotide was omitted, is shown in Fig. 6C. Specificity of the binding reaction was demonstrated by competition (Fig. 6B) with 10‐fold (10×) excess of non‐labelled in vitro translated wild‐type protein pNishedV5 product. Then, each mutant was evaluated for their repression activity on the pMutIRELuc, and we observed that NΔ1 lacking the first 34 aa although binds to the CSS motifs, yet fails to repress the promoter activity suggesting that the trans‐repression domain might be located within the first 34 aa region of the protein (Fig. 6D).
Figure 6.
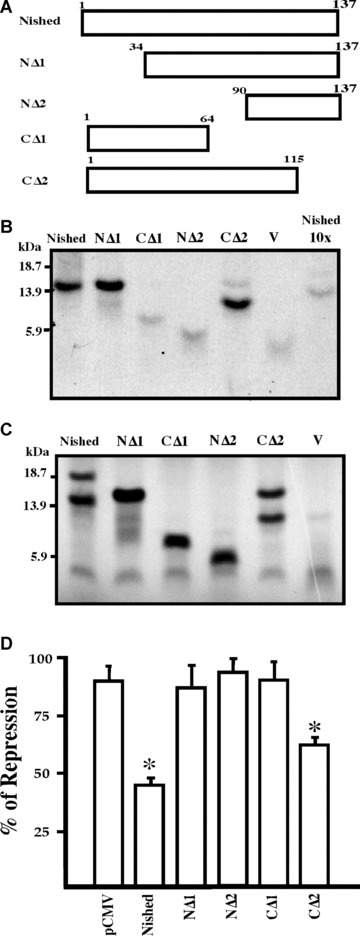
Schematic representation of Nished and its C‐and N‐terminal deletion mutants. (A) The deletions were made and cloned in the mammalian expression vector, pcDNA6V5/HisB, as described in ‘Material and Methods’. Numbers denote the amino acids in the constructs. (B) microaffinity isolation of S35 methionine labelled Nished and its N and C‐terminal deletion proteins generated by coupled transcription‐translation reaction with biotinylated CSS oligonucleotide as described in ‘Material and Methods’. (C) One tenth of reaction mixture volume of S35 methionine labelled Nished and its N‐and C‐terminal deletions proteins were electrophoresed on an 18% SDS gel. (D) Functional analysis of Nished deletion mutants. Primary skeletal muscle cells were co‐transfected with the IRE mutant (pMutIRELuc and Nished or the N and C terminal deletions mutants). The luciferase activity was normalized against the internal control, Renilla luciferase activity. (n= 7 in triplicate, □ one sample t‐test with Bonferroni correction, P < 0.05). Nished full length protein; NΔ1 (34–137 a.a); NΔ2 (90137 a.a.); CΔ1 (1–64 a.a); CΔ2 (1–115 a.a); V, Vector alone (negative control); Nished 10X, competition with lysate contouring vector alone.
Nished associates with HDACs co‐repressor proteins
To evaluate the mechanism by which Nished inhibits MLC2v gene transcription in non‐cardiac cells, we examined the involvement of HDAC activity in MLC‐2v gene repression. Upon exposure to Trichostatin A (TSA), a Class I‐II HDAC inhibitor, the promoter activity of the pMutIRELuc reporter in skeletal muscle cells was activated in a dose‐dependent manner (5, 10, 25 nM), to nearly 2.5 fold (Fig. 7A), whereas the pMutIRELuc was not activated in primary cardiac cells exposed to TSA (Fig. 7B). The differential response of MLC‐2v gene to TSA in cardiac and skeletal muscle cells indicates the specific requirement of HDAC activity for active repression in skeletal muscle cells. The selective requirement of classes I and II HDACs was demonstrated by lack of release of inhibition by the HDAC class III inhibitors, sirtinol [17] and M15 [18]. To identify the HDAC, which interacts with Nished, tissue extracts of heart and skeletal muscle were immunoprecipitated with anti‐Nished antibody followed by Western blotting with antibodies against HDAC3, HDAC5 and Nished. We observed interaction of Nished with HDAC 3 and HDAC 5 in extracts from skeletal muscle only (Fig. 8A), indicating that Nished’s association with HDAC3 and 5 might be important in repression of cardiac MLC2v gene in skeletal muscle. In an attempt to identify the putative HDAC3/5 binding domain(s) in Nished, we tested the deletion mutants of Nished for Nished/HDAC3/5 binding assay as above. Figure 8B shows that NΔ2 does not bind with HDAC3 and 5, whereas NΔ1 binds with HDAC 3 but not with HDAC5, suggesting that the optimal binding activity, at least for HDAC3, resides in CΔ2, and involves amino acids 37–64 in Nished.
Figure 7.
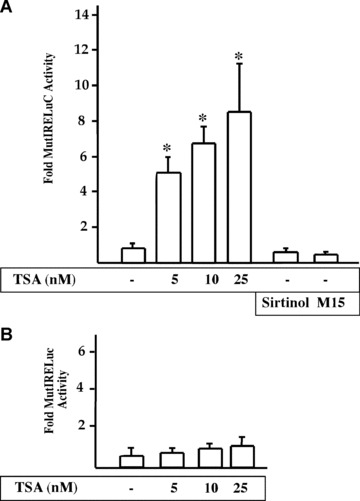
Tricostatin A releases CSS mediates repression of MLC2v promoter in C2C12 cells. (A) C2C12 cells were transfected with MLC2v promoter containing mutated IRE (pMutIRELuc). After 24 hrs, cells were treated with Tricostation A (TSA), an inhibitor of Class I and II HDACs, and 25 μM each of Sirtinol and M15, inhibitors of Class III HDACs. The HDACs inhibitors were dissolved in DMSO. Luciferase assays were performed 18 hrs after treatment. Nished repression of MLC2v promoter was relieved by TSA in a dose‐dependent manner, whereas Sirtionol and M15 had no effect. (B) Similar transient transfection experiment was performed in chicken primary cardiac cells, however, TSA treatment failed to modify the MLC2v promoter activity (n= 4 in triplicate, □ one sample t‐test, P < 0.05).
Figure 8(a).
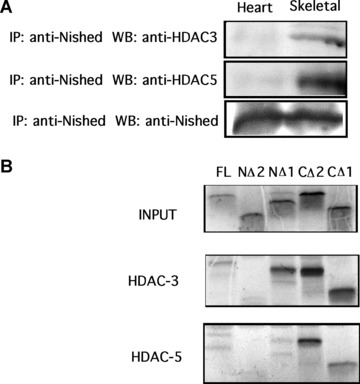
Total cell lystate of mouse cardiac and skeletal muscle tissue was co‐immunoprecipicated with anti‐Nished antibodies and immunoblotted with anti‐HDAC3, anti‐HDAC5 and anti‐Nished antibody (loading control). (B) In vitro transcription/translation pull‐down assay was done using pcNishedV5, CΔ1, CΔ2, NΔ1, NΔ2, pcHDAC3′ flag, pcHDAC5′ flag as described in ‘Material and Methods’. Nished proteins were labelled with S35 methionine and combined with HDAC3 or HDAC5. Proteins were pulled down using M2 anti‐flag agarose, and run on a 18% polyacrylamide gel. Panel 1 (INPUT) shows translated Nished proteins. Panel 2 shows HDAC3 binding to Nished mutants, no binding is seen with NΔ2; panel 3 shows HDAC5 binding to Nished mutant proteins (no binding is seen with NΔ2).
Discussion
Our laboratory has characterized the MLC2v gene extensively and identified several proximal and distal regulatory elements and their cognate DNA binding proteins. More recently, we have demonstrated the activation role of IRE bound Nished in conjunction with co‐activators in hypertrophy agonist‐induced up‐regulation of the MLC2v gene [15]. The function of IRE and the role of Nished as an activator becomes further evident by the finding that the αBE‐4 element in B‐crystallin gene, which shares sequence homology with IRE, is needed for the maximal expression of the B‐crystallin gene in myocardial cells [19, 20]. Nished or an analogous DNA binding protein is likely to be involved in this context in activation of this gene in cardiomyocytes. The question that remains unresolved, however, is the mechanism by which Nished causes the inhibition of expression of cardiac‐specific MLC2v gene in skeletal muscle.
The regulatory association of muscle‐specific proteins with activators and repressors of cardiac genes have been amply documented in a large number of studies, yet the precise mechanism involved in silencing the cardiac genes in skeletal muscle remains unsolved. Post‐translational modifications of chromatin such as acetylation, deacetylation and methylation have been shown to be involved in regulation of gene transcription. Deacetylation of the histones results in chromatin compaction blocking gene transcription. Our data presented in this study suggest that the repression of MLC2v in skeletal muscle involves two steps; one where the IRE sequence becomes inaccessible to the putative activator(s) of transcription, and second where CSS/Nished tissue‐specific association with the deacetylases HDAC3/5 mediates suppression of the MLC2v gene expression in skeletal muscle, perhaps via inhibition of the MEF‐2 protein. Indeed, MEF‐2 interaction with element B is essential for the transcriptional activity of the MLC2v promoter [21], and this Element B‐protein interaction is disrupted in presence of CSS binding protein (data not shown). The repression of MLC2v transcription in skeletal muscle cells, however, is underscored by the selectively of the CSS site and the lack of access of the IRE site in physiological conditions. These findings are consistent with the reported role of other co‐repressors such as SMRT, SiN3, NuRD and their functional association with class I and II HDACs [22]. The association of HDACs with these distinct corepressors constitutes a family of transcription repression complexes that interfere with the activation role of transcription factors such as MEF‐2 [23, 24].
Our results here suggest that the antithetical role of Nished, i.e. to potentiate the promoter activity of MLC2v gene in cardiac cells and to inhibit the same gene in skeletal muscle is afforded by the ability of Nished to interact with activators [15] or repressors (this report). Moreover, the association of DNase hypersensitivity and histone modifications in specific cell types suggests that developmentally established chromatin changes dictate the spatial and temporal expression of genes. In higher eukaryotes, histone modification of the type that involves methylation of histone H3 at lysines 4 and 9 (K4 and K9) leads to complementary functions; methyl K4 is enriched in transcriptionally active regions and methyl K9 in silent regions [25, 26]. Our data show that the histone H3 K4 residue is methylated around the IRE sequence in cardiac cells, suggesting that IRE is accessible to the transcriptional machinery in the cardiac tissue and inaccessible in non‐cardiac tissues. IRE must, therefore, reside in a developmentally established instructive chromatin conformational environment that directs the transcription of MLC2v gene in cardiac cells. This is consistent with the methylation profile of H3 K9 and K4 residues across the chicken βglobin locus that similarly dictates the erythroid development stage‐specific §‐globin gene expression [27]. Likewise, chromatin modification at the myogenin loci is marked by the interaction of Pbx/Meis complex with MyoD, which, in association with other factors, regulates the temporal expression of myogenin during muscle differentiation through a feed‐forward mechanism [28].
We have previously reported that IRE–Nished interaction serves as the target of hypertrophy‐induced signalling in expression of MLC2v gene [15]. Nished interacts with transcription factor, NFATc4, and recruits p300 at the IRE site. The DNA–protein interaction is stimulated by the hypertrophic agonists angiotensin II. In this context, the dual role of Nished is significant given the identical Nished binding sequence present in the two disparate elements (CSS and IRE) in the MLC2v promoter. The association of Nished with HDAC and with p300 thus identifies the antithetical role of Nished in mediating transcription regulation. Our data identify Nished as a regulator of both positive and negative transcription in the target muscle cells of distinct lineages. That Nished is expressed in both adult cardiac and skeletal muscle cells, as well as in non‐muscle cells, makes it likely that it has a role(s) in processes other than that in myogenesis.
Acknowledgement
This work was supported by Grant No HL073399 from the National Institutes of Health.
References
- 1. Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays. 1998; 20: 615–26. [DOI] [PubMed] [Google Scholar]
- 2. Xu L, Glass CK, Rosenfeld MG. Coactivator and corepressor complexes in nuclear receptor function. Curr Opin Genet Dev. 1999; 9: 140–7. [DOI] [PubMed] [Google Scholar]
- 3. McKinsey TA, Zhang CL, Lu J, et al . Signal‐dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000; 408: 106–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McKinsey TA, Zhang CL, Olson EN. MEF2: a calcium‐dependent regulator of cell division, differentiation and death. Trends Biochem Sci. 2002; 27: 40–7. [DOI] [PubMed] [Google Scholar]
- 5. Han A, He J, Wu Y, et al . Mechanism of recruitment of class II histone deacetylases by myocyte enhancer factor‐2. J Mol Biol. 2005; 345: 91–102. [DOI] [PubMed] [Google Scholar]
- 6. Mal A, Sturniolo M, Schiltz RL, et al . A role for histone deacetylase HDAC1 in modulating the transcriptional activity of MyoD: inhibition of the myogenic program. EMBO J. 2001; 20: 1739–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goswami S, Qasba P, Ghatpande S, et al . Differential expression of the myocyte enhancer factor 2 family of transcription factors in development: the cardiac factor BBF‐1 is an early marker for cardiogenesis. Mol Cell Biol. 1994; 14: 5130–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dalla Libera L. A comparative study of atrial and ventricular myosin light subunits from different species. Comp Biochem Physiol B. 1986; 83: 751–5. [DOI] [PubMed] [Google Scholar]
- 9. O’Brien TX, Lee KJ, Chien KR. Positional specification of ventricular myosin light chain 2 expression in the primitive murine heart tube. Proc Natl Acad Sci USA. 1993; 90: 5157–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shen RA, Goswami SK, Mascareno E, et al . Tissue‐specific transcription of the cardiac myosin light‐chain 2 gene is regulated by an upstream repressor element. Mol Cell Biol. 1991; 11: 1676–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Qasba P, Lin E, Zhou MD, et al . A single transcription factor binds to two divergent sequence elements with a common function in cardiac myosin light chain‐2 promoter. Mol Cell Biol. 1992; 12: 1107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Qasba P, Danishefsky K, Gadot M, et al . Functional analysis of a CArG‐like promoter element in cardiac myosin light chain 2 gene. Cell Mol Biol. 1992; 38: 561–9. [PubMed] [Google Scholar]
- 13. Zhou MD, Wu Y, Kumar A, et al . Mechanism of tissue‐specific transcription: interplay between positive and negative regulatory factors. Gene Expr. 1992; 2:127–38. [PMC free article] [PubMed] [Google Scholar]
- 14. Dhar M, Mascareno EM, Siddiqui MAQ. Two distinct factor‐binding DNA elements in cardiac myosin light chain 2 gene are essential for repression of its expression in skeletal muscle. Isolation of a cDNA clone for repressor protein Nished. J Biol Chem. 1997; 272: 18490–7. [DOI] [PubMed] [Google Scholar]
- 15. Mathew S, Mascareno E, Siddiqui MAQ. A ternary complex of transcription factors, Nished and NFATc4, and co‐activator p300 bound to an intronic sequence, intronic regulatory element, is pivotal for the up‐regulation of myosin light chain‐2v gene in cardiac hypertrophy. J Biol Chem. 2004; 279: 41018–27. [DOI] [PubMed] [Google Scholar]
- 16. Arnold HH, Klapthor H, Winter B. The cardiac myosin light chain (MLC‐2A) gene in chicken is methylated in both expressing and nonexpressing tissues. Cell Biol Toxicol. 1984; 1: 41–53. [DOI] [PubMed] [Google Scholar]
- 17. Grozinger CM, Chao ED, Blackwell HE, et al . Identification of a class of small molecule inhibitors of the sirtuin family of NAD‐dependent deacetylases by phenotypic screening. J Biol Chem. 2001; 276: 38837–43. [DOI] [PubMed] [Google Scholar]
- 18. Hassig CA, Fleischer TC, Billin AN, et al . Histone deacetylase activity is required for full transcriptional repression by mSin3A. Cell. 1997; 89: 341–7. [DOI] [PubMed] [Google Scholar]
- 19. Gopal‐Srivastava R, Haynes JI 2nd, Piatigorsky J. Regulation of the murine alpha B‐crystallin/small heat shock protein gene in cardiac muscle. Mol Cell Biol. 1995; 15: 7081–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hoover HE, Thuerauf DJ, Martindale JJ, et al . alpha B‐crystallin gene induction and phosphorylation by MKK6‐activated p38. A potential role for alpha B‐crystallin as a target of the p38 branch of the cardiac stress response. J Biol Chem. 2000; 275: 23825–33. [DOI] [PubMed] [Google Scholar]
- 21. Zhou MD, Goswami SK, Martin ME, et al . A new serum‐responsive, cardiac tissue‐specific transcription factor that recognizes the MEF‐2 site in the myosin light chain promoter. Mol Cell Biol. 1993; 13: 1222–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Downes M, Ordentlich P, Kao HY, et al . Identification of a nuclear domain with deacetylase activity. Proc Natl Acad Sci USA. 2000; 97: 10330–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang CL, McKinsey TA, Lu JR, et al . Association of COOH‐terminal binding protein (CtBP) and MEF2‐interacting transcription repressor (MITR) contributes to transcriptional repression of the MEF2 transcription factor. J Biol Chem. 2001; 276: 35–9. [DOI] [PubMed] [Google Scholar]
- 24. Youn HD, Grozinger CM, Liu JO. Calcium regulates transcriptional repression of myocyte enhancer factor 2 by histone deacetylase 4. J Biol Chem. 2000; 275: 22563–7. [DOI] [PubMed] [Google Scholar]
- 25. Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 2001; 15: 2343–60. [DOI] [PubMed] [Google Scholar]
- 26. Richards EJ, Elgin SC. Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell. 2002; 108: 489–500. [DOI] [PubMed] [Google Scholar]
- 27. Litt MD, Simpson M, Gaszner M, et al . Correlation between histone lysine methylation and developmental changes at the chicken beta‐ globin locus. Science. 2001; 293: 2453–5. [DOI] [PubMed] [Google Scholar]
- 28. Berkes CA, Bergstrom DA, Penn BH, et al . Pbx marks genes for activation by MyoD indicating a role for a homeodomain protein in establishing myogenic potential. Mol Cell. 2004; 14: 465–77. [DOI] [PubMed] [Google Scholar]