

Abstract
In order to understand the transcriptional mechanism that underlies cell protection to stress, we evaluated the role of CLP‐1, a known inhibitor of the transcription elongation complex (pTEFb), in CLP‐1 +/− mice hearts. Using the isolated heart model, we observed that the CLP‐1+/− hearts, when subjected to ischaemic stress and evaluated by haemodynamic measurements, exhibit significant cardioprotection. CLP‐1 remains associated with the pTEFb complex in the heterozygous hearts, where as it is released in the wild‐type hearts suggesting the involvement of pTEFb regulation in cell protection. There was a decrease in Cdk7 and Cdk9 kinase activity and consequently in phosphorylation of serine‐5 and serine‐2 of Pol II CTD in CLP‐1 +/− hearts. However, the levels of mitochondrial proteins, PGC‐1α and HIF‐1α, which enhance mitochondrial activity and are implicated in cell survival, were increased in CLP‐1+/− hearts subjected to ischaemic stress compared to that in wild‐type CLP‐1+/+ hearts treated identically. There was also an increase in the expression of pyruvate dehydrogenase kinase (PDK‐1), which facilitates cell adaptation to hypoxic stress. Taken together, our data suggest that regulation of the CLP‐1 levels is critical to cellular adaptation of the survival program that protects cardiomyocytes against stress due collectively to a decrease in RNA Pol II phosphorylation but an increase in expression of target proteins that regulate mitochondrial function and metabolic adaptation to stress.
Keywords: ischaemia, pTEFb, Hexim1, CLP‐1, cardioprotection
Introduction
Over the past decade considerable progress was made in understanding the molecular changes that occur in heart in response to stress [1, 2, 3, 4, 5]. Specific changes in transcription, for example, were attributed to cardioprotection as an early response to stress in a number of animal models. Recent findings implicate the transcription elongation P‐TEFb complex in the compensatory response of the heart to stress [6, 7, 8]. Phosphorylation of carboxyl termination domain (CTD) of RNA polymerase II (Pol II) is essential in mediating RNA chain elongation in transcription of eukaryotic genes. In order for RNA Pol II to enter the transition into the elongation phase, the serine kinase activity of P‐TEFb, composed of Cyclin‐dependent kinase 9 (Cdk9) and Cyclin T, is required [9, 10]. The P‐TEFb complex activity is, however, inhibited by its association with the inhibitory protein Hexim1 [11, 12, 13], which upon its release from the complex promotes phosphorylation of Pol II by Cdk 9 at CTD and consequently the RNA chain elongation continues [14, 15]. Initial support for the role of Hexim1 in cardiac function comes from our recent finding that the dissociation of CLP‐1, the mouse homolog of human Hexim1 protein, from the pTEFb complex, allows Cdk‐mediated phosphorylation of Pol II and promotes cardiac growth and hypertrophy [16]. Recent findings, however, suggested that the increase in Cdk9 as well as Cyclin‐dependent kinase 7 (Cdk7) activity cause inhibition of peroxisome proliferator‐activated receptor γ coactivator‐1α (PGC‐1α) [17, 18], a key regulator of mitochondrial function and essential in metabolic adaptation of the heart in cardiovascular diseases [19, 20]. These observations prompted us to ask the question whether the decrease in CLP‐1 protein level in heterozygous CLP‐1 +/− mice would influence the response to stress distinct from that in the wild‐type CLP‐1 +/+ heart.
To this end, we used an animal model developed in our laboratory where the CLP‐1 gene was ablated [16, 21]. CLP‐1 −/− mice die in utero during day 16–17 p.c, however, the heterozygous CLP +/− mice survive and appear normal phenotypically and serve as a useful model for investigating the role of CLP‐1. We, therefore, subjected the CLP‐1 +/− hearts to stress induced by preconditioning, ischemia and ischemia/reperfusion as described before [22] and observed that the hearts of CLP‐1+/− mice exhibit pronounced resistance to ischaemic stress. In order to understand the underlying cardioprotective mechanisms, we examined the components of the pTEFb complex as well as the levels of mitochondrial proteins such as, PGC‐1α and the hypoxia inducible factor 1α (HIF‐1α). Both proteins, PGC‐1α and HIF‐1α, play a significant role in mitochondrial function and cell survival during stress conditions [23, 24, 25]. We found that when the CLP‐1 wild‐type hearts were subjected to ischaemic stress, the pTEFb complex was activated by releasing the inhibitor CLP‐1 protein; however, in the CLP‐1 heterozygous mice hearts it remained associated and consequently Pol II phosphorylation was decreased. CLP‐1 +/− hearts also exhibited an increase in the levels of HIF‐1α during ischemia and ischemia/reperfusion when compared with wild‐type CLP‐1 hearts. The increase in HIF‐1α levels is known to trigger the expression of pyruvate dehydrogenase kinase‐1 (PDK‐1) [26, 27], as such, we observed an increase in PDK‐1 protein in extracts from CLP‐1 +/− hearts. Collectively, these data suggest that CLP‐1 heterozygocity in hearts is an important facet of cell protection that is mediated apparently by changes in nuclear transcriptional activity and regulation of genes critical to mitochondrial function.
Material and methods
Animals
The mice used in this investigation conform with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996).
Isolated perfused heart preparation
Isolated mice hearts from wild‐type littermates and CLP‐1 +/− subjected to ischaemic stress as before [22] were divided into two groups (see Fig. 1A): (1) isolated hearts were perfused with Krebs‐Henseleit bicarbonate (KHB) buffer for 15 min and then subjected to 30 min of ischemia followed by 15, 30, 60 or 120 min of reperfusion (control); (2) the hearts were preconditioned to ischaemic stress by four cyclic episodes of 5 min ischemia each followed by another 10 min reperfusion followed by 30 min of ischemia and 15, 30, 60 or 120 min of reperfusion (PC). The isolated hearts from CLP‐1 +/− or wild‐type mice were obtained after injecting the mice with pentobarbital sodium (80 mg/kg body wt ip) and anticoagulant heparin sodium (500 IU/kg body wt ip). The hearts were excised and transferred to ice‐cold modifiedKHB, containing in mM (NaCl 118; KCl 4.7; CaCl2 1.7; NaHCO3 24; KH2PO4 1.2; MgSO4 12; Glucose 10). The aorta and pulmonary vein were cannulated and perfused in retrograde Langendorff mode. Perfusate KHB temperature was maintained at 37°C and saturated with 95% O2 and 5% CO2. The perfusion was then switched to anterograde mode where the buffer enters the cannulated left atrium at pressure equivalent to 10 cm of water (10 kPa). The heart measurements were recorded by Gould p23XL transducer (Gould Instrument System, Inc., Valley View, OH, USA). The signal was amplified by using Gould 6600 6600 series signal conditioner (Gould Instrument System, Inc.) and monitored on a Cordat II real‐time acquisition system (Triton technologies, San Diego, CA, USA). The aortic flow was measured by a flow meter. Once baseline measurements of heart rate, coronary flow, aortic flow, left ventricular developed pressure and its maximum first derivative were performed, the anterograde perfusion line was closed, and the heart was subjected to 30 min of ischemia. Before the initiation of the 2 hrs reperfusion the heart was perfused in a retrograde mode to avoid the development of ventricular fibrillation. Hearts that showed any cardiac disturbance such as ventricle arrhythmia and fibrillation during the experiment, were excluded from the study.
Figure 1.
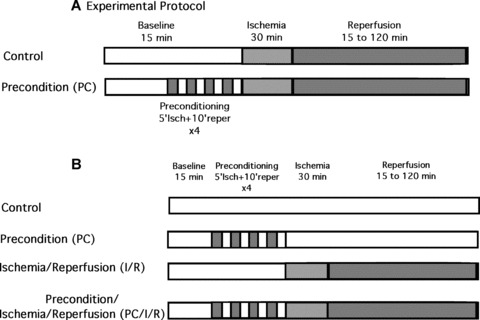
(A) Schematic representation of the experimental protocol used for the evaluation of haemodynamic parameters and infarct size. (B) A diagram of the experimental protocols for stress application and analysis of wild‐type and CLP‐1 +/− hearts (see Figs. 4 and 5).
Measurement of Infarct Size
Following global ischemia, the heart was infused with 10% solution of the triphenyl tetrazolium (TTC) in phosphate buffer through the aortic cannula for 20 min. The left ventricle was removed and sliced into 1‐mm thickness of cross‐sectional pieces and weight. Each slice was scanned with computer‐assisted scanner (Hewlett Packard Scanjet 5370C Flat bed scanner). The risk area of the whole myocardium was stained in red by TTC while the infarct zone remained unstained by TTC. These were measured with computerized software (Scion Image); and areas were multiplied by the weight of each section and summed up to obtain the total of the risk zone and an infarct zone. The infarct size was expressed as the ratio of the infarct zone to the risk zone.
Western blotting and Cdk9 kinase assay
The isolated mice hearts used are from four groups (see Fig. 1B): (i) isolated hearts were perfused with KHB buffer for 3 hrs and 45 min. (control); (ii) the hearts were preconditioned (PC) to ischaemic stress by four cyclic episodes of 5 min ischemia each followed by another 10 min reperfusion; (iii) the hearts were subjected to 30 min of ischemia followed by 2 hrs reperfusion (I/R) and (iv) the hearts were subjected first to PC followed by to 30 min of ischemia, followed by 2 hrs of reperfusion (PC/I/R). Heart tissues were homogenized in lysis buffer (50 mm Tris‐HCl pH 8.0, 10 mm MgCl2, 120 mm NaCl, 0.5% Nonidet P‐40, 5 mm dithiothreitol, 2.5 mm MnCl2, protease inhibitors and RNAsin 40 U/ml (Promega Corporation). The lysates were incubated overnight with specific antibodies (rabbit anti‐CLP‐1, mouse anti‐Cdk9, or goat anti‐cyclin‐T1) as described in the catch and release protocol from Upstate Bitechnology. The Cdk9 kinase reactions were performed using the anti‐Cdk9 immuno‐complexes and following the protocol by Upstate Biotechnology. The Cdk9/cyclinT1 immunocomplex was incubated with 8 mm MOPS (pH 7.0), 1 mm EDTA, 10 mm MgAc, 5 mm dithiothreitol, protease inhibitors, RNAsin 40 U/ml, 5 μM ATP, 5 μCi [γ‐32‐P]ATP (500–800 cpm/pmol), 200 ng GST‐CTD (YSPTSPS)4 to the beads and incubating at 30°C for 20 min. The reaction was stopped with 5 μl of 3% phosphoric acid, and aliquots were transferred to P30 filtermat, washed with 0.75% phosphoric acid, dried and 4 ml of scintillation cocktail was added and read in a scintillator counter. Immunoprecipitates were evaluated by Western blot analysis. Proteins were separated on a 7.5% or 10% SDS/polyacrylamide gel and transferred to nitrocellulose membranes, and probed with specific antibodies (monoclonal mouse anti‐Cdk9, monoclonal mouse anti‐ubiquitin (P4D1), goat anti‐cyclin‐T1, anti‐SUMO1, monoclonal anti‐HIF‐1α (MAB5382), anti‐phosphoserine 2 H5 and anti‐phosphoserine 5 H14 (Covance), rabbit polyclonal anti‐CLP‐1 (developed in our laboratory), and polyclonal anti‐GAPDH for loading control from ABCAM). Following the incubation with primary antibodies and the corresponding HRP‐conjugated secondary antibody, the Western blot was developed according to the Chemiluminiscence protocol (Amersham).
Statistics
The values for myocardial functional parameters, and infarct sizes were all expressed as the mean ± standard error of mean (SEM). The statistical analysis was performed by one‐way ANOVA for any differences between the mean values of all groups. Differences between data were analysed for significance by performing Student’s t ‐test after Bonferroni’s correction. The results were considered significant if P < 0.05.
Results
Heterozygous CLP‐1 hearts are resistant to ischaemic stress
Isolated hearts from CLP‐1+/+ and CLP‐1 +/− mice were subjected to the experimental stress protocol of ischemia/reper fusion, as shown in Figure 1 and described in ‘Materials and Methods’. Functional haemodynamic measurements such ascoronary flow and LVdp/dt were made as before [22]. After reperfusion, the control hearts of CLP‐1+/+ showed a decrease in the absolute values of aortic flow, developed pressure, and the first derivative of developed pressure (dP/dt max). As expected, preconditioned CLP‐1+/+ hearts display a recovery following ischemia/reperfusion (see Fig. 2A, D and E). However, following reperfusion, the hearts of the CLP‐1+/− mice showed haemodynamic values of developed pressure, the first derivative of developed pressure (dP/dt max) and aortic flow similar to hearts from wild‐type CLP‐1 subjected to PC, suggesting that the CLP‐1+/− hearts harbour an intrinsic cardioprotective program. Indeed, when CLP‐1 +/− mice hearts were subjected to PC followed by I/R, the degree of cardioprotection was similar to CLP‐1 +/+ subjected to PC, i.e. no further protection was observed. Coronary flow and heart rate did not exhibit significant changes between genotypes (Fig. 2B and C). Cardioprotection was also evident when the infarct sizes were measured. In the wild‐type hearts, the infarct size was reduced upon PC, whereas in the CLP‐1+/− mice the infarct size was the same with or without PC (Fig. 3).
Figure 2.

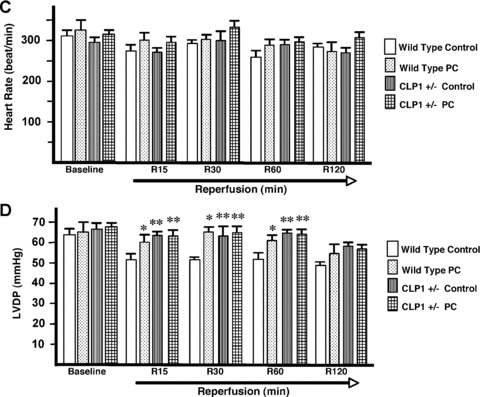
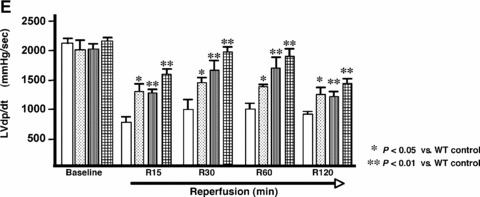
CLP‐1 +/− mice hearts subjected to ischemia/reperfusion show a cardioprotective phenotype. (A) aortic flow, (B) coronary flow, (C) Heart rate, (D) Developed pressure and (E) first derivative of developed pressure, respectively reveal that the haemodynamic parameters in CLP‐1 +/− mice hearts are similar to those of preconditioned wild‐type heart during ischemia/reperfusion. Ischemia, reperfusion and preconditioning were done as described in ‘Materials and Methods’. Results are shown as mean ± S.E.M. of six mice per group. *P < 0.05 versus control, **P < 0.01 versus control.
Figure 3.
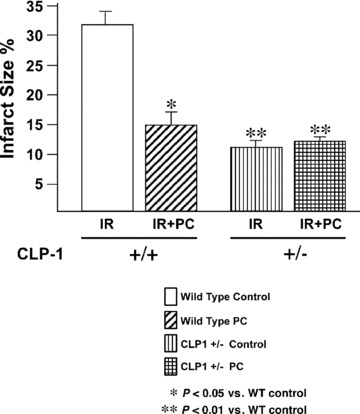
CLP‐1 +/− mice hearts exhibit a decreased infarct size. Infarct size was measured as described in Materials and Methods. Infarct size is significantly reduced in CLP‐1 +/− heart mice during ischemia/reperfusion without preconditioning. Results are shown as mean ± S.E.M. of six mice per group. *P < 0.05 versus control, **P < 0.01 versus control.
Transcription elongation factor‐b (P‐TEFb) complex and its components
In order to understand the molecular basis of the stress‐induced cardioprotection in CLP‐1 +/− hearts, we evaluated the putative changes in the pTEFb complex activity. First the CLP‐1 protein levels in CLP‐1 +/+ and +/− hearts were examined in control mice by Western blot as shown in Figure 4A. As expected, the heterozygote hearts have a reduced level of CLP‐1 protein (approx. 40%) as compared to wild‐type hearts. GAPDH expression served as loading control. We then examined the levels of CLP‐1 associated with cyclin‐T1 in the P‐TEFb complex. Immunoprecipitation with anti‐Cdk9 or anti‐CLP‐1 antibodies was followed by Western blot with anti‐cyclin T1 antibody (Fig. 4B). The cyclin‐T1‐Cdk9 association in the pTEFb complex remained unchanged after each stress in both wild‐type and heterozygous hearts. However, the interaction of CLP‐1 with cyclin T1 decreased in the heart of wild‐type mice subjected to stress, but not in heterozygote hearts under identical conditions. Because CLP‐1 is an intrinsic inhibitor of cyclin‐T1‐mediated Cdk9 activity, its dissociation is required for Cdk9 activity in CLP‐1 +/+ mice subjected to stress. The lack of CLP‐1 dissociation in CLP‐1 +/− hearts suggests that cdk9 activity would be inhibited. We, therefore, examined the Cdk9 activity in extracts of the same hearts of heterozygous CLP‐1 +/− mice and found that it was markedly reduced compared to the activity level in wild‐type CLP‐1 +/+ hearts subjected to identical stress (Fig. 4C) confirming the functional association of CLP‐1 with cyclin T1. This would also raise the question whether the kinase activity of both Cdk7 and Cdk9 are altered in the heterozygous heart. We, therefore, examined the phosphorylation status of serine 5 and serine 2 of the CTD in RNA Pol II, as they reflect the activation of Cdk7 and Cdk9, respectively [17, 18]. Figure 4D shows that there was a significant decrease in both serine5 and serine2 phosphorylation in the heart extracts from CLP‐1 +/− mice subjected to IR or PC and IR induced stress (see Discussion).
Figure 4.
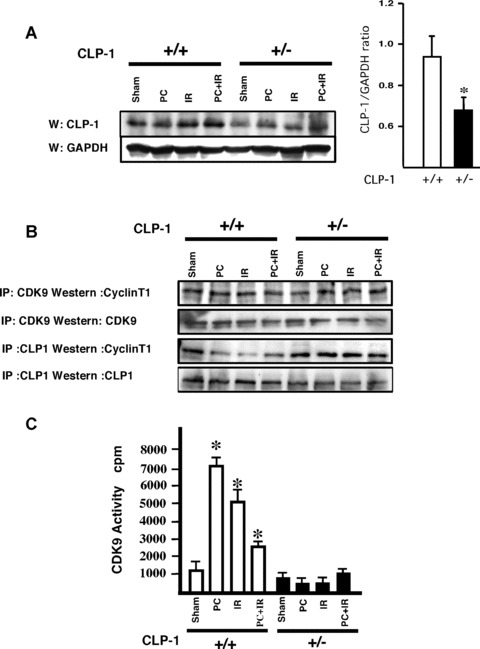

Changes in CLP‐1 expression level modulates the pTEFb complex activity. (A) Western blot shows the level of CLP‐1 protein expression in CLP‐1 +/+ and CLP‐1 +/− hearts during each stress. The bar graph indicates the ratio between CLP‐1 and GAPDH expression, and shows a decrease in expression level of CLP‐1 protein in CLP‐1 +/− hearts. This measurement was performed in quadruplicate and the results are shown as mean ± S.E.M. per group. *P < 0.05 versus CLP‐1+/+. (B) Association of CLP‐1 with cyclin T was determined by immunoprecipitation of extracts with antibodies against Cdk9 or CLP‐1 followed by Western blotting with cyclin T1 antibody. (C) Cdk activity was measured as described in Materials and Methods. The Cdk9 activity measurement was performed in triplicate and the results are shown as mean ± SEM per group. *P < 0.05 versus control. (D) Extracts as above were used for Western blotting using antibodies for phosphoserine 2 and 5 and Pol II. Graph bars show the relative increase or decrease in phosphoserine 5 and 2 versus total Pol II expression. At least three experiments were performed and a representative figure is shown. *P < 0.05 versus control CLP‐1+/+, **P < 0.05 versus control CLP‐1 +/−, #P < 0.01 versus control CLP‐1+/+.
CLP‐1 regulates mitochondrial genes
It was reported recently that the increase in Cdk7 and Cdk9 activities such as that occurs in compensatory cardiac hypertrophy, causes down‐regulation of PGC‐1α, a master regulator of mitochondrial genes [17, 18]. We, therefore, examined whether the converse is true, i.e. whether the repressed level of Cdk9 activity in CLP‐1 +/− mice would enhance PGC‐1α level. As seen in Figure 5A, the PGC‐1 expression was noticeably enhanced in CLP‐1 +/− heart subjected to stress. The increase in the level of PGC‐1 suggests that the mitochondrial viability and function must have been enhanced. Mitochondria are intimately involved in cellular oxygen sensing. Since the activation of the hypoxia‐induced transcription factor, HIF‐1α, serves as an index of mitochondrial activity [26, 27] we examined the protein level of HIF‐1α in CLP‐1 +/− heart subjected to stress. Western blot analysis showed that there was a significant increase in the levels of HIF‐1α protein in CLP‐1 +/− hearts subjected to ischaemic stress as compared to CLP‐1 +/+ hearts treated identically (Fig. 5B). HIF‐1α degradation is mediated by ubiquitination [23]. Therefore, it is likely that the enhanced level of HIF‐1α is due to inhibition of ubiquitination. We, therefore, immunoprecipitated the heart extracts using anti‐ubiquitin antibody followed by Western blot with anti‐HIF‐1α antibody (Fig. 5C). We observed the characteristic profile of ubiquitination of HIF‐1α under each stress condition in CLP‐1 +/+ hearts, but the same was significantly diminished in heterozygous CLP‐1 +/− hearts. It was suggested that sumoylation is an additional post‐translational mechanism that targets HIF‐1α protein and influences its expression level in hypoxic condition [27]. When HIF‐1 was immunoprecipitated followed by Western blotting with antibody against SUMO‐1, there was an increase in HIF‐1α/SUMO‐1 interaction in extracts from heterozygous CLP‐1 +/− hearts (Fig. 5D). The lower panel shows HIF‐1α as loading control when equal amounts of HIF‐1 were used for each stress column. There was also an increase in the expression of SUMO‐1 protein in the heart of CLP‐1 +/− mice (Fig. 5E). Since HIF‐1 expression during hypoxic conditions is known to induce pyruvate dehydrogenase kinase‐1 (PDK‐1), which facilitates cell adaptation to low oxygen pressure by repressing mitochondrial function and reactive oxygen species (ROS) production [28, 29], we examined PDK‐1 expression and found it to be increased in CLP‐1 +/− hearts (Fig. 5F).
Figure 5.
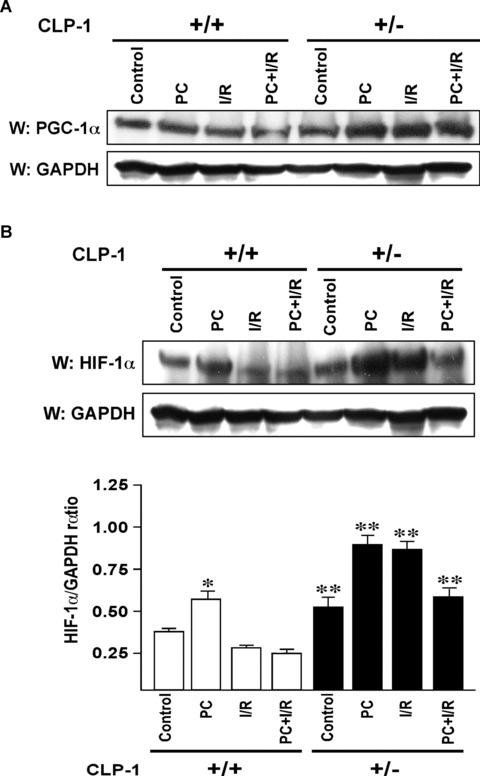
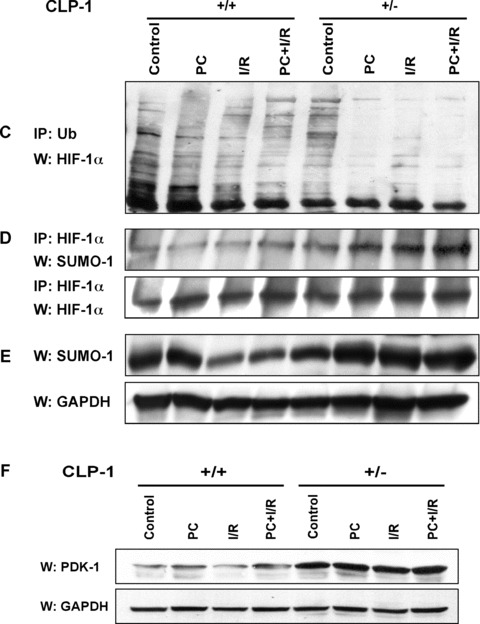
Expression of PGC‐1α and HIF‐1α in heart‐tissue of CLP‐1 heterozygous mice. (A) The expression levels of PGC‐1α in the protein heart extracts from control, preconditioning (PC), ischemia/reperfusion (I/R) and I/R with PC groups of wild‐type and in CLP‐1 +/− hearts was determined by Western blot analysis. GAPDH was used as loading control. (B) We use the same extracts as in Figure 4A to determine the expression levels of HIF‐1α, and as before GAPDH was used as loading control. The experiment was repeated three times and a representative experiment is shown. *P < 0.05 versus wild‐type control, **P < 0.01 versus wild‐type control. (C) The ubiquitination profile of HIF‐1 was examined by using heart extracts from each group. The extracts were used for immunoprecipitation with anti‐ubiquitin antibody followed by Western blot with an antibody against HIF‐1α. (D) The interaction between HIF‐1α and SUMO‐1 was determined by immunoprecipitations using anti− HIF‐1α antibody followed by Western blotting with anti‐SUMO‐1 antibody. Western blot with anti− HIF‐1α antibody served as loading control. (E) Western blot was performed as in (A) to evaluate expression of SUMO‐1. GAPDH was used as loading control. (F) Expression of pyruvate dehydrogenase kinase‐1 (PDK‐1) was evaluated by Western blot using antibody against PDK‐1. The representative figure shows increase in PDK‐1 expression in extracts from CLP‐1 +/− mice hearts subjected to stress. GAPDH expression was used as loading control.
Discussion
In this report, we provide several lines of evidence that indicate that CLP‐1 is a potent mediator of cardioprotection. The stress‐induced association of CLP‐1 with the components of the PTEFb complex, the overall decrease in Pol II phosphorylation and the enhanced expression of mitochondrial proteins crucial for the cell survival program implicate CLP‐1 in induction of the cellular response to ischaemic stress. Our results would suggest that the CLP‐1+/− hearts have acquired functional characteristics similar to those of pre‐conditioned heart [30, 31] as revealed by measurements of the haemodynamic parameters. At the molecular level, however, we observed distinct changes in Cdk9 kinase activity, and modulation in the expression of mitochondrial genes, suggesting that down‐regulation of CLP‐1 protein triggers a cardioprotective program with molecular changes that are distinct from that in pre‐conditioning.
We observed that the interaction of CLP‐1 with cyclin T1 inhibits the Cdk kinase activity and consequently the phosphorylation of Pol II in CLP‐1 +/− hearts, consistent with previous findings where overexpression of cyclin T1 in transgenic mice triggered an increase in Cdk9 activity [7]. Phosphorylation of Pol II at the serine 5 and 2 residues occurs sequentially and is known to be associated with the transition from initiation to productive elongation of RNA chain. Serine 5 is phosphorylated by Cdk7 and serine 2 by Cdk9. Serine 5 phosphorylation is linked to capping of the messenger RNA, whereas serine 2 is linked to elongation and polyadenylation. Since both the serine 2 and serine 5 residues were under‐phosphorylated upon application of stress to the CLP‐1+/− hearts, it would appear that CLP‐1 acts on all sequential steps of transcription elongation, and possibly beyond the elongation steps to coordinate transcription.
There is apparently a linkage between the suppression of Cdk9 kinase activity and the expression of genes for mitochondrial function via expression of the master regulator PGC‐1α. Overexpression of PGC‐1α in heart is known to up‐regulate a repertoire of genes important for fatty acid oxidation, TCA cycle and mitochondrial functions and, as such, PGC‐1α is considered to be an important player in coordination of gene expression controlling mitochondrial biogenesis and function. The stress‐induced association of CLP‐1 with cyclin T1 and consequent down‐regulation of Cdk9 kinase activity is likely to stimulate an adaptation response in mitochondrial function. Given the suppression of nuclear transcriptional activity, it is not surprising that the expression of PGC‐1α is provoked to provide the critical energy needs as a contribution to the initial steps in the cardioprotection program.
Mitochondria as an essential source for cellular energy would be critical to cardiac cell survival following ischaemic injury, as such, the cardio‐protection against stress in CLP‐1 +/− hearts might be due to up‐regulation of PGC‐1α as observed in our studies above. Likewise, the transcription factor HIF‐1α that increased in CLP‐1+/− hearts is likely to influence the mitochondrial activity and is known to be pivotal in cell survival [24, 26]. It is known that HIF‐1α regulates hypoxic gene expression changes that are thought to be adaptive for cells exposed to reduce oxygen environment. For instance, HIF‐1α induces PDK‐1, which then facilitates the glycolytic pathway. Expression of genes for glycolysis and pyruvate catabolism is critical during the cellular metabolic adaptation to hypoxia [27, 28]. Enhanced expression of HIF‐1α is thus expected to provide protection against ischaemic injury [32]. Our results on the increase in HIF‐1α in CLP‐1 +/− hearts validate the onset of cell survival due to ischaemic stress and implicate the CLP‐1‐mediated regulation of PGC‐1α and HIF‐1 expression as part of an important network in triggering cardioprotection. We also show that there was a marked decrease in ubiquitination of HIF‐1α possibly due to an interaction of HIF‐1 with SUMO‐1 in the CLP‐1 +/− mice hearts. Consistent with the expected role of HIF‐1α protein in ischaemic stress, we found that the hearts from CLP‐1 +/− mice have high levels of PDK‐1 expression that suggests that pyruvate utilization by the mitochondrial TCA cycle was inhibited, and perhaps the ROS production was decreased.
Collectively, these observations suggest that fine tuning of CLP‐1 protein level is critical to transcriptional regulation in nuclear and mitochondrial genes that lead to important alterations in cellular functions. The role of CLP‐1‐mediated modulation of target gene expression is perhaps dictated by the ability of CLP‐1 to recruit different regulatory partner proteins and co‐factors influenced by the nature of stress and/or signalling components [33]. Indeed, CLP‐1 protein is known to interact with both positive and negative transcription factors in addition to the components of the pTEFb complex [34]. Our data here point to a novel mechanism for cardioprotection against ischaemic stress that conceptually involves specific alterations rendered by the Pol II inhibitor protein CLP‐1 in the nuclear and mitochondrial gene expression program.
Acknowledgement
This work was supported by Grant No. HL073399 from the National Institutes of Health.
References
- 1. Ago T, Sadoshima J. Thioredoxin and ventricular remodeling. J Mol Cell Cardiol. 2006; 41: 762–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ananthakrishnan R, Hallam K, Li Q, et al . JAK‐STAT pathway in cardiac ischaemic stress. Vascul Pharmacol. 2005; 43: 353–6. [DOI] [PubMed] [Google Scholar]
- 3. Willis MS, Patterson C. Into the heart: the emerging role of the ubiquitin‐ proteasome system. J Mol Cell Cardiol. 2006; 41: 567–9. [DOI] [PubMed] [Google Scholar]
- 4. Hall G, Hasday JD, Rogers TB. Regulating the regulator: NF‐kappaB signaling in heart. J Mol Cell Cardiol. 2006; 41: 580–91. [DOI] [PubMed] [Google Scholar]
- 5. Das DK, Maulik N. Conversion of death signal into survival signal by redox signaling. Biochemistry. 2004; 69: 10–7. [DOI] [PubMed] [Google Scholar]
- 6. Sano M, Abdellatif M, Oh H, et al . Activation and function of cyclin T‐Cdk9 positive transcription elongation factor‐b in cardiac muscle‐cell hypertrophy. Nat Med. 2002; 8: 1310–7. [DOI] [PubMed] [Google Scholar]
- 7. Sano M, Schneider, MD . Cyclins that don’t cycle–cyclin T/cyclin‐dependent kinase‐9 determines cardiac muscle cell size. Cell Cycle. 2003; 2: 99–104. [PubMed] [Google Scholar]
- 8. Schneider A, Fischer A, Kruger C, et al . Identification of regulated genes during transient cortical ischemia in mice by restriction‐mediated differential display (RMDD). Brain Res Mol Brain Res. 2004; 124: 20–8. [DOI] [PubMed] [Google Scholar]
- 9. Peterlin BM, Price DH. Controlling the elongation phase of transcription with P‐TEFb. Mol Cell. 2006; 23: 297–305. [DOI] [PubMed] [Google Scholar]
- 10. Marshall RM, Grana X. Mechanisms controlling CDK9 activity. Front Biosci. 2006; 11: 2598–613. [DOI] [PubMed] [Google Scholar]
- 11. Yang Z, Zhu Q, Luo K, et al . The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature. 2001; 414: 317–22. [DOI] [PubMed] [Google Scholar]
- 12. Chen R, Yang Z, Zhou Q. Phosphorylated positive transcription elongation factor b (P‐TEFb) is tagged for inhibition through association with 7SK snRNA. J Biol Chem. 2004; 279: 4153–60. [DOI] [PubMed] [Google Scholar]
- 13. Yik JH, Chen R, Nishimura R, et al . Inhibition of P‐TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol Cell. 2003; 12: 971–82. [DOI] [PubMed] [Google Scholar]
- 14. He N, Pezda AC, Zhou Q. Modulation of a P‐TEFb functional equilibrium for the global control of cell growth and differentiation. Mol Cell Biol. 2006; 26: 7068–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang Z, Yik JH, Chen R, et al . Recruitment of P‐TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005; 19: 535–45. [DOI] [PubMed] [Google Scholar]
- 16. Espinoza‐Derout J, Wagner M, Shahmiri K, et al . Pivotal Role of CLP‐1 in the P‐TEFb complex formation in cardiac hypertrophy. Cardiovascular Res. 2007; 75: 129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sano M, Izumi Y, Helenius K, et al . Menage‐a‐trois 1 is critical for the transcriptional function of PPARgamma coactivator 1. Cell Metab. 2007; 5: 129–42. [DOI] [PubMed] [Google Scholar]
- 18. Sano M, Wang S C, Shirai M, et al . Activation of cardiac Cdk9 represses PGC‐1 and confers a predisposition to heart failure. EMBO J. 2004; 23: 3559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Finck BN, Kelly DP. PGC‐1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006; 116: 615–22.Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Handschin C, Spiegelman BM. Peroxisome proliferator‐activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. 2006; 27: 728–35. [DOI] [PubMed] [Google Scholar]
- 21. Huang F, Wagner M, Siddiqui MAQ. Ablation of the CLP‐1 gene leads to down regulation of the HAND1 gene and abnormality of the left ventricle of the heart and fetal death. Mech Dev. 2004; 121: 559–72. [DOI] [PubMed] [Google Scholar]
- 22. Mascareno E, El‐Shafie, M , Maulik N, et al . Jak/STAT signaling is associated with cardiac dysfunction during ischemia and reperfusion. Circulation. 2001; 104: 325–9. [DOI] [PubMed] [Google Scholar]
- 23. Ratan RR, Siddiq A, Aminova L, et al . Translation of ischaemic preconditioning to the patient: prolyl hydroxylase inhibition and hypoxia inducible factor‐1 as novel targets for stroke therapy. Stroke. 2004; 35: 2687–9. [DOI] [PubMed] [Google Scholar]
- 24. Semenza GL. Hypoxia‐inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol Med. 2001; 7: 345–50. [DOI] [PubMed] [Google Scholar]
- 25. Mansfield KD, Guzy RD, Pan Y, et al . Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF‐alpha activation. Cell Metab. 2005; 1: 393–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chandel NS. Mitochondrial regulation of oxygen sensing. Mitochondrion. 2005; 5: 322–32. [DOI] [PubMed] [Google Scholar]
- 27. Shao R, Zhang FP, Tian F, et al . Increase of SUMO‐1 expression in response to hypoxia: direct interaction with HIF‐1alpha in adult mouse brain and heart in vivo . FEBS Lett. 2004; 569: 293–300. [DOI] [PubMed] [Google Scholar]
- 28. Papandreou I, Cairns RA, Fontana L, et al . HIF‐1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006; 3: 187–97. [DOI] [PubMed] [Google Scholar]
- 29. Kim JW, Tchernyshyov I, Semenza GL, et al . HIF‐1‐mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006; 3: 177–85. [DOI] [PubMed] [Google Scholar]
- 30. Kloner RA, Jennings RB. Consequences of brief ischemia: stunning, preconditioning, and their clinical implications: part 1. Circulation. 2001; 104: 2981–89. [DOI] [PubMed] [Google Scholar]
- 31. Bolli R. Preconditioning: a paradigm shift in the biology of myocardial ischemia. Am J Physiol Heart Circ Physiol . 2007; 292: H19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Simon MC. Mitochondrial reactive oxygen species are required for hypoxic HIF alpha stabilization. Adv Exp Med Biol. 2006; 588: 165–70. [DOI] [PubMed] [Google Scholar]
- 33. Dey A, Chao SH, Lane DP. HEXIM1 and the control of transcription elongation: from cancer and inflammation to AIDS and cardiac hypertrophy. Cell Cycle. 2007; 6: 1856–63. [DOI] [PubMed] [Google Scholar]
- 34. Zhou Q, Yik JH. The Yin and Yang of P‐TEFb regulation: implications for human immunodeficiency virus gene expression and global control of cell growth and differentiation. Microbiol Mol Biol Rev. 2006; 70: 646–59. [DOI] [PMC free article] [PubMed] [Google Scholar]