

Abstract
Background Virus‐inducible reporter genes have been used as the basis of virus detection and quantitation assays for a number of viruses. A strategy for influenza A virus‐induction of a reporter gene was recently described. In this report, we describe the extension of this strategy to influenza B virus, the generation of stable cell lines with influenza A and B virus‐inducible reporter genes, and the use of these cells in various clinically relevant viral assays. Each of the cell lines described herein constitutively express an RNA transcript that contains a reporter gene coding region flanked by viral 5′‐ and 3′‐untranslated regions (UTR) and therefore mimics an influenza virus genomic segment. Upon infection of the cells with influenza virus the virus‐inducible reporter gene segment (VIRGS) is replicated and transcribed by the viral polymerase complex resulting in reporter gene expression.
Findings Reporter gene induction occurs after infection with a number of laboratory strains and clinical isolates of influenza virus including several H5N1 strains. The induction is dose‐dependent and highly specific for influenza A or influenza B viruses.
Conclusions These cell lines provide the basis of simple, rapid, and objective assays that involve virus quantitation such as determination of viral titer, assessment of antiviral susceptibility, and determination of antibody neutralization titer. These cell lines could be very useful for influenza virus researchers and vaccine manufacturers.
Keywords: Influenza virus, neutralizing antisera, reporter cell lines, virus detection
Introduction
Influenza causes considerable morbidity and mortality in the human population worldwide. Annual outbreaks in the United States claim over 36 000 lives and cost billions of dollars. 1 , 2 Antiviral therapy is available, 3 but the emergence of human H1N1 viruses that are resistant to oseltamivir and human H3N2 viruses resistant to amantadine 4 have raised concerns about the use of these antivirals. Vaccination remains the most effective defense against influenza 5 , 6 but the process of vaccine strain selection and antigenic drift of human influenza A virus isolates has led to mismatches between vaccines and circulating virus strains. 7 The threat of another influenza A virus pandemic is a continued concern, particularly because of increasing exposure of humans to avian and swine strains of influenza A virus. 8 , 9 , 10
Accurate, specific, and rapid detection and quantification of influenza virus are critical for successful development of diagnostic tests, antiviral drugs and vaccines. Assays that are based on virus‐induced cytopathic effects such as the plaque forming unit (pfu) and the tissue culture infectious dose (TCID50) are still commonly used to quantify influenza virus, but such assays are time‐consuming and labor‐intensive. 11 Antibody neutralization assays which are used for studies of vaccine efficacy include the hemagglutination inhibition assay and the microneutralization assay, share similar limitations. 12 , 13 Considerable effort has been devoted to improve methods of cell‐based antiviral influenza drug screening using a colorimetric or fluorometric readout that is based on inhibition of virus‐induced cytotoxicity. In one such method a cell viability assay, which measures ATP via a luciferase luminescence system, was shown to give comparable results to the use of a neutral red assay. 14
Cell lines containing a virus inducible reporter gene have been used successfully to detect retroviruses such as Human Immunodeficiency virus (HIV), 15 , 16 , 17 DNA viruses such as Herpes Simplex Virus (HSV) 18 , 19 , 20 , 21 , 22 and RNA viruses such as respiratory syncytial virus (RSV). 23 Recently, a detection system for influenza A virus was developed based on a virus‐inducible reporter gene segment (VIRGS). 24 These preliminary studies with transiently transfected cells demonstrated the potential of this approach to detect and quantify influenza A virus using a luciferase or green fluorescent protein (GFP) reporter gene system. These findings prompted us to generate permanent cell lines using linearized plasmid DNA integrated into the host cell chromosome. In this study, we demonstrate the utility of these cell lines in objective, rapid, and quantitative assays for virus quantification, virus neutralization assays, and drug screening assays. We have applied the acronym enzyme‐linked virus inhibitor reporter assay (ELVIRA) for this technology and our cell lines as ELVIRA® Flu A or Flu B (Diagnostic Hybrids, Inc., Athens, OH, USA).
Materials and methods
Plasmid construction
Plasmids that contain virus‐inducible reporter genes were constructed as described in Lutz et al. 24 Briefly, the 5′‐and 3′‐UTRs of the influenza A virus (A/WSN/33 H1N1) NP genomic segment were inserted between the human polymerase I promoter and the terminator elements in plasmid pHH21 25 using the following two partially complementary oligonucleotides that contain the 5′‐ (red) and 3′‐ (blue) untranslated regions of the NP segment:
-
1
5′‐TATTAGTAGAAACAAGGGTATTTTTCTGTTTAAACTTTACCGATGTCACTCAGTGAGTGATTATCTACCCTGTTTCTAC‐3′
-
2
5′‐GGGAGTAGAAACAGGGTAGATAATCACTCACTGAGTGACATCGGTAAAGTTTAAACAGAAAAATACCCTTGTTTCTACT‐3′
The oligonucleotides were designed so that after annealing there would be a 3 or 4‐base 5′‐overhang on each end to allow ligation to the Bsm BI site in pHH21 and a Pme I restriction site between the two UTR elements for insertion of the reporter genes. The firefly luciferase (pGL‐2‐Basic; Promega, Madison, WI, USA) or GFP (pEGFP‐1; Clontech, Mountain View, CA, USA) reporter genes were inserted into the Pme I site of the resultant pHH21/UTR plasmid. The Pol I promoter/UTR/reporter gene cassette was removed from the above vector and inserted into the pRep4 vector. Finally, the EBV origin of replication and EBNA‐1 gene were deleted from the plasmid with restriction endonucleases Ssp I and Spe I to generate plasmids pHH21influenza A virus/Luc‐Hyg and pHH21influenza A virus/GFP‐Hyg. A similar strategy was used for the influenza B virus UTRs from the NP segment of the B/Yamanashi strain. Four oligomers were used and linked to make a 174 base pair oligonucleotide shown below as two partially complementary oligonucleotides:
-
1
5′‐GGGAGCAGAAGCACAGCATTTTCTTGTGAACTTCAAGTACCAATAAAAAGAACTGAAAAATCAAAGTTTAAACAGCAACAAAATAGACACTATGGCTGTGATTGTTTCAATACGTTTGGAATGTGGGTGTTTACTCTTATTAAAATAAATATAAAAAATGCTGTTGTTTCTACT‐3′
-
2
5′‐TATTAGTAGAAACAACAGCATTTTTTATATTTATTTTAATAAGAGTAAACACCCACATTCCAAACGTATTGAAACAATCACAGCCATAGTGTCTATTTTGTTGCTGTTTAAACTTTGATTTTTCAGTTCTTTTTATTGGTACTTGAAGTTCACAAGAAAATGCTGTGCTTCTGCT‐3′
Red‐colored letters are B/Yamanashi NP 5′‐UTR (62 bases). Blue color letters are B/Yamanashi NP 3′‐UTR (101 bases). As with the influenza A virus cloning strategy after annealing the duplex oligonucleotide has a 3 or 4‐base 5′‐overhang on each end to allow ligation to the Bsm BI site in pHH21 and a Pme I restriction site between the two UTR elements for insertion of the reporter genes renilla luciferase (phRL‐TK; Promega) and GFP to generate plasmids pHH21influenza B virus/Luc‐Hyg, and pHH21influenza B virus/GFP‐Hyg.
Cells
Human kidney 293T cells (American Type Culture Center, Manassas, VA, USA) were grown in Dulbecco’s modified Eagle’s medium containing 4% or 10% fetal bovine serum, 2 mm of glutamine and 50 μg/ml of gentamcin at 37°C with 5% CO2. Madin‐Darby Canine kidney (MDCK) cells (ATTC) were grown in the same medium as the 293T cells.
Transfection and selection of the ELVIRA® Flu A and Flu B cell lines
293T cells, plated in two wells of a 24‐well plate, were transfected with linearized pHH21influenza A virus/Luc‐Hyg, pHH21influenza B virus/Luc‐Hyg, pHH21influenza A virus/GFP‐Hyg and pHH21influenza B virus/GFP‐Hyg using Hyfect (Denville Scientific Inc., Metuchen, NJ, USA) according to the manufacturer’s instructions. After incubation for 24 hours at 37°C the cells were trypsinized and serially diluted into 96‐well plates. The transfected cells were cultured in media containing 150 μg/ml hygromycin B (Invitrogen Corporation Carlsbad, CA, USA). After colonies were observed, they were transferred into a single well of a 24‐well plate. When the monolayer was confluent, the cells of each colony were passed into four wells in two separate 24‐well plates. One plate was maintained in the culture medium and incubated at 37°C. One well from each colony of the other plate was either infected with influenza A virus or influenza B virus or used as an uninfected control. Twenty‐four hours after virus infection, luciferase activity or GFP fluorescence was measured. The cells from colonies with a high infected‐to‐uninfected ratio of luciferase or GFP activity were selected for further subcloning. Selected clones were plated in 96‐well plates at an average 0·5 cell/well and cultured in medium containing 150 μg/ml hygromycin B to obtain isolated clonal cell lines.
Virus strains and virus infection
Virus strains including influenza A and B and other respiratory viruses were obtained from ATCC; CDC, Atlanta, GA, USA; Ohio Department of Health, Alliance, OH, USA; Seattle & King County Public Health Laboratory, Seattle, WA, USA; Iowa Methodist Hospital Des Moines, Iowa; and Riley Hospital for Children, Indianapolis, Indiana. The H5N1 strains were supplied by Battelle and experiments with these strains was performed in their biosafety level 3 facility in Columbus, OH, USA.
Nearly, confluent monolayers of ELVIRA® Flu A or Flu B cells were infected with influenza A or B or other respiratory virus with the indicated multiplicity‐of‐infection (MOI). Virus infections were performed in the presence of Opti‐MEM medium containing 0·5 μg/ml trypsin. Following virus inoculation, the cells were centrifuged for 1 hour at 700g and incubated in 37°C with 5% CO2 for 20–24 hours. The amount of the virus inocula were confirmed by simultaneous infection of R‐mix monolayers (Diagnostic Hybrids Inc.) which contain Mv1Lu and A549 cells or by infection of the ELVIRA® Flu A‐luc cell line followed by immunostaining with anti‐influenza antibodies (Diagnostic Hybrids Inc.) after 20–24 hours.
Luciferase assays
Virus‐infected ELVIRA® Flu A‐luc cells were lysed and analyzed for firefly luciferase activity using the Bright‐Glo Luciferase Assay System (Promega). ELVIRA® Flu B‐Rluc cells were lysed and analyzed for activity using the Renilla Luciferase Assay System (Promega). A Veritas Luminometer (Turner BioSystems, Sunnyvale, CA, USA) was used to measure relative light units (RLU) according to the manufacturers’ instructions.
GFP time course study
The ELVIRA® Flu A‐GFP and Flu B‐GFP cell lines were assessed for reporter gene expression by visual examination of the monolayer at 6, 19, 48, and 72 hours post‐infection under a Nikon Eclipse TS100 fluorescent light microscope at 100× magnification (Nikon, Melville, NY, USA). The light source for the microscope was a Mercury‐100W (Chiu Technical Corporation, Kings Park, NY, USA) and the image was captured with QCapture 2·70·0 software (Quantitative Imaging Corporation, Surrey, BC, Canada).
Neutralization assay
Influenza A/WSN/33 virus (5000 infectious units) or A/Wisconsin/56/2005 (500 infectious units/well) were incubated for 2 hours at 37°C with twofold serially diluted mouse anti‐A/WSN/33 26 and human sera, respectively. All the sera samples were heated at 56°C for 30 minutes prior to the incubation with virus. The virus‐serum mixture was then added to monolayers of ELVIRA® Flu A‐luc cells grown in 96‐well plates. Cells were lysed and the cell lysates were analyzed for luciferase activity 24 hours post‐infection. The per cent reduction of luciferase activity, measured as RLU of wells with serum‐treated virus compared with the virus‐only controls, was calculated. The reciprocal of the dilution that resulted in a reduction of 99% or greater was recorded as the neutralization titer.
Hemagglutination Inhibition assay
Influenza A/Wisconsin/56/2005 (4 HA units = 115,000 infectious units/well) was incubated with twofold serially diluted human sera for 1–2 hours at 37°C in 96‐well plates. Then 1% guinea pig red blood cells (RBCs) (Colorado Serum Co., Denver, CO, USA) were added to the virus‐serum solution and mixed. The plates were read after 30 minutes incubation at room temperature.
Determination of virus titer
ELVIRA® Flu A‐luc and MDCK cells were grown in 96‐well plates to 90–100% confluence. The cells were washed once with serum‐free medium or phosphate‐buffered saline. Nine serial 10‐fold dilutions of influenza A virus were made with serum free medium. Dilutions 10−4 through 10−9 were used to inoculate 10 wells of each cell type. The plates were centrifuged for 1 hour at 700g after infection and then incubated at 37°C with 5% CO2. ELVIRA® Flu A‐luc cell lysates were analyzed for luciferase activity 24–48 hours after infection. MDCK cells were observed for cytopathic effects (CPE) 4–6 days after infection. ELVIRA® Flu A‐luc wells were considered positive for viral infection if their RLU value was >3 times the mean background RLU value. The 50% tissue culture infectious dose (TCID50) was determined by the Reed & Muench method. 27
Measurement of antiviral activity using ELVIRA® Flu A cells
The ELVIRA® Flu A‐luc cells were used to assess the inhibition of influenza A by various concentrations of amantadine (Sigma‐Aldrich, St Louis, MO, USA) or bafilomycin. The cells were plated in 96‐well plates as described above and incubated with serial dilutions of the drugs for 15 minutes (amantadine) or 30 minutes (bafilomycin) at 37°C. Influenza A virus (10 000 infectious units) was added to wells containing amantadine (A/ODH 154 infected cells) or bafilomycin (A/Wisconsin56/2005 infected cells). Plates were centrifuged at 700g for 1 hour and then incubated at 37°C with 5% CO2 for 24 hours followed by measurement of luciferase activity. The concentration of drug that reduced the RLU by 50% compared with the no‐drug control was calculated and reported as the IC50 value.
Results
Detection of influenza A and B viruses using ELVIRA® cells
Stable human kidney 293T cell lines that constitutively express a negative‐sense RNA encoding the firefly luciferase gene (luc) or the GFP gene flanked by the untranslated regions (UTR) of the NP segment of influenza A virus (A/WSN/33) UTRs or influenza B virus (B/Yamanashi) were generated as described in the Materials and methods section. The virus‐inducible reporter gene in these cells is expressed under the control of the human RNA polymerase I promoter/terminator sequences. 28 , 29 The human polymerase I promoter is species‐specific which required that the parental cell line be of human origin. These cell lines have been cultured for 50 passages with no appreciable loss of inducible reporter gene expression.
Virus‐inducible reporter gene expression from the ELVIRA® Flu A‐luc and ELVIRA® Flu B‐Rluc cell lines was assessed after infection with influenza A and B viruses respectively at high and low MOI. At various times after infection cells were lysed and luciferase activity was measured (Figure 1). The graphic display of the data reveals classic single and multistep viral growth kinetics and a time course very similar to infectious virus production on these cells. These results reveal that measurement of luciferase activity provides a valid surrogate marker of virus replication.
Figure 1.
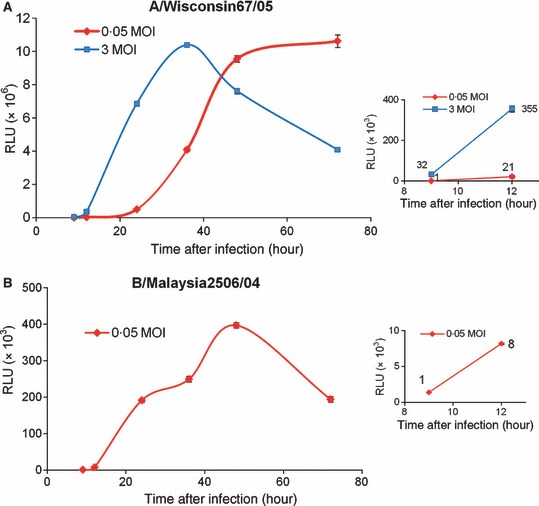
Time course of luciferase expression of ELVIRA® Flu A‐luc or Flu B‐Rluc cells following infection with influenza viruses. ELVIRA® Flu A‐luc or Flu B‐Rluc cells were infected with influenza A/Wisconsin67/05 or influenza B/Malaysia2506/04 at the indicated MOI. Luciferase activity was measured at various time points after virus infection and expressed as RLU/well. Results shown are mean of 3 replicates. (A) Luciferase activity of ELVIRA® Flu A‐luc cells infected with influenza A/Wisconsin67/05. The insert shows data from the early time points. (B) Luciferase activity of ELVIRA® Flu B‐Rluc cells infected with influenza B Malaysia2506/04.
A virus dose–response was performed by infecting the ELVIRA® Flu A‐luc and ELVIRA® Flu B‐Rluc cell lines with various amounts of influenza A and B viruses and measuring luciferase activity after 24 hours (Figure 2). For both cell lines there was a linear dose–response curve with a wide dynamic range. This result suggested that this methodology can be used to accurately titer virus stocks. We next determined the titer of stocks of several influenza A and B virus strains using the ELVIRA® Flu A‐luc and ELVIRA® Flu B‐Rluc cells and determined the titers at the same time with MDCK cells using a TCID50 method. We included in this analysis four strains of highly pathogenic avian (H5N1) influenza A virus which all were able to induce luciferase in the ELVIRA® Flu A‐luc cells (data not shown). As shown in Table 1 the titers generated with both cell lines were very similar.
Figure 2.
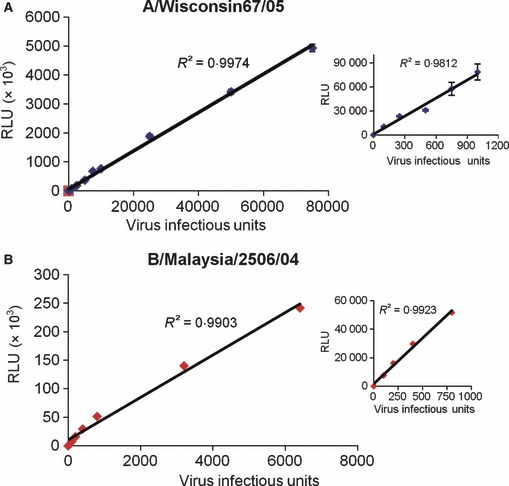
Dose–response of luciferase expression of ELVIRA® Flu A‐luc or ELVIRA® Flu B‐Rluc detecting cell line following infection with either Influenza A or B virus. Cell lines were infected with their corresponding virus and analyzed for luciferase activity 24 hours after infection (RLU/well). Results are the average of triplicates ± SD. (A) Luciferase activity of ELVIRA® Flu A‐luc cell line after infection with 12 different amounts of Influenza A/Wisconsin67/05 from 100–75 000 infectious units. (B) Luciferase activity of ELVIRA® Flu B‐Rluc cell line infected with seven different concentrations of influenza B/Malaysia/2506/04 ranging from 100 to 7500 infectious units.
Table 1.
Comparison of TCID50 titers of influenza A and B virus on ELVIRA Flu A‐luc and Flu B‐Rluc cells and MDCK cells
| Virus strain | Method* | |
|---|---|---|
| ELVIRA Flu A Luc | MDCK | |
| A | ||
| A/Brisbane/59/2007 (H1N1) | 1·08E + 07 | 1·26E + 07 |
| A/Solomon island/03/2006 (H1N1) | 1·58E + 07 | 1·99E + 07 |
| A/Wisconsin/56/2005 (H3N2) | 1·80E + 07 | 1·19E + 07 |
| A/Mal/302/54 (H1N1) | 2·59E + 07 | 8·89E + 06 |
| A/Whooperswan/Mongolia/ 244/05 (H5N1) | 1·78E + 08 | 1·2E + 08 |
| A/Hong Kong/483/97 (H5N1) | 5·01E + 07 | 2·51E + 06 |
| A/Hong Kong/486/97 (H5N1) | 2·09E + 08 | 1·17E + 07 |
| A/Vietnam/1203/04 (H5N1) | 2·57E + 07 | 3·31E + 07 |
| B | ||
| B/Malaysia2506/04 | 5·00E + 07 | 2·32 E + 07 |
| B/GL/1739/54 | 5·00E + 05 | 1·25 E + 06 |
| B/Florida04/2006 | 5·00E + 06 | 2·59 E + 06 |
*Ten‐fold serial dilutions of influenza A or B virus stocks were made in serum free medium. ELVIRA Flu A and MDCK cells were planted in 96‐well plates and then infected with the 10−4 through 10−9 dilutions (10 wells/dilution). After infection, the cells were incubated at 37°C with 5% CO2 for 24 or 48 hours (ELVIRA cells) or 4 days (MDCK cells). ELVIRA Flu A luc and ELVIRA Flu B Rluc wells with a RLU reading of >3 times the background mean were considered positive for influenza infection. MDCK cells were inspected microscopically for cytopathic effects. TCID50 was calculated by the Reed & Muench method.
In addition to cell lines that express virus‐inducible luciferase, we generated cell lines that express GFP following influenza virus infection using the same strategy. Figure 3 shows the absence of GFP expression in mock‐infected cells and a high level of expression in the cytoplasm of influenza A virus‐infected ELVIRA® Flu A‐GFP cells and influenza B virus‐infected Flu B‐GFP cells. No cross‐induction was observed. These cells could be useful in methods for which there is an advantage to using either live or intact cells (i.e., cells that are not lysed).
Figure 3.
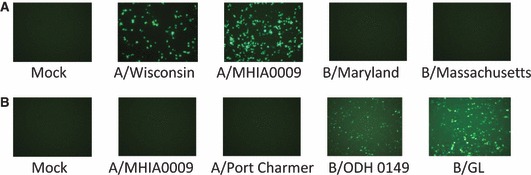
Photomicrographs (100×) showing GFP expression from ELVIRA® Flu‐GFP cell lines infected with influenza A or B viruses. GFP‐expressing cells were detected by fluorescent microscopy at 24 or 48 hours after infection. (A) The ELVIRA® Flu A‐GFP cell line was infected with influenza A virus strains A/Wisconsin/67/05 and A/MHIA0009, and influenza B virus strains B/Maryland/1/59 (ATCC VR‐296) and B/Massachusetts (ATCC VR‐523) at 1 MOI. Photomicrographs were taken at 24 hours after infection. (B) The ELVIRA® Flu B‐GFP cell line was infected with influenza B virus B/ODH0149 and B/GL/1739/54 at 0·5 MOI and influenza A virus influenza A virus A/MIHA0009 and A/PortChalmers/1/73(H3N2) at 1 MOI. Photomicrographs were taken at 48 hours after infection.
Specificity of virus‐induced luciferase expression from ELVIRA® Flu A‐luc cells
We tested a panel of influenza A virus and influenza B virus isolates including H3N2 and H1N1 types on the ELVIRA® Flu A‐luc cells. All influenza A virus isolates induced high levels of luciferase with signal‐to‐noise ratios ranging from 20 to over 1000 (Table 2). Influenza A virus triggered luciferase in the ELVIRA® Flu B‐Rluc cells, but at a low level. One isolate of influenza B virus (B/Taiwan) induced a low level of luciferase, but two others did not induce activity in the ELVIRA® Flu A‐luc cells above background (Table 2). Both cell lines were also infected with a panel of unrelated respiratory viruses at a high MOI. Twenty‐four hours after infection, luciferase expression was essentially the same as background and the signal‐to‐noise was never higher than two (Table 2).
Table 2.
Specificity of ELVIRA® Flu A* (A) or Flu B** (B) cell lines following infection with influenza A, influenza B, or unrelated respiratory viruses
| Virus | Strain/sample | Source | Ave RLU (n = 3) | STDEV | S/B |
|---|---|---|---|---|---|
| A | |||||
| Parainfluenza 1 | Clinical isolate | Health alliance | 195 | 10 | 1·3 |
| Parainfluenza 2 | Clinical isolate | Health alliance | 280 | 6 | 1·9 |
| Parainfluenza 3 | Clinical isolate | Health alliance | 222 | 15 | 1·3 |
| hMPV*** | Clinical isolate (A1) | Italy | 135 | 25 | 0·8 |
| hMPV | Clinical isolate (A2) | Italy | 217 | 8 | 1·3 |
| hMPV | Clinical isolates (B1) | Italy | 126 | 8 | 0·7 |
| Parainfluenza 3 | C‐243 | ATCC VR‐93 | 141 | 20 | 0·9 |
| Parainfluenza 1 | C‐35 | ATCC VR‐94 | 182 | 20 | 1·2 |
| Parainfluenza 4b | CH 19503 | ATCC VR‐1377 | 166 | 12 | 1·1 |
| Parainfluenza 2 | Greer | ATCC VR‐92 | 193 | 11 | 1·3 |
| RSV† | Long | ATCC VR‐26 | 151 | 16 | 0·5 |
| Parainfluenza 4a | M25 | ATCC VR‐1378 | 143 | 17 | 1 |
| Adenovirus | Clinical isolate (MHIA‐0021) | Methodist Hospital | 160 | 18 | 1·1 |
| Adenovirus | Type 5 | ATCC VR‐5 | 114 | 13 | 0·8 |
| RSV | WC‐192 | West Chester | 134 | 10 | 0·5 |
| Coronavirus | 229E | ATCC VR‐740 | 147 | 38 | 1·1 |
| Coronavirus | OC34 | ATCC VR‐1558 | 139 | 5 | 1 |
| Influenza B | B/Massachusetts | ATCC VR‐523 | 154 | 10 | 0·5 |
| Influenza B | Clinical isolate B/MHIA‐0023 | Methodist Hospital | 160 | 18 | 1·1 |
| Influenza B | B/Taiwan | ATCC VR‐295 | 5247 | 222 | 16·7 |
| Influenza A | A/ODH 0023 | Ohio Department of Health | 455481 | 26225 | 1098 |
| Influenza A | A/Hong Kong/8/68 (H3N2) | ATCC VR‐544 | 541988 | 18149 | 240 |
| Influenza A | A/MaI/302/54 (H1N1) | ATCC VR‐98 | 922701 | 17436 | 409 |
| B | |||||
| Adenovirus | Type 5 | ATCC VR‐5 | 27 | 2 | 2·1 |
| Parainfluenza I | C‐35 | ATCC VR‐94 | 10 | 1 | 0·8 |
| Parainfluenza II | Greer | ATCC VR‐92 | 12 | 1 | 0·9 |
| RSV | Long | ATCC VR‐26 | 22 | 2 | 1·7 |
| Influenza A | A/Denver/1/57 (H1N1) | ATCC VR‐546 | 38 | 7 | 2 |
| Influenza A | A/PortChalmers/1/73(H3N2) | ATCC VR‐810 | 1004 | 27 | 41 |
| Influenza A | A/WS/33 (H1N1) | ATCC VR‐1520 | 270 | 74 | 11 |
| Influenza B | Clinical isolate B/ODH0149 | Ohio Department of Health | 245825 | 2140 | 18910 |
| Influenza B | B/Malaysia2506/04 | CDC | 134920 | 6839 | 9637 |
| Influenza B | B/GL/1739/54 | ATCC VR‐823 | 151492 | 8412 | 10821 |
*ELVIRA® Flu A‐luc cells were infected with seventeen non‐influenza respiratory viruses and three strains of influenza B viruses (MOI = 1) and three strains of influenza A (MOI = 0·1) were used as a positive control. The cells were assayed for luciferase activity 24 hours post‐infection.
**ELVIRA® Flu B‐Rluc cells were infected with four non‐influenza respiratory viruses and three strains of influenza A viruses (MOI = 1) and three strains of influenza B virus (MOI = 0·5). The cells were analyzed for luciferase activity 24 hours post‐infection.
***Human metapneumovirus.
†Respiratory syncytial virus.
Measurement of neutralizing antibody using ELVIRA® cells
The results of Figure 2 show that luciferase activity is proportional to the virus inoculum and suggest that this system could be applied to any influenza assay that depends on a quantitative assessment of virus infectivity. We therefore tested the usefulness of this system to titer neutralizing serum. Dilutions of immune or non‐immune mouse sera were added to a fixed amount of influenza virus. As shown in Figure 4 immune serum reduced luciferase activity in a dose‐dependent manner to nearly background levels. We recorded the titer as the reciprocal of the lowest dilution of serum that resulted in at least a 99% reduction in luciferase compared with the virus‐only control. We then compared the use of this assay to a hemagglutination inhibition assay to assess neutralization titer of nine human sera against influenza A virus (A/Wisconsin/56/2005) and five sera against influenza B virus (B/Malaysia2506/04). As shown in Table 3, the titers were comparable, although not identical which is not uncommon when comparing microneutralization to HI. 30
Figure 4.
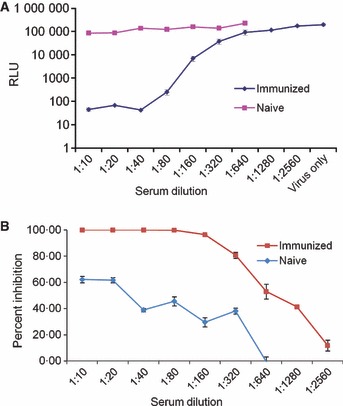
The effect of neutralizing antibody on influenza A virus‐induced luciferase activity of ELVIRA® Flu A‐luc cells. Naive and immunized mouse sera were heat‐treated at 56°C for 30 minutes and then serial diluted from 1:10 to 1:640 (non‐immune serum) and 1:10 to 1:2560 (immune serum). Influenza A virus (WS/33; H1N1) (5000 infectious units) was incubated 1:1 (vol/vol) with each dilution of the sera for 60 minutes at room temperature. The virus was then used to infect ELVIRA® Flu A‐luc cells and luciferase activity was measured after 24 hours. (A) Data expressed as total RLU per well. (B) Data expressed as a percentage of luciferase activity in virus‐only wells.
Table 3.
Comparison of antibody serum neutralization titers against influenza A virus (A) and influenza B virus (B) obtained using ELVIRA® Flu A‐luc and Flu B‐Rluc cells and Hemagglutination inhibition
| A. | |||||||||
| Serum | Neg | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| ELVIRA Flu A* | <10 | 1280 | 160 | 1280 | 80 | 640 | 1280 | 1280 | 1280 |
| HI** | <10 | 1280 | 40 | 1280 | 20 | 320 | 320 | 320 | 1280 |
| B. | |||||
| Serum | 1 | 5 | 8 | 9 | 10 |
| ELVIRA Flu B*** | <10 | 40 | 80 | 80 | 40 |
| HI† | <10 | 80 | 160 | 80 | 80 |
*ELVIRA Flu A‐luc cells were infected with A/Wisconsin/56/2005 (500 infectious units/well) that had been incubated for 2 hours with dilutions (1:10 followed by twofold serial dilutions) of human serum samples from eight individuals with an unknown vaccination history and one sample that was negative (Neg). Twenty‐four hours after incubation, the ELVIRA cells were lysed and the luciferase assay was performed. Dilutions with greater than 99% inhibition of luciferase activity relative to virus‐only wells were considered neutralized.
**For the HI assay 1 15 000 infectious units/well were incubated with the same dilutions of the serum samples and 1% guinea pig RBCs was added to the serum/virus mixture. The results were evaluated after 30 minutes of incubation at room temperature by comparing the formation of hemagglutination in serum + virus samples with that in control wells that contained RBCs only.
***ELVIRA Flu B Rluc cells were infected with B/Malaysia2506/04 (500 infectious units/well) that had been incubated for 2 hours with dilutions (1:10 followed by twofold serial dilutions) of five human serum samples from individuals with an unknown vaccination history.
†For the HI assay 60 300 infectious units/well were incubated were incubated with the same dilutions of the serum samples and 1% guinea pig RBCs added to the serum/virus mixture. The results were evaluated after 30 minutes of incubation at room temperature by comparing the formation of hemagglutination in serum + virus samples with that in control wells that contained RBCs only.
Antiviral susceptibility testing of influenza A and B viruses using ELVIRA® cells
The applicability of this methodology for antiviral susceptibility testing was also evaluated. Figure 5 shows the effect of various concentrations of two known anti‐influenza compounds on virus‐induced luciferase expression. The graphic display of the results shows classic sigmoidal curves and the IC50 determinations calculated using this method were similar to results reported in the literature for amantadine‐sensitive influenza strains. 31 Influenza B virus is resistant to amantadine and this is reflected in the lack of reduction of luciferase expression following amantadine treatment of infected ELVIRA® Flu B‐Rluc cells (Figure 5D).
Figure 5.
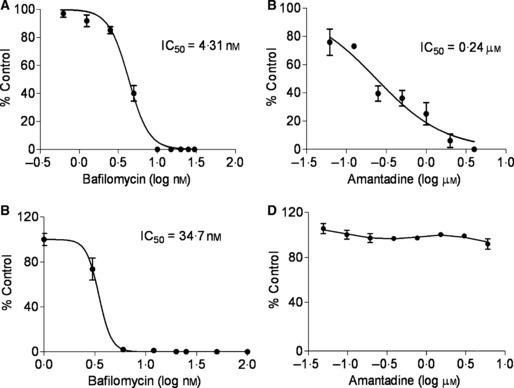
Antiviral susceptibility testing using ELVIRA Flu A‐luc and ELVIRA Flu B‐Rluc cells Bafilomycin (0·78–30 nm) and amantadine (4 –62 μm) were added to ELVIRA® Flu A‐luc cells and incubated at 37°C for 30 minutes and then the cells were infected with influenza A or B virus. Luciferase activity was measured after 24 hours. Data expressed as per cent no‐drug control. (A) Bafilomycin inhibition curve of influenza A virus (A/MHIA0009) ELVIRA® Flu A‐luc cells. (B) Amantadine inhibition curve of influenza A virus (A/MHIA0009) on ELVIRA® Flu A‐luc cells. (C) Bafilomycin inhibition of influenza B virus (B/Malaysia/2506/04) on ELVIRA® Flu B‐Rluc cells. (D) Amantadine inhibition of influenza B virus (B/Malaysia/2506/04) on ELVIRA® Flu B‐Rluc cells.
Discussion
Accurate, specific, and rapid identification and quantification of influenza virus is an important component of influenza research, antiviral drug discovery, and vaccine development. New technologies that promote these goals will result in the development of better drugs and vaccines to control seasonal influenza and hopefully prevent or lessen the impact of a pandemic. In this study, we describe the successful establishment of stable cell lines engineered to contain influenza virus‐inducible reporter genes. These cells form the basis of rapid and objective assays that can be used to measure the titer of infectious virus, the titer of neutralizing antibody, and the susceptibility of influenza virus strains to antiviral drugs.
The concept of VIRG was described a number of years ago 17 , 20 , 32 and was first successfully applied to influenza viruses by Lutz et al. 24 In this proof‐of‐concept report influenza virus‐dependent reporter gene expression was demonstrated using an RNA segment that was transcribed from a human polymerase I promoter and contained a reporter gene flanked by untranslated regions (UTRs) from the influenza nucleoprotein (NP) RNA segment. Unfortunately, the transient and the episomal expression systems used in this previous work have drawbacks when attempting to apply this technology to robust and reliable assays that could be used for industrial or clinical applications. In this report, we generated stably transformed cell lines and demonstrated that these cell lines can be maintained for many passages without loss of their phenotype with respect to virus‐induction of the reporter gene. We have also extended the technology to influenza B virus which is a component of most influenza vaccines and which contributes to significant morbidity and mortality during many influenza seasons.
We have shown that a number of isolates of influenza A and B viruses induce a high signal‐to‐noise on their respective cell line. A linear dose–response relationship between reporter enzyme activity and virus inoculum was observed. This observation allows the use of reporter enzyme levels as a surrogate marker for virus amount in assays that depend on knowing how much virus is in a sample. Of course, a standard curve must be generated for each strain, but for many applications the strain of virus is known and this approach will be easily applied. For detection of unknown strains an empiric assessment of the ability of the virus to induce reporter enzyme levels will have to be performed.
We assessed the specificity of reporter enzyme induction using a panel of respiratory viruses. None of the respiratory viruses we inoculated onto the cell lines induced significant luciferase activity above background levels. There was some level of cross‐induction between influenza A and B viruses, but the induction by the homologous virus was many‐fold higher than by the heterologous virus. In a recent report, it was shown that activity for the type B polymerase machinery with a type A promoter depends on the specific RNA segment and that the levels of activity differ between type B strains. 33 Using plasmid transfection they observed that type B polymerase machinery that is composed of homologous subunits is functional with a type A promoter albeit to a variable extent depending upon the segments from which the regions downstream of the promoter sequence were derived. These authors did not assess delivery of the polymerase genes by virus infection which may explain the greater specificity we observed.
Titration of influenza virus is often performed on MDCK cells by endpoint dilution, observation of CPE and calculation of the TCID50. We determined the titer of stocks of a number of strains of influenza A virus including five H5N1 strains by using the TCID50 method and by using either ELVIRA® Flu A‐luc cells with luciferase as the readout or MDCK cells with CPE as the readout. The titers determined with the two methods were very similar despite the fact that there may be differences in susceptibility of the two cell lines to the virus strains tested. More strains need to be tested to support the use of the ELVIRA® Flu A‐luc cells for this purpose, but there are clear advantages in terms reduced time to result (e.g., 1–2 days versus 4–6 days), greater objectivity, and less labor.
Titration of neutralizing antibody is an assay that can potentially benefit from use of these cell lines. It is clear that the induction of reporter enzyme depends on infectious virus. Using immunized mouse serum we demonstrated that the neutralizing activity of antiserum can be identified as a function of the reduction in luciferase activity and that titers can be calculated based on a 99% inhibition. The HI assay is currently the gold standard for measurement of immunity following vaccination. Comparison of neutralization titers between the two methodologies showed a reasonable correlation though it is not surprising that the titers were not identical given that each assay measures neutralization in a very different manner. The assay described in this report offers a number of advantages over other virus neutralization assays that are based on various endpoints. 34 , 35
Finally, we applied this technology to an assessment of antiviral susceptibility. We observed excellent dose–response when measuring luciferase activity versus drug concentration for two antiviral compounds that have a mechanism of action that acts after virus entry. 31 Calculated IC50 values were very similar to published reports using the amantadine‐sensitive virus strains. We have not used this method for neuraminidase inhibitors. However, the results suggest that the ELVIRA® Flu A‐luc cells could be used for screening of novel antiviral agents and for quantifying antiviral potency. Identification of new antiviral lead compounds depends on robust primary assays for high‐throughput screening of large libraries of compounds. Methods have also been developed for rapid evaluation of compounds for antiviral activity in 96‐well microplates, which include visual quantitation of antiviral activity based upon inhibition of virus‐induced CPE or by less subjective colorimetric or fluorometric means. 36 A cell‐based screen for potential influenza antivirals that measures the CPE induced by influenza virus (A/Udorn/72, H3N2) infection in MDCK cells using the luminescent‐based CellTiter Glo system was recently described. 14 Recently, a viral inhibition assay based on neuraminidase (NA) activity was described and shown to be suitable for high throughput screening to identify potential antiviral compounds or to identify drug‐resistant influenza strains. 37 , 38 Fluorometric and chemiluminescence NA assays have also been described. 39 , 40 The application of the technology described in this report may also be used for primary antiviral screening. One advantage is that the readout is a direct function of viral RNA replication and transcription.
In summary, we have engineered stable cell lines that contain an influenza A virus or influenza B virus‐inducible reporter gene that can be used in assays to objectively measure infectious virus titer, antibody neutralization titer, and antiviral drug potency. The cell lines have been maintained for more than 50 passages without loss of inducible reporter enzyme activity. Although further studies are needed to verify the accuracy and precision of this methodology, there is great potential for these assays to replace traditional techniques.
Acknowledgements
We thank the staff of Diagnostic Hybrids, Inc. for providing seasonal influenza virus strains. We gratefully acknowledge the expert technical assistance provided by Melicia Gainey and Ann Wasko of Battelle in Columbus, OH who performed the titer assays on the H5N1 influenza strains. We thank Drs Yung Huang, Dave Scholl, Miguel Quiñones‐Mateu for review of the manuscript and helpful suggestions.
This work was supported by NIAID R44AI060312 to PDO and R01AI053629 to AP.
References
- 1. Rothberg MB, Haessler SD, Brown RB. Complications of viral influenza. Am J Med 2008; 121:258–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Molinari NA, Ortega‐Sanchez IR, Messonnier ML et al. The annual impacrt of seasonal influenza in the US: measuring disease burden and costs. Vaccine 2007; 25:5086–5096. [DOI] [PubMed] [Google Scholar]
- 3. Lipsitch M, Cohen T, Murray M, Levin BR. Antiviral resistance and the control of pandemic influenza. PLoS Medicine 2007; 4:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Update: influenza activity ‐ United States, September 28, 2008 – January 31, 2009. MMWR 2009; 58:115–119. [PubMed] [Google Scholar]
- 5. Kreijtz JH, Osterhaus AD, Rimmelzwaan GF. Vaccination strategies and vaccine formulations for epidemic and pandemic influenza control. Hum Vaccin 2009; 5(3):126–135. [DOI] [PubMed] [Google Scholar]
- 6. Simonsen L, Reichert TA, Viboud C, Blackwelder WC, Taylor RJ, Miller MA. Impact of influenza vaccination on seasonal mortality in the US elderly population. Arch Intern Med 2005; 165:265–272. [DOI] [PubMed] [Google Scholar]
- 7. Carrat F, Flahault A. Influenza vaccine: the challenge of antigenic drift. Vaccine 2007; 25:6852–6862. [DOI] [PubMed] [Google Scholar]
- 8. Swine Influenza A (H1N1) infection in two children – Southern California, March‐April 2009. MMWR 2009; 58:400–402. [PubMed] [Google Scholar]
- 9. Update: infections with a swine‐origin influenza A (H1N1) virus United States and other countries, April 28, 2009. MMWR 2009; 58:431–433. [PubMed] [Google Scholar]
- 10. Katz JM, Veguilla V, Belser JA et al. The public health impact of avian influenza viruses. Poult Sci 2009; 88:872–879. [DOI] [PubMed] [Google Scholar]
- 11. Tobita K, Sugiura A, Enomote C, Furuyama M. Plaque assay and primary isolation of influenza A viruses in an established line of canine kidney cells (MDCK) in the presence of trypsin. Med Microbiol Immunol 1975; 162:9–14. [DOI] [PubMed] [Google Scholar]
- 12. Yoon KJ, Janke BH, Swalla RW, Erickson G. Comparison of a commercial H1N1 enzyme‐linked immunosorbent assay and hemagglutination inhibition test in detecting serum antibody against swine influenza viruses. J Vet Diagn Invest 2004; 16:197–201. [DOI] [PubMed] [Google Scholar]
- 13. Okuno Y, Tanaka K, Baba K, Maeda A, Kunita N, Ueda S. Rapid focus reduction neutralization test of influenza A and B viruses in microtiter system. J Clin Microbiol 1990; 28:1308–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Noah JW, Severson W, Noah DL, Rasmussen L, White EL, Jonsson CB. A cell‐based luminescence assay is effective for high‐throughput screening of potential influenza antivirals. Antiviral Res 2007; 73:50–59. [DOI] [PubMed] [Google Scholar]
- 15. Bacheler LT, Strehl LL, Neubauer RH, Petteway SR Jr, Ferguson BQ. Stable indicator cell lines exhibiting HIV‐1 tat function. AIDS Res Hum Retroviruses 1989; 5:275–278. [DOI] [PubMed] [Google Scholar]
- 16. Dorsky DI, Wells M, Harrington RD. Detection of HIV‐1 infection with a green fluorescent protein reporter system. J Acquir Immune Defic Syndr Hum Retrovirol 1996; 13:308–313. [DOI] [PubMed] [Google Scholar]
- 17. Rocancourt D, Bonnerot C, Jouin H, Emerman M, Nicolas JF. Activation of a beta‐galactosidase recombinant provirus: application to titration of human immunodeficiency virus (HIV) and HIV‐infected cells. J Virol 1990; 64:2660–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Olivo PD. Detection of herpes simplex virus by measurement of luciferase activity in an infected‐cell lysate. J Virol Methods 1994; 47:117–128. [DOI] [PubMed] [Google Scholar]
- 19. Olivo PD, Frolov I, Schlesinger S. A cell line that expresses a reporter gene in response to infection by Sindbis virus: a prototype for detection of positive strand RNA viruses. Virol 1994; 198:381–384. [DOI] [PubMed] [Google Scholar]
- 20. Stabell EC, Olivo PD. Isolation of a cell line for rapid and sensitive histochemical assay for the detection of herpes simplex virus. J Virol Methods 1992; 38:195–204. [DOI] [PubMed] [Google Scholar]
- 21. Stabell EC, O’Rourke SR, Storch GA, Olivo PD. Evaluation of a genetically engineered cell line and a histochemical beta‐galactosidase assay to detect herpes simplex virus in clinical specimens. J Clin Microbiol 1993; 31:2796–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Proffitt MR, Schindler SA. Rapid detection of HSV with an enzyme‐linked virus inducible system (ELVIS) employing a genetically modified cell line. Clin Diagn Virol 1995; 4:175–182. [DOI] [PubMed] [Google Scholar]
- 23. Olivo PD, Collins PL, Peeples ME, Schlesinger S. Detection and quantitation of human respiratory syncytial virus (RSV) using minigenome cDNA and a Sindbis virus replicon: a prototype assay for negative‐strand RNA viruses. Virol 1998; 251:198–205. [DOI] [PubMed] [Google Scholar]
- 24. Lutz A, Dyall J, Olivo PD, Pekosz A. Virus‐inducible reporter genes as a tool for detecting and quantifying influenza A virus replication. J Virol Methods 2005; 126:13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Neumann G, Zobel A, Hobom G. RNA polymerase I‐mediated expression of influenza viral RNA molecules. Virol 1994; 202:477–479. [DOI] [PubMed] [Google Scholar]
- 26. Wu WH, Pekosz A. Extending the cytoplasmic tail of the influenza a virus M2 protein leads to reduced virus replication in vivo but not in vitro. J Virol 2008; 82:1059–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reed LJ, Muench H. A simple method of estimating fifty percent endpoints. Am J Hyg 1938; 27:493–497. [Google Scholar]
- 28. Neumann G, Watanabe T, Kawaoka Y. Plasmid‐driven formation of influenza virus‐like particles. J Virol 2000; 74:547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zobel A, Neumann G, Hobom G. RNA polymerase I catalysed transcription of insert viral cDNA. Nucleic Acids Res 1993; 21:3607–3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stephenson I, Das RG, Wood JM, Katz JM. Comparison of neutralising antibody assays for detection of antibody to influenza A/H3N2 viruses: an international collaborative study. Vaccine 2007; 25:4056–4063. [DOI] [PubMed] [Google Scholar]
- 31. Ochiai H, Sakai S, Hirabayashi T, Shimizu Y, Terasawa K. Inhibitory effect of bafilomycin A1, a specific inhibitor of vacuolar‐type proton pump, on the growth of influenza A and B viruses in MDCK cells. Antiviral Res 1995; 27:425–430. [DOI] [PubMed] [Google Scholar]
- 32. Felber BK, Pavlakis GN. A quantitative bioassay for HIV‐1 based on trans‐activation. Science 1988; 239:184–187. [DOI] [PubMed] [Google Scholar]
- 33. Iwatsuki‐Horimoto K, Hatta Y, Hatta M et al. Limited compatibility between the RNA polymerase components of influenza virus type A and B. Virus Res 2008; 135:161–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bachmann MF, Ecabert B, Kopf M. Influenza virus: a novel method to assess viral and neutralizing antibody titers in vitro. J Immunol Methods 1999; 225:105–111. [DOI] [PubMed] [Google Scholar]
- 35. Frank AL, Puck J, Hughes BJ, Cate TR. Microneutralization test for influenza A and B and parainfluenza 1 and 2 viruses that uses continuous cell lines and fresh serum enhancement. J Clin Microbiol 1980; 12:426–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Smee DF, Morrison AC, Barnard DL, Sidwell RW. Comparison of colorimetric, fluorometric, and visual methods for determining anti‐influenza (H1N1 and H3N2) virus activities and toxicities of compounds. J Virol Methods 2002; 106:71–79. [DOI] [PubMed] [Google Scholar]
- 37. Eichelberger MC, Hassantoufighi A, Wu M, Li M. Neuraminidase activity provides a practical read‐out for a high throughput influenza antiviral screening assay. Virol J 2008; 5:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gubareva LV, Webster RG, Hayden FG. Detection of influenza virus resistance to neuraminidase inhibitors by an enzyme inhibition assay. Antiviral Res 2002; 53:47–61. [DOI] [PubMed] [Google Scholar]
- 39. Wetherall NT, Trivedi T, Zeller J et al. Evaluation of neuraminidase enzyme assays using different substrates to measure susceptibility of influenza virus clinical isolates to neuraminidase inhibitors: report of the neuraminidase inhibitor susceptibility network. J Clin Microbiol 2003; 41:742–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kiseleva I, Su Q, Toner TJ et al. Cell‐based assay for the determination of temperature sensitive and cold adapted phenotypes of influenza viruses. J Virol Methods 2004; 116:71–78. [DOI] [PubMed] [Google Scholar]